Introduction
Targeted therapies for lung cancer harboring the
epidermal growth factor receptor (EGFR) fusion gene generally
involve the use of EGFR-tyrosine kinase inhibitors (TKIs) and have
a large influence on patient outcome. The EGFR-TKI gefitinib is the
first-line treatment for EGFR-positive non-small cell lung cancer
(NSCLC) patients, however, almost all patients invariably acquire
resistance to the drug (1,2). The mechanisms underlying this acquired
resistance to gefitinib have been extensively explored and mainly
stem from secondary mutations, such as Thr790Met, Asp761Tyr and
Thr854Ala, as well as the activation of bypass or alternative
pathways, including MET gene amplification and
histological/phenotypic transformation (3,4).
Although the T790M mutation and MET gene amplification have been
identified as the major mechanisms of resistance, the full
signaling pathway involved in the process is largely unknown.
The calpain (CAPN) protein family is an important
group of proteases that are activated by calcium, and serves an
important role in cell migration, survival and apoptosis (5–7).
CAPN2, also known as m-CAPN, is one of the most widely investigated
members of this family. Experimental and clinical evidence
indicates that CAPN2 is aberrantly expressed in various tumors, and
has a pivotal function in tumorigenesis, disease progression and
therapy resistance (8–10). It has also been demonstrated that
CAPNs are responsible for activating various downstream proteins
that further facilitate cancer progression. In breast cancer, for
instance, CAPN has been associated with the cleavage of human
EGFR2, which is responsible for cancer pathogenesis (11). Furthermore, the expression of CAPN
was associated with relapse-free survival in HER2-positive breast
cancer patients treated with trastuzumab following adjuvant
chemotherapy (12). A more recent
study supported these functions (13), indicating that high CAPN2 expression
had a clear adverse effect in ovarian cancer patients and was also
important in platinum-based therapy resistance. These results
highlight the predictive significance of CAPN2 in metastasis and
progression in multiple types of cancer. However, the function of
CAPN2 in NSCLC, particularly in EGFR-TKI resistance, has not yet
been elucidated.
In our previous studies, we identified that PLIN2
and Annexin A5 confer gefitinib resistance (12,14).
In the present study, CAPN2 expression was investigated in
gefitinib-resistant lung adenocarcinoma cells. CAPN2 function in
these cells was further evaluated using gene knockdown in
vitro and in vivo. To the best of our knowledge, this is
the first investigation of CAPN2 expression and function during
EGFR-TKI resistance.
Materials and methods
Cell lines and culture
Gefitinib-sensitive PC9 and HCC4006 human lung
adenocarcinoma cells were purchased from the American Type Culture
Collection (Manassas, VA, USA). To induce gefitinib resistance, the
cells were exposed to increasing concentrations of gefitinib
(ranging from 0.01 to 10.0 mM; Selleck Chemicals, Houston, TX,
USA). The viability of gefitinib-resistant PC9R and HCC4006R cells
was unaffected up to 10 µM gefitinib (Fig. 1J and K), whereas the viability of
gefitinib-sensitive PC9 and HCC4006 cells was significantly reduced
in the presence of 0.1 µM gefitinib (Fig. 1D). Gefitinib-sensitive and
-resistant cells were cultured in RPMI-1640 medium (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) supplemented with 10%
heat-inactivated fetal bovine serum and 100 U/ml
penicillin/streptomycin. In order to maintain gefitinib resistance,
1 µM gefitinib was added to the cells every 2 weeks. The cells were
grown as monolayers in a humidified atmosphere of 5% CO2
at 37°C.
Cell proliferation and viability
assay
A Cell Counting Kit-8 (CCK-8; Dojindo Molecular
Technologies, Inc., Kumamoto, Japan) was used to assess cell
proliferation. Briefly, cells were plated in 96-well plates at
~1,000 cells/well with 200 µl culture medium. After 24 h, 10 µl of
the CCK-8 solution was added to each well, and the plates were
incubated for 1 h at 37°C. Finally, the absorbance values at 450 nm
were determined using a microplate reader (Multiskan; Thermo Fisher
Scientific, Inc.), with a reference wavelength of 650 nm. All the
experiments were conducted at least in triplicate.
Lentiviral construction and
infection
For gene expression silencing, two short hairpin RNA
(shRNA) vectors against the CAPN2 genes, namely shCAPN2-1
(TRCN0000272831) and shCAPN2-2 (TRCN0000003543), were obtained from
the RNAi Consortium (Broad Institute, Cambridge, MA, USA).
Lentiviral plasmids, containing GV112-shCAPN2-1 and shCAPN2-2, and
negative control (shNEG) plasmids were obtained from GeneChem Co.,
Ltd. (Shanghai, China). For overexpression of EGFR, lentiviruses
carrying human EGFR (GenBank accession no. NM_005228) and empty
vector were also purchased from GeneChem Co., Ltd. The lentiviral
particles were produced by transfecting 293T cells with the
lentiviral plasmids. For viral infection, gefitinib-resistant PC9R
or HCC4006R cells (5×105 cells/well) were plated in
6-well plates, grown to 50–70% confluence in a humidified
atmosphere of 5% CO2 at 37°C, and subsequently incubated
with RPMI-1640 medium containing the virus and 4 µg/ml polybrene at
a multiplicity of infection of 10. After incubation for 6 h, the
transfection medium was replaced with normal RPMI-1640 medium
supplemented with 10% heat-inactivated fetal bovine serum and 100
U/ml penicillin/streptomycin. After incubation for another 18 h,
the infected cells were then incubated with gefitinib (ranging from
0 to 20.0 µM) for 96 h. The transfection protocol was used in all
the following experiments. For EGFR overexpression rescue
experiments, EGFR overexpressed (PC9R-EGFR) and empty vector
overexpressed (PC9R-vector) PC9R cells were transfected with shNEG
and shCAPN2-1. For survivin or PI3K/AKT inhibition experiments,
PC9R cells following transfection with shNEG, shCAPN2-1 or
shCAPN2-2, were followed by incubation with 10 nM YM155 (a survivin
inhibitor) or 20 µM LY294002 (a phosphoinositide 3-kinase/AKT
inhibitor) for 24 h before CCK8 measurement.
5-Ethynyl-2′-deoxyuridine (EdU)
incorporation assay
Following transfection for 96 h, gefitinib-resistant
PC9R and HCC4006R cells were incubated with 10 µM EdU (Thermo
Fisher Scientific, Inc.) for 4 h before being fixed in 3.7%
formaldehyde in phosphate-buffered saline (PBS) for 15 min at room
temperature. EdU incorporation was detected according to
manufacturer's protocol. After EdU staining, Hoechst 33342 (Thermo
Fisher Scientific, Inc.) staining was detected according to
manufacturer's protocol. The cells were then imaged using a Nikon
A1R confocal laser scanning microscope system (Nikon Corporation,
Tokyo, Japan). Gefitinib-resistant PC9R and HCC4006R cells that
were positive for EdU incorporation and Hoechst 33342 staining were
counted using ImageJ software (version 1.42; National Institutes of
Health, Bethesda, MD, USA), and then the percentage of EdU-positive
cells was calculated.
Detection of apoptosis by terminal
deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)
staining
A TUNEL assay was performed to measure cell
apoptosis using the In Situ Cell Death Detection Kit,
Fluorescein (Roche Molecular Diagnostics, Pleasanton, CA, USA).
Briefly, transfected PC9R cells (5×105 cells/well) were
seeded on glass coverslips coated with poly-L-lysine (ScienCell
Research Laboratories, Inc., San Diego, CA, USA) and treated with 1
µM gefitinib. Following transfection for 96 h, cells were fixed in
3.7% formaldehyde in PBS for 15 min at room temperature. TUNEL
staining was performed according to manufacturer's protocol. After
TUNEL staining, DAPI staining (Thermo Fisher Scientific, Inc.) was
performed. Slides were scanned (Pannoramic P250; 3DHistech Ltd.,
Budapest, Hungary) and viewed using the Pannoramic Viewer software
(3DHistech Ltd.). PC9R cells positive for TUNEL and DAPI staining
were counted using ImageJ software (version 1.42), and the
percentage of TUNEL-positive cells was calculated.
Detection of apoptosis by flow
cytometry
An Annexin V-APC and DAPI double staining kit
(Thermo Fisher Scientific, Inc.) was used to analyze cellular
apoptosis. Transfected PC9R cells were seeded in 6-well plates
(5×105 cells/well) and treated with 1 µM gefitinib.
Cells were then digested with trypsin (Gibco®
trypsin-EDTA; Thermo Fisher Scientific, Inc.), washed with PBS
three times, suspended in 500 µl binding buffer and then incubated
with 5 µl APC-conjugated Annexin V and 3 µl DAPI for 15 min at room
temperature in the dark. The stained cells were detected using a BD
FACSAria II flow cytometer (BD Biosciences, San Jose, CA, USA).
Cell cycle analysis
Transfected PC9R cells were seeded in 6-well plates
(5×105 cells/well) and treated with 1 µM gefitinib.
Subsequently, cells were collected, washed with PBS and fixed in
70% ethanol for 24 h at 4°C. The fixed cells were then stained with
propidium iodide and RNase (FS9527-100; Cell Cycle Fast detecting
kit; Fusion Biotech, Shanghai, China) in the dark for 30 min at
room temperature. Finally, the cell cycle distribution was analyzed
by flow cytometry using a BD FACSAria II device (BD
Biosciences).
Measurement of mitochondrial membrane
potential
In order to examine changes in the mitochondrial
membrane potential, a MitoProbe™ JC-1 assay kit (Thermo Fisher
Scientific, Inc.) was used, according to the manufacturer's
protocol. A BD FACSAria II flow cytometer was used to obtain the
results. In healthy mitochondria, JC-1 forms J-aggregates emitting
red fluorescence at 590 nm, while J-monomers emit green
fluorescence at 490 nm in depolarized mitochondria; thus,
mitochondria damage was indicated by an increase in the ratio of
J-monomers.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted using TRIzol reagent (Thermo
Fisher Scientific, Inc.) and quantified using the NanoDrop 2000
spectrophotometer (Thermo Fisher Scientific, Inc.). An amount of 1
µg RNA was used for reverse transcription by PrimeScript™ RT
Reagent Kit (RR037A; Takara, Osaka, Japan). The cDNA (20 ng) was
subsequently used as the template for qPCR. The amplification
cycling parameters (40 cycles) were as follows: 15 sec at 95°C, 15
sec at 60°C and 45 sec at 72°C. The following primer sequences were
used in the present study: CAPN2 sense, 5′-CGAGAGGGCCATCAAGTACC-3′
and antisense, 5′-TAGGGCCCCAACTCCTTGAA-3′; cyclin-dependent kinase
inhibitor 1A (CDKN1A) sense, 5′-CTGGGGATGTCCGTCAGAAC-3′ and
antisense, 5′-CATTAGCGCATCACAGTCGC-3′; growth arrest and DNA damage
inducible α (GADD45A) sense, 5′-CCATGCAGGAAGGAAAACTATG-3′ and
antisense, 5′-CCCAAACTATGGCTGCACACT-3′; cyclin-dependent kinase 1
(CDK1) sense, 5′-TAGCGCGGATCTACCATACC-3′ and antisense,
5′-CATGGCTACCACTTGACCTG-3′; CDK2 sense, 5′-GCCCTATTCCCTGGAGATTC-3′
and antisense, 5′-CAAGCTCCGTCCATCTTCAT-3′; and β-actin sense,
5′-CTGGCACCCAGCACAATG-3′ and antisense,
5′-CCGATCCACACGGAGTACTTG-3′. Gene expression was normalized to that
of β-actin and calculated with the 2−ΔΔCq method
(15). The RT-qPCR assay was
performed at least three separate times in triplicate.
Western blot assay
Total protein from PC9, PC9R, HCC4006 and HCC4006R
cells was extracted using RIPA lysis buffer and the protein
concentration was determined using BCA assay (Shanghai Zhuoli
Biotechnology Co., Ltd., Shanghai, China). Next, total protein was
separated on polyacrylamide gels (5% stacking gel and 12%
separating gel), and transferred to polyvinylidene difluoride
membranes (PVDF). The membranes were then incubated at 4°C
overnight with anti-CAPN2 (1:2,000) and keratin-5 (KRT5, 1:1,000)
antibodies purchased from Abcam (Cambridge, MA, USA), as well as
EGFR (1:1,000), protein kinase B (AKT, 1:1,000), phosphorylated AKT
(pAKT; Ser473, 1:1,000), surviving (1:1,000), cleaved caspase-3
(Asp175, 1:1,000), cleaved poly-ADP-ribose polymerase (PARP;
Asp214, 1:1,000) and β-actin (1:2,000) antibodies obtained from
Cell Signaling Technology, Inc. (Danvers, MA, USA). Subsequently,
membranes were incubated with horseradish peroxidase-conjugated
goat anti-rabbit (1:2,000) or anti-mouse immunoglobulin G secondary
antibodies (1:2,000) obtained from Jackson ImmunoResearch, Inc.
(West Grove, PA, USA) at room temperature for 1 h. Proteins were
visualized using Pierce ECL western blotting substrate (Thermo
Fisher Scientific, Inc.) and autoradiography. The blots were
analyzed using Quantity One, version 4.6 software (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
cDNA array screening
Total RNA was extracted using RNeasy Plus Mini Kit
(Qiagen, Valencia, CA, USA) from PC9R cells expressing shCAPN2-1 or
negative control shRNA, reverse transcribed, and amplified using
OneArray Plus RNA Amplification Kit (Phalanx Biotech Group,
Taiwan). Cy5-labeled amplicons were hybridized to Human Whole
Genome OneArray (Phalanx Biotech Group), imaged on a G2505C Agilent
Microarray Scanner (Agilent Technologies, Santa Clara, CA, USA),
and analyzed in Resolver (Rosetta Biosoftware, Seattle, WA,
USA).
Evaluation of tumorigenicity
The animal experiments performed in the present
study were approved by the Institutional Animal Care and Use
Committee at Zhongshan Hospital, Fudan University (Shanghai,
China). Eight male BALB/c nude mice (age, 4–6 weeks; weight, 18–20
g) were obtained from the Shanghai Experimental Animal Center of
the Chinese Academy of Sciences (Shanghai, China), and were housed
in cages with access to food and water in a temperature-controlled
room with a 12 h dark/light cycle. PC9R cells transfected with
shNEG or shCAPN2-1 were subcutaneously injected into the right
flanks of nude mice without any treatment. Tumor volume was
monitored and calculated according to the following formula:
Volume=length × width2/2. After ~1 month, nude mice were
sacrificed by cervical dislocation after anesthesia, and the tumors
were isolated, weighed and embedded in paraffin. Ki67
immunohistochemical staining was performed on the sections of the
paraffin-embedded tissue to identify the proliferating cells in the
xenograft tumors. Ki67-positive cells were quantified in randomly
selected fields from each tissue section using ImageJ software.
Statistical analysis
Data are expressed as the mean ± standard deviation
of at least three independent experiments. One-way analysis of
variance followed by Bonferroni's multiple comparison test was used
to compare different groups, while Student's t-test was used for
comparisons between two groups. A P-value of <0.05 was
considered to denote a statistically significant difference.
Results
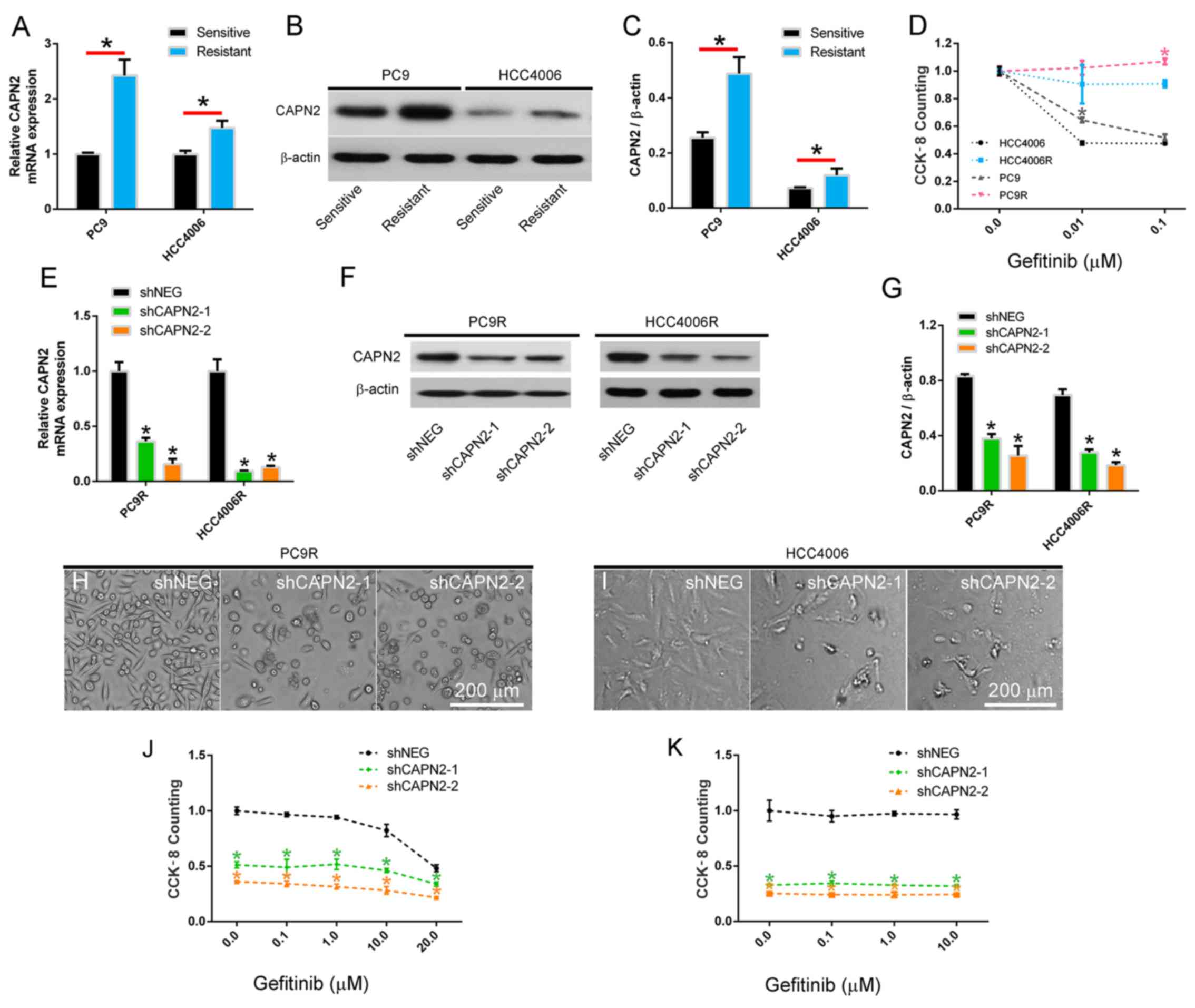
CAPN2 is upregulated in
gefitinib-resistant cells and mediates gefitinib resistance
Two gefitinib resistant NSCLC cell lines, designated
PC9R and HCC4006R, were established by continuously exposing PC9
and HCC4006 cells to increasing concentrations of gefitinib. Next,
CAPN2 mRNA and protein expression was investigated, and the results
revealed that CAPN2 was significantly upregulated in the PC9R and
HCC4006R cell lines compared with the normal PC9 and HCC4006 cell
lines, respectively (Fig. 1A-C),
indicating that CAPN2 may serve an important role in gefitinib
resistance. To confirm this, gefitinib resistance was examined in
these four cell lines by CCK-8 assay, and it was observed that PC9
cells, which expressed relatively higher CAPN2 protein in
comparison with HCC4006 cells, had a higher proliferation rate
compared with that of HCC4006 cells in the presence of 0.01 µM
gefitinib. By contrast, PC9R cells, which expressed relatively
higher CAPN2 protein in comparison with HCC4006R cells, had a
higher proliferation rate than that of HCC4006R cells in the
presence of 0.1 µM gefitinib (Fig.
1D).
To further evaluate the role of CAPN2 in gefitinib
resistance, PC9R and HCC4006R cells were transfected with shCAPN2-1
or shCAPN2-2 to silence CAPN2 expression. shNEG was transfected
into cells as a control. The RT-qPCR and western blot analyses
demonstrated that shCAPN2-1 and shCAPN2-2 transfection
significantly inhibited CAPN2 expression compared with that in
shNEG transfected cells (Fig.
1E-G). Furthermore, a morphological examination indicated that
CAPN2 knockdown reduced the number of gefitinib-resistant cells, as
observed by the increase in gefitinib effectiveness (1 µM, for 96
h) in PC9R and HCC4006R cells infected (24 h) with shCAPN2-1 or
shCAPN2-2 compared with that in the shNEG (Fig. 1H and I). It was also confirmed that
CAPN2 knockdown reduced the viability of gefitinib-resistant cells
by treating silenced and control cells with different gefitinib
concentrations (ranging between 0.1 and 20.0 µM; Fig. 1J and K).
CAPN2 knockdown inhibits
gefitinib-resistant cell proliferation in vitro and in vivo
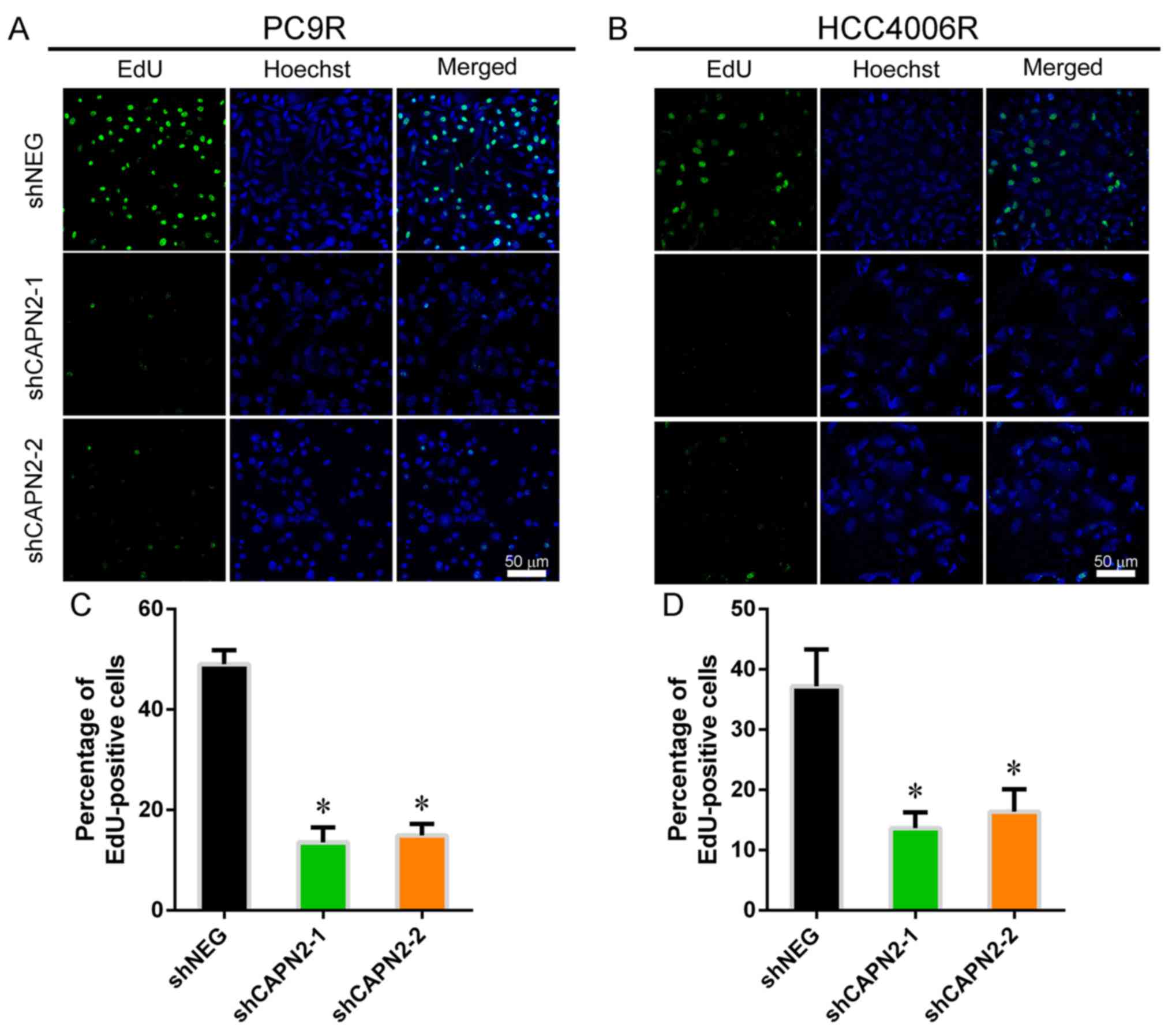
To investigate the effect of CAPN2 on
gefitinib-resistant cell proliferation, EdU incorporation assay was
performed (Fig. 2A and B). CAPN2
knockdown appeared to suppress the proliferation of
gefitinib-resistant cells in the presence of gefitinib (Fig. 2C and D), implying that CAPN2
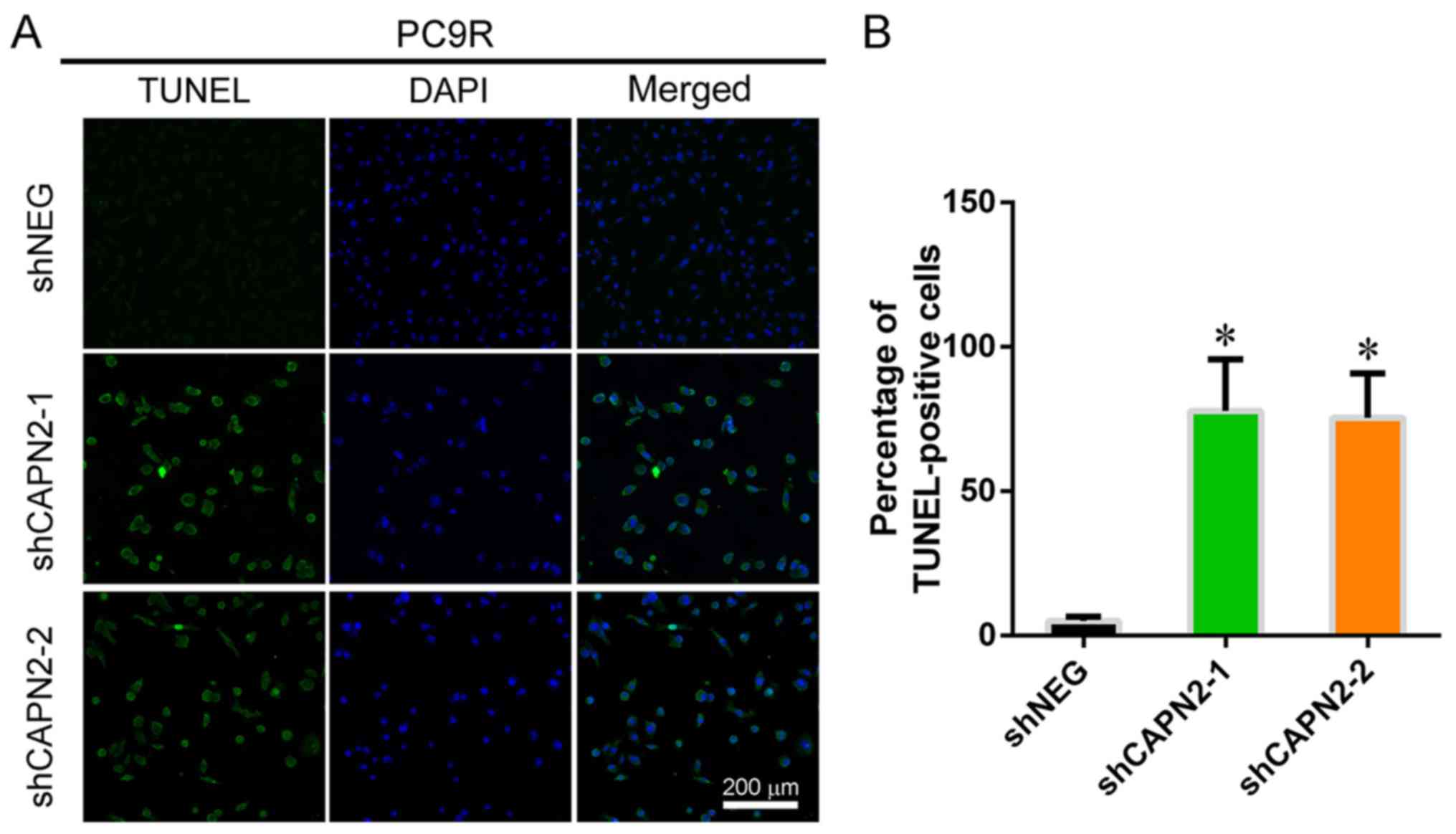
promotes proliferation in gefitinib-resistant cells. Furthermore,
TUNEL staining demonstrated that CAPN2 knockdown significantly
increased the apoptosis of gefitinib-resistant PC9R cells in the
presence of gefitinib (Fig. 3A and
B).
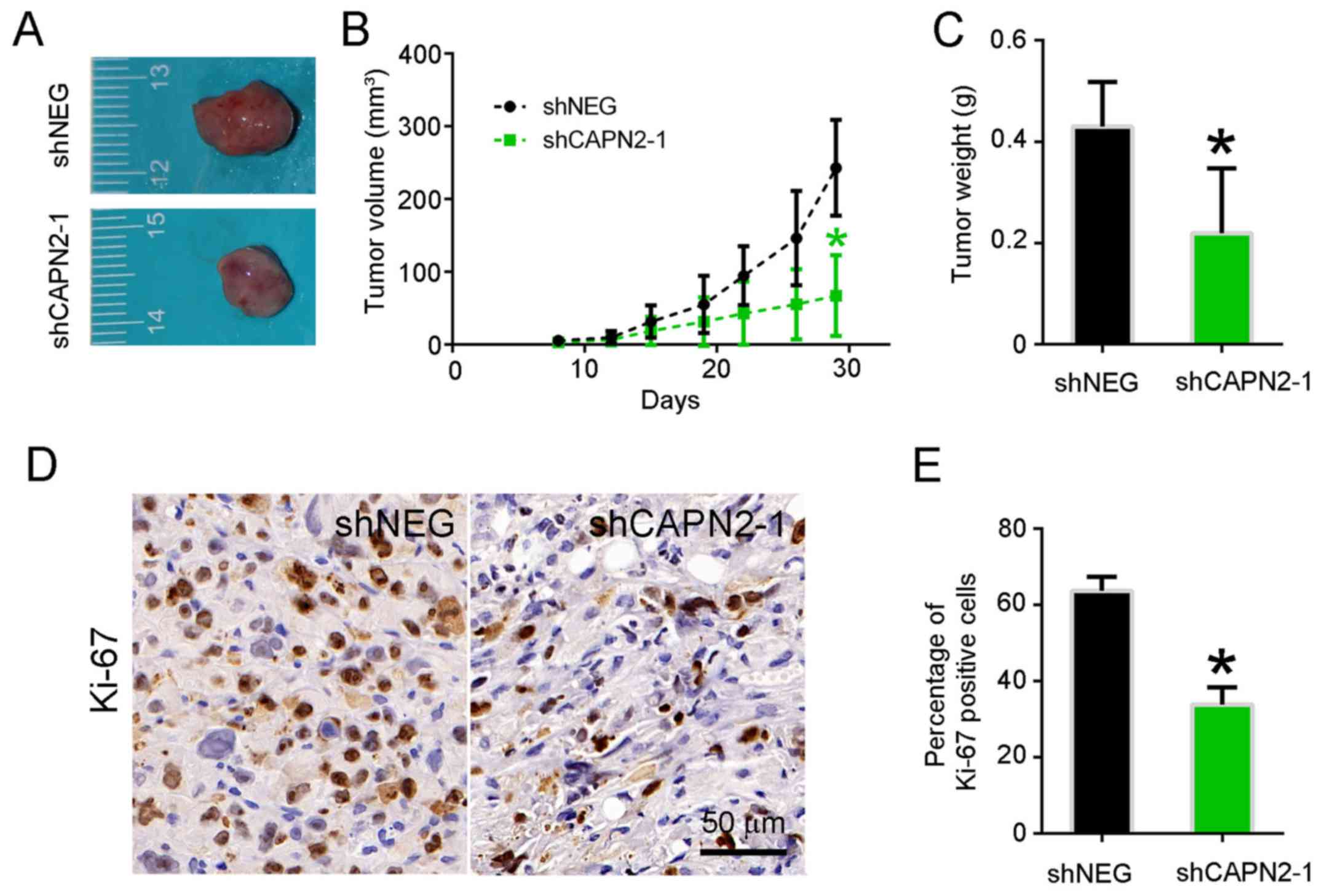
To determine whether CAPN2 knockdown suppresses lung
tumor growth in vivo, PC9R cells transfected with shCAPN2-1
and shNEG were injected into nude mice. Visible tumors were
observed in animals after 1 week; however, the volume of tumors
derived from CAPN2-silenced cells was evidently smaller in
comparison with that of tumors derived from control cells (Fig. 4A). A similar trend was observed at 1
month after transfection in terms of the tumor volume and weight
(Fig. 4B and C). Accordingly, the
abundance of proliferating Ki67-positive cells was significantly
lower in CAPN2-deficient tumors as compared with that in control
xenografts (Fig. 4D and E).
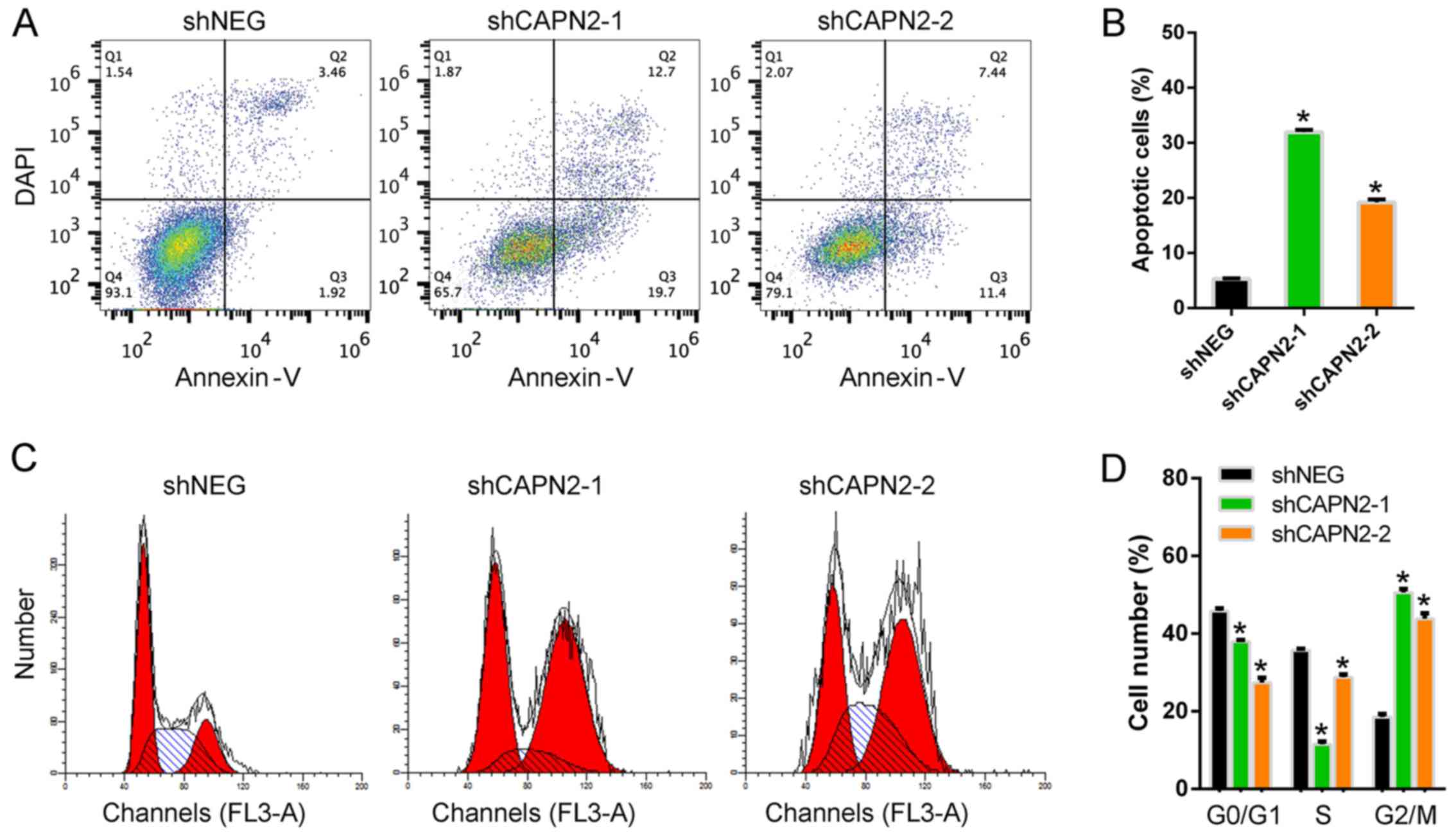
CAPN2 silencing promotes apoptosis and
cell cycle arrest in gefitinib-resistant cells via caspase
activation and mitochondrial dysfunction
To further confirm the function of CAPN2 during
gefitinib resistance, the cell apoptosis levels were analyzed. Upon
treatment with gefitinib, the apoptotic rate in CAPN2-silenced PC9R
cells was significantly higher than that of control cells (Fig. 5A and B). Furthermore, the number of
cells in the G0/G1 and S phases was markedly
decreased upon CAPN2 silencing, while the number of cells in the
G2/M phase was increased, indicating cell cycle arrest
(Fig. 5C and D).
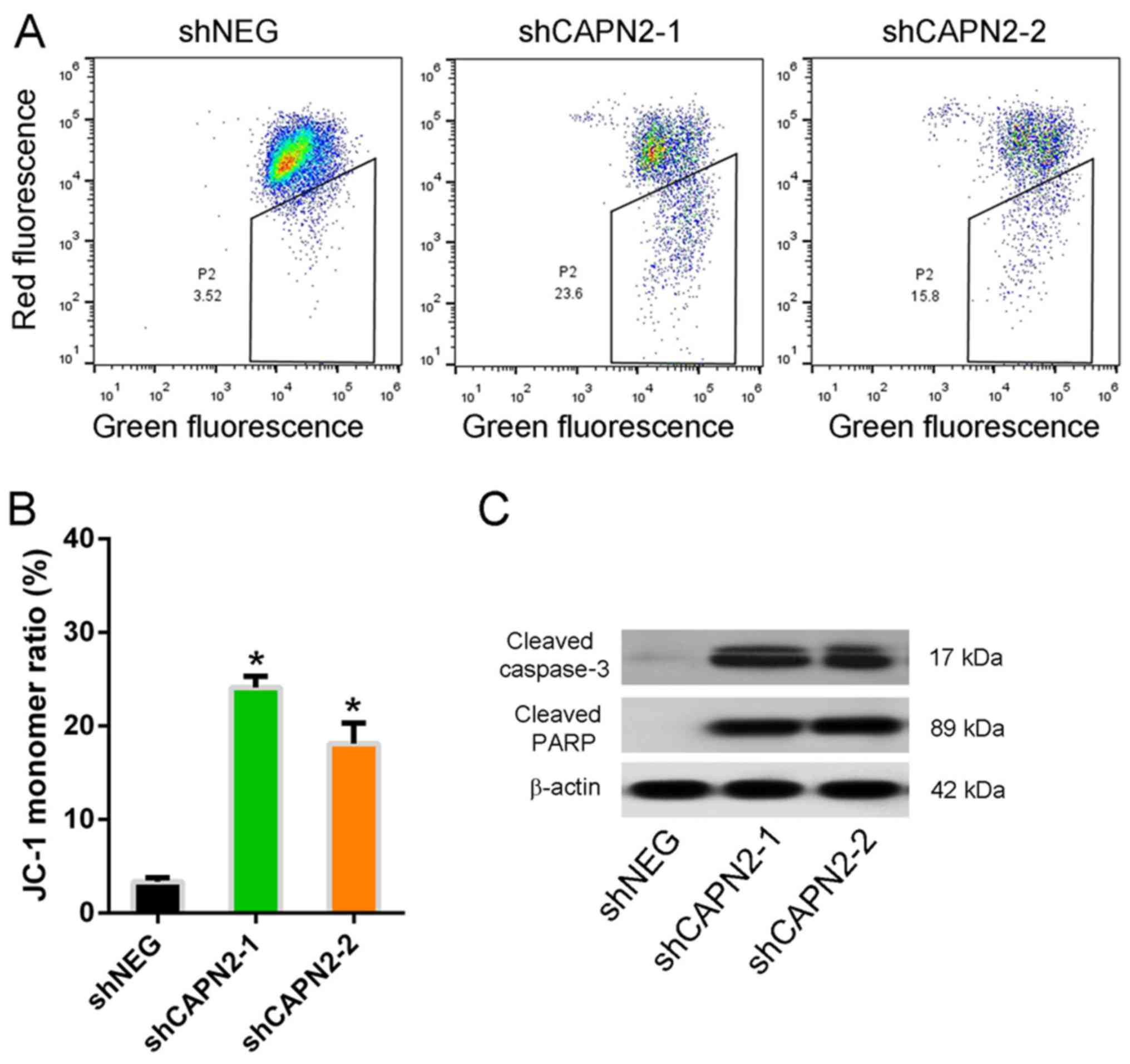
The mitochondrial integrity was also assessed by
staining cells with JC-1, a dye that emits red fluorescence as it
aggregates in intact mitochondria, but emits green fluorescence as
a monomer in damaged mitochondria. The JC-1 monomer ratio in
CAPN2-silenced PC9R cells was higher compared with that of control
cells in the presence of gefitinib (Fig. 6A and B), indicating mitochondrial
dysfunction. Furthermore, cleaved caspase-3 and cleaved PARP, two
apoptotic proteins, were upregulated in CAPN2-depleted cells
compared with the levels in control cells (Fig. 6C), indicating that CAPN2-depleted
cells undergo apoptosis (16).
CAPN2 regulates gefitinib-resistance
via the EGFR/AKT/survivin signaling pathway
To evaluate the molecular mechanism underlying
CAPN2-mediated gefitinib resistance, a whole human cDNA array was
used, and changes in CAPN2-associated downstream signaling pathways
upon CAPN2 silencing were investigated (Fig. 7A). Western blotting confirmed that
EGFR, pAKT, survivin and KRT5 levels were evidently decreased in
cells transfected with shCAPN2-1 or shCAPN2-2 compared with those
in cells transfected with shNEG (Fig.
7B). Furthermore, RT-qPCR analysis confirmed that the
expression levels of the cell cycle genes CDKN1A and GADD45A were
significantly increased, whereas CDK1 and CDK2 levels were
decreased in PC9R cells transfected with shCAPN2-1 or shCAPN2-2
compared with those in cells transfected with shNEG (Fig. 7C). Given that the EGFR/AKT/survivin
pathway serves a crucial role in NSCLC progression (17,18),
it is likely that CAPN2 confers gefitinib resistant through EGFR
signaling.
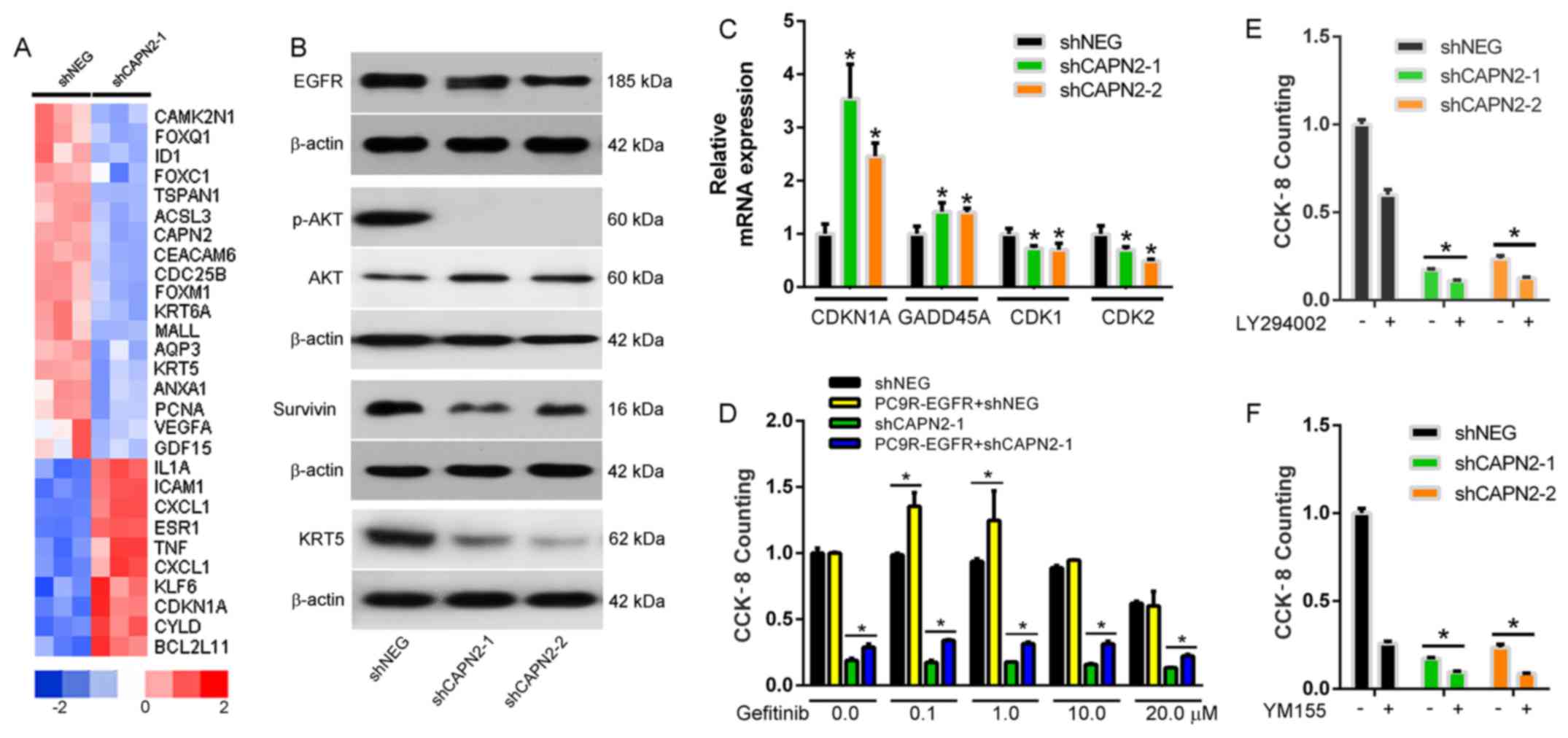 | Figure 7.CAPN2 regulates gefitinib-resistance
via the EGFR/AKT/survivin signal pathway. (A) Heatmap of downstream
CAPN2 target proteins determined by a whole human cDNA array. (B)
EGFR, pAKT, AKT, survivin and KRT5 protein expression was measured
by western blotting. (C) CDKN1A, GADD45A, CDK1 and CDK2 gene
expression was measured by reverse transcription-quantitative
polymerase chain reaction. *P<0.05 vs. negative control. (D) The
viability of PC9R cells transfected with EGFR overexpressing vector
and/or shCAPN-1 in the presence of various concentrations of
gefitinib was determined by cell counting kit-8 assay. Viability of
PC9R cells following transfection with shNEG, shCAPN2-1 or
shCAPN2-2, followed by incubation with (E) 20 µM LY294002 (a
phosphoinositide 3-kinase/AKT inhibitor) or (F) 10 nM YM155 (a
survivin inhibitor). *P<0.05 between two groups. CAPN2, calpain
2; sh, short hairpin RNA; NEG, negative control; EGFR, epidermal
growth factor receptor; AKT, protein kinase B; KRT5, keratin-5;
CDKN1A, cyclin-dependent kinase inhibitor 1A; GADD45A, growth
arrest and DNA damage inducible α; CDK, cyclin-dependent
kinase. |
To evaluate this possibility, a rescue assay was
performed in the PC9R cells. It was observed that overexpression of
EGFR in PC9R cells increased cell viability in the presence of
gefitinib (Fig. 7D). However,
overexpression of EGFR in the PC9R-shCAPN2 cells also rescued the
cell viability in the presence of gefitinib. These results suggest
that EGFR overexpression partly reverses CAPN2 depletion-induced
apoptosis. Furthermore, inhibition of phosphoinositide 3-kinase
(PI3K)/AKT (Fig. 7E) or survivin
(Fig. 7F) in the PC9R-shCAPN2 cells
was able to further induce cell apoptosis. Taken together, all
these findings indicated that CAPN2 stimulates gefitinib resistance
mainly via activation of the EGFR/AKT/survivin signaling
pathway.
Discussion
Drug resistance is a major cause of cancer treatment
failure. Targeted EGFR drugs, such as gefitinib, appear to be
effective for approximately 1 year, but resistance is inevitably
developed shortly after this period. CAPN2 has been demonstrated to
serve a role in altering the cellular response to certain cancer
therapies (9,10,19).
However, the events and mechanisms involved in these processes
remain incompletely understood. In the present study, it was
observed that CAPN2 is strongly associated with
gefitinib-resistance, with CAPN2 mRNA and protein expression levels
being significantly increased in gefitinib-resistant cell lines. In
addition, CAPN2 was found to regulate gefitinib-resistant cell
proliferation, mitochondrial function and apoptosis, largely
involving dysregulation of the PI3K/AKT/survivin pathway.
Previously, high CAPN2 expression was reported to be
significantly associated with platinum resistant tumors in ovarian
cancer (13), while CAPN2
degradation mediated irinotecan secondary resistance in colorectal
cancer xenografts (20). Consistent
with these previous studies, CAPN2 was observed to be upregulated
in gefitinib-resistant cells in the present study. Furthermore,
CAPN2 knockdown inhibited gefitinib-resistant cell proliferation
in vitro and in vivo.
In addition to its effect on proliferation in drug
resistant cells, CAPN2 knockdown also appears to affect the
mitochondrial function. The mitochondria are essential during
various cellular processes and function to protect cells from
apoptosis, genomic instability, inflammation, abnormal
bioenergetics, oxidative stress and metastasis (21). Cleaved caspase-3 and cleaved PARP
are two critical targets of the mitochondria-mediated apoptosis
pathway (22). Indeed, a previous
study has demonstrated that mitochondria-mediated caspase-dependent
apoptosis is involved in the anticancer activity against human lung
cancer cells (23). In the present
study, the results revealed that CAPN2 knockdown induced
gefitinib-resistant cell apoptosis and cell cycle arrest in
conjunction with caspase activation and increased mitochondrial
dysfunction. This is not altogether surprising as CAPNs are known
to be responsible for mitochondrial membrane permeabilization
(24), which subsequently alters
various mitochondrial processes.
Notably, using a whole human cDNA array in the
current study, EGFR was identified to be a downstream signaling
molecule of CAPN2. The EGFR downstream signaling route includes the
MAPK, JAK/STAT and PI3K/AKT/mTOR pathways among others (6). Previous studies have demonstrated that
AKT activation contributes to EGFR-TKI resistance in EGFR
mutation-positive lung cancer (3,20,25),
indicating that the PI3K/AKT pathway is an important target for
overcoming EGFR-TKI resistance. It has also been reported that
CAPN2 activates AKT signaling in pulmonary artery smooth muscle
cells (9), which is consistent with
the changes in AKT phosphorylation observed in the present study.
Survivin is an apoptosis inhibitor involved in tumorigenesis, and
inhibition of the EGFR/PI3K/AKT pathway appears to downregulate its
expression in EGFR mutation-positive NSCLC cells (17). This is consistent with the findings
of the present study, which demonstrated that CAPN2 knockdown in
gefitinib-resistant cells repressed survivin expression.
Furthermore, KRT5 is a well-recognized marker for basal cells
(26,27) and is overexpressed in various cancer
types (7). Similar to surviving
expression, the current study observed that CAPN2 inhibition
reduced KRT5 expression, suggesting that CAPN2 may regulate cell
differentiation via KRT5 inhibition.
In conclusion, the present study investigated the
role of CAPN2 in gefitinib-resistant lung adenocarcinoma cells and
revealed that CAPN2 expression is upregulated during drug
resistance. CAPN2 appeared to regulate PC9R cell proliferation and
apoptosis by triggering mitochondrial caspase-dependent cell death
pathways and mainly targeting EGFR/AKT/survivin signaling.
Furthermore, downregulation of CAPN2 was observed to inhibit PC9R
cell proliferation and induce cell apoptosis both in vitro
and in vivo. These data demonstrate that CAPN2 functions as
a facilitator of gefitinib resistance, largely via the activation
of EGFR/AKT/survivin signaling pathway. Thus, the present study
supports the development of drugs targeting CAPN2 as a means to
enhance the effectiveness of cancer therapy and reduce drug
resistance.
Acknowledgements
The authors would like to thank Mr. Lei Gao, a
member of Bai Lab, for suggestions and discussions.
Funding
This study was supported by grants from the National
Key R&D Plan (no. 2016YFC1304104), the National Major
Scientific and Technological Special Project for ‘Significant New
Drugs Development’ (no. 2018ZX09201002-006), the Natural Science
Foundation of China (nos. 81400018, 81570028 and 81770039), the
Shandong Province Natural Science Foundation (no. ZR2017PH066), the
Shanghai Science and Technology Committee Grant (15DZ1941100), the
National Key Technology Research and Development Program of the
Ministry of Science and Technology of China (no. 2013BAI09B00), the
Zhongshan Hospital Clinical Research Foundation (no. 2016ZSLC05),
and the Shanghai Three-Year Plan of the Key Subjects Construction
in Public Health-Infectious Diseases and Pathogenic Microorganism
(no. 15GWZK0102).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ, CB and QH were involved in the conception of the
study and supervised the conduction of the entire project. GZ, TF,
MC and JL performed the research, analyzed the data and prepared
the manuscript. All authors have read the final version of the
manuscript and are in agreement for publication upon
acceptance.
Ethics approval and consent to
participate
Experiments were approved by the Research Ethics
Committee of Zhongshan Hospital, Fudan University (Shanghai, China)
and performed according to relevant guidelines and regulations.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tan CS, Gilligan D and Pacey S: Treatment
approaches for EGFR-inhibitor-resistant patients with
non-small-cell lung cancer. Lancet Oncol. 16:e447–e459. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhong WZ, Zhou Q and Wu YL: The resistance
mechanisms and treatment strategies for EGFR-mutant advanced
non-small-cell lung cancer. Oncotarget. 8:71358–71370. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Calvayrac O, Mazieres J, Figarol S,
Marty-Detraves C, Raymond-Letron I, Bousquet E, Farella M,
Clermont-Taranchon E, Milia J, Rouquette I, et al: The RAS-related
GTPase RHOB confers resistance to EGFR-tyrosine kinase inhibitors
in non-small-cell lung cancer via an AKT-dependent mechanism. EMBO
Mol Med. 9:238–250. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang L and Fu L: Mechanisms of resistance
to EGFR tyrosine kinase inhibitors. Acta Pharm Sin B. 5:390–401.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Moreira JB, Wohlwend M, Alves MN, Wisløff
U and Bye A: A small molecule activator of AKT does not reduce
ischemic injury of the rat heart. J Transl Med. 13:762015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sordella R, Bell DW, Haber DA and
Settleman J: Gefitinib-sensitizing EGFR mutations in lung cancer
activate anti-apoptotic pathways. Science. 305:1163–1167. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Camilo R, Capelozzi VL, Siqueira SA and
Del Carlo Bernardi F: Expression of p63, keratin 5/6, keratin 7,
and surfactant-A in non-small cell lung carcinomas. Hum Pathol.
37:542–546. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ho WC, Pikor L, Gao Y, Elliott BE and
Greer PA: Calpain 2 regulates Akt-FoxO-p27(Kip1) protein signaling
pathway in mammary carcinoma. J Biol Chem. 287:15458–15465. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li P, Miao C, Liang C, Shao P, Wang Z and
Li J: Silencing CAPN2 expression inhibited castration-resistant
prostate cancer cells proliferation and invasion via AKT/mTOR
signal pathway. Biomed Res Int. 2017:25936742017.PubMed/NCBI
|
|
10
|
Liu B, Zhou Y, Lu D, Liu Y, Zhang SQ, Xu
Y, Li W and Gu X: Comparison of the protein expression of
calpain-1, calpain-2, calpastatin and calmodulin between gastric
cancer and normal gastric mucosa. Oncol Lett. 14:3705–3710. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li ST, Chen NN, Qiao YB, Zhu WL, Ruan JW
and Zhou XZ: SC79 rescues osteoblasts from dexamethasone though
activating Akt-Nrf2 signaling. Biochem Biophys Res Commun.
479:54–60. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou J, Chang M, Li J, Fang T, Hu J and
Bai C: Knockdown of annexin A5 restores gefitinib sensitivity by
promoting G2/M cell cycle arrest. Respir Res. 19:962018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Storr SJ, Safuan S, Woolston CM,
Abdel-Fatah T, Deen S, Chan SY and Martin SG: Calpain-2 expression
is associated with response to platinum based chemotherapy,
progression-free and overall survival in ovarian cancer. J Cell Mol
Med. 16:2422–2428. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chang M, Hu J, Fang T, Li J, Song Y, Bai C
and Zhou J: PLIN2 confers gefitinib resistance by inhibiting cell
apoptosis via activation of EGFR/AKT/survivin in PC9R cells.
Oncotarget. Jan 10–2018.(Epub ahead of print). doi:
10.18632/oncotarget.24153.
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oliver FJ, de la Rubia G, Rolli V,
Ruiz-Ruiz MC, de Murcia G and Murcia JM: Importance of
poly(ADP-ribose) polymerase and its cleavage in apoptosis: Lesson
from an uncleavable mutant. J Biol Chem. 273:33533–33539. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Okamoto K, Okamoto I, Hatashita E, Kuwata
K, Yamaguchi H, Kita A, Yamanaka K, Ono M and Nakagawa K:
Overcoming erlotinib resistance in EGFR mutation-positive non-small
cell lung cancer cells by targeting survivin. Mol Cancer Ther.
11:204–213. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Okamoto K, Okamoto I, Okamoto W, Tanaka K,
Takezawa K, Kuwata K, Yamaguchi H, Nishio K and Nakagawa K: Role of
survivin in EGFR inhibitor-induced apoptosis in non-small cell lung
cancers positive for EGFR mutations. Cancer Res. 70:10402–10410.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miao C, Liang C, Tian Y, Xu A, Zhu J, Zhao
K, Zhang J, Hua Y, Liu S, Dong H, et al: Overexpression of CAPN2
promotes cell metastasis and proliferation via AKT/mTOR signaling
in renal cell carcinoma. Oncotarget. 8:97811–97821. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jacobsen K, Bertran-Alamillo J, Molina MA,
Teixidó C, Karachaliou N, Pedersen MH, Castellví J, Garzón M,
Codony-Servat C, Codony-Servat J, et al: Convergent Akt activation
drives acquired EGFR inhibitor resistance in lung cancer. Nat
Commun. 8:4102017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lim W, Yang C, Jeong M, Bazer FW and Song
G: Coumestrol induces mitochondrial dysfunction by stimulating ROS
production and calcium ion influx into mitochondria in human
placental choriocarcinoma cells. Mol Hum Reprod. 23:786–802. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sivalingam KS, Paramasivan P, Weng CF and
Viswanadha VP: Neferine potentiates the antitumor effect of
cisplatin in human lung adenocarcinoma cells Via a
mitochondria-mediated apoptosis pathway. J Cell Biochem.
118:2865–2876. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Losuwannarak N, Sritularak B and
Chanvorachote P: Cycloartobiloxanthone induces human lung cancer
cell apoptosis via mitochondria-dependent apoptotic pathway. In
Vivo. 32:71–78. 2018.PubMed/NCBI
|
|
24
|
Molina JR, Yang P, Cassivi SD, Schild SE
and Adjei AA: Non-small cell lung cancer: Epidemiology, risk
factors, treatment, and survivorship. Mayo Clinic Proc. 83:584–594.
2008. View Article : Google Scholar
|
|
25
|
Wang B, Jiang H, Wang L, Chen X, Wu K,
Zhang S, Ma S and Xia B: Increased MIR31HG lncRNA expression
increases gefitinib resistance in non-small cell lung cancer cell
lines through the EGFR/PI3K/AKT signaling pathway. Oncol Lett.
13:3494–3500. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rock JR, Onaitis MW, Rawlins EL, Lu Y,
Clark CP, Xue Y, Randell SH and Hogan BL: Basal cells as stem cells
of the mouse trachea and human airway epithelium. Proc Natl Acad
Sci USA. 106:12771–12775. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zuo W, Zhang T, Wu DZ, Guan SP, Liew AA,
Yamamoto Y, Wang X, Lim SJ, Vincent M, Lessard M, et al:
p63+Krt5+ distal airway stem cells are
essential for lung regeneration. Nature. 517:616–620. 2015.
View Article : Google Scholar : PubMed/NCBI
|