Introduction
First-generation epidermal growth factor receptor
(EGFR)-tyrosine kinase inhibitors (TKIs), gefitinib and erotinib,
are used to treat patients with advanced non-small cell lung cancer
(NSCLC) that harbors an EGFR-activating mutation, especially a
deletion in exon 19 and an L858R mutation in exon 21. However,
despite their initial response to EGFR-TKI treatment, most patients
eventually develop resistance and consequently relapse. The
mechanisms by which resistance to EGFR-TKIs is acquired include a
mutation of EGFR T790M within exon 20. Although EGFR T790M-specific
EGFR-TKIs have been developed, resistance also occurs with this
class of EGFR-TKIs. The distinct mechanisms by which resistance to
EGFR-TKIs is acquired are the activation of bypass signaling (MET,
AXL and/or ERBb2) and their downstream pathways (PI3K, AKT and/or
MEK). Therefore, new strategies to overcome multifactorial
resistance to EGFR-TKIs are needed to improve the efficacy of this
treatment.
Heat shock protein 90 (HSP90), which is highly
conserved evolutionarily and ubiquitously expressed, has been
attributed to the folding, stabilization, and proteolytic
degradation of client oncoproteins involved in the proliferation of
tumors (1). Inhibition of HSP90
results in degradation of its clients, including receptor tyrosine
kinases (RTKs), and its downstream signaling molecules, such as
AKT, MEK and Src (2). Therefore,
HSP90 represents an engaging molecular target for anticancer
therapy and has been under preclinical and clinical development for
the treatment of NSCLC (3).
Although several HSP90 inhibitors have shown promising results in
preclinical research and 17-AAG, the first HSP90 inhibitor, has
entered into a phase I clinical trial, this drug might not be
approved to treat lung cancer because of its poor drug-like
properties, such as poor solubility, poor patient enrichment,
suboptimal target inhibition, and off-target toxicities (4).
The function of HSP90 is regulated by
post-translational modification. Acetylation of HSP90 has been
observed in cancer cells treated with histone deacetylase
inhibitors (HDACIs), leading to destabilization of its client
proteins. First, HDACIs emerged as potential multifunctional agents
that regulate chromatin remodeling and are crucial to the
epigenetic regulation of various genes, such as tumor suppressors
and oncogenes. Later, it was discovered that HDAC6 is located in
the cytosol and modulates the acetylation of cytosolic proteins,
including HSP90, p53 and tubulin. Recent data suggest that HDACIs
can increase sensitivity and reverse resistance to EGFR-TKIs in
lung cancer cells by inducing E-cadherin expression in those cells
(5). Thus, ongoing research on
incorporating HDACIs into NSCLC treatment concentrates on combining
HDACIs with EGFR-TKIs (6).
Vorinostat, also called suberoylanilide hydroxamic acid (SAHA), is
an inhibitor of class I and II histone deacetylases that regulate
the transcription of genes involved in cell survival and apoptosis
and has demonstrated considerable antigrowth effects on NSCLC cells
(7). Given the potential synergy
between HDACIs and EGFR-TKIs, we conducted a study in which
gefitinib and vorinostat were combined to target HSP90 in NSCLC
with an EGFR mutation.
Materials and methods
Cell culture and reagents
The human NSCLC cell line PC9 and
gefitinib-resistant PC9 (PC9GR) cells were provided by Dr Rho (Asan
Medical Center, Seoul, Korea) (8,9). All
cell lines were cultured in Gibco® RPMI-1640 medium
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) containing 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) and
1% penicillin/streptomycin (Gibco; Thermo Fisher Scientific, Inc.)
at 37°C in an atmosphere containing 5% CO2. The
following compounds were used in this study: Gefitinib (Iressa,
AstraZeneca, London, UK); vorinostat (Crystal Genomics, Inc.,
Seoul, Korea); 4-(2-aminoethyl)-benzenesulfonyl fluoride
hydrochloride (AEBSF, Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany); and Z-VAD-FMK, Z-LEHD-FMK, Z-DEVD-FMK and Z-IETD-FMK
(R&D Systems, Minneapolis, MN, USA). All reagents were
purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany)
unless otherwise specified.
Drug treatment
The cells were seeded at a density of
3×103 cells/well in 96-well plates. After overnight
incubation, cells were pretreated with AEBSF, Z-VAD, Z-IETD or
Z-LEHD for 2 h, followed by treatment with gefitinib and/or
vorinostat in RPMI-1640 medium containing 10% FBS.
Cell viability assay and combination
index analysis
The percentage of viable cells was determined using
the CellTiter-Glo luminescent cell viability assay according to the
manufacturer's recommendations (Promega Corp., Madison, WI, USA).
The combination index (CI) was calculated using with CalcuSyn v.
2.1.1 (BioSoft, Cambridge, UK), which is based on the Chou-Talalay
method and provides qualitative information on drug interaction.
Antagonism is defined as CI>1.0, an additive effect is CI=1.0,
and synergism is CI<1.0. After achieving a maximum effect from
the drugs tested on cancer cells, a mean CI was obtained from the
value of the fraction of cell growth that is affected (Fa) (e.g.,
Fa=0.5 is equivalent to a 50% reduction in cell growth).
Western blot analysis
Cell lysates were prepared from 1×107
cells by dissolving cell pellets in 200 µl lysis buffer containing
20 mM Tris-HCl (pH 7.4), 150 mM NaCl, 1% (v/v) Nonidet P-40, 0.5%
(w/v) sodium deoxycholate, 0.1% (w/v) sodium dodecyl sulfate, and a
protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany).
Cell lysates were centrifuged at 20,000 × g for 20 min, and the
protein concentrations were determined using a BCA Protein Assay
kit (Pierce, Rockford, IL, USA). Lysates with sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer
were heated for 5 min at 100°C and 50 µl proteins were resolved
using SDS-PAGE 6–10% gels. The gels were transferred to
polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA,
USA) and the blots were probed with specific antibodies. Primary
antibodies against the various proteins were obtained from the
following sources: Mcl-1 (cat. no. sc-819) and GAPDH (cat. no.
sc-20357) were obtained from Santa Cruz Biotechnology (Dallas, TX,
USA) and HSP90 (cat. no. 4874), caspase-3 (cat. no. 9662),
caspase-8 (cat. no. 9746), caspase-9 (cat. no. 9502), PARP (cat.
no. 9542), Bax (cat. no. 2772), HER2/ErbB2 (cat. no. 2242), pEGFR
(Y1068) (cat. no. 2234), EGFR (cat. no. 2646), pMET (T1234/1235)
(cat. no. 3129), MET (cat. no. 4560), pAKT (S473) (cat. no. 9271)
and AKT (cat. no. 9272) were obtained from Cell Signaling
Technology (Danvers, MA, USA). The dilution ratio of these
antibodies was 1:1,000. Secondary antibodies purchased from as
follows: goat anti-mouse IgG-horseradish peroxidase (HRP) (1:5,000;
cat. no. 32430), goat anti-rabbit IgG-HRP (1:5,000; cat. no. 32460)
obtained from Pierce Biotechnology (Rockford, IL, USA) and mouse
anti-goat IgG-HRP (1:5,000; cat. no. sc-2354) were obtained from
Santa Cruz Biotechnology. The membranes were developed using
Immobilon Western ECL solution (Millipore) and detected using the
Kodak Image Station 4000MM system (Kodak, Rochester, NY, USA). The
blots were analyzed using Kodak Molecular Imaging, version 4.0.5
software (Eastman Kodak Company, Rochester, NY, USA).
Labeling of the cell nuclei with
Hoechst 33258 to detect nuclear fragmentation
After treatment, the cells were fixed with 4%
paraformaldehyde for 10 min at room temperature and washed twice
with phosphate-buffered saline (PBS). The nuclei were then labeled
with 2.5 µg/ml Hoechst 33258 for 15 min and washed again three
times with PBS. Fluorescent micrographs of the labeled nuclei were
captured using an Olympus IX71 fluorescence microscope (Olympus
Corp., Tokyo, Japan). Five images per well were captured and the
fragmented nuclei were counted.
Flow cytometry
After treatment, apoptosis was measured using the
FITC Annexin V apoptosis detection kit II (BD Pharmingen; BD
Biosciences, San Diego, CA, USA) in accordance with the
manufacturer's instructions. The stained cells were detected using
a BD FACSCanto 2 flow cytometer and analyzed by BD FACSDiva
(version 8.0; BD Biosciences).
Assessment of reactive oxygen species
production
After the drug-containing medium was removed, the
cells were washed with serum-free medium and incubated with 10 µM
2′,7′-dichlorodihydrofluorescin diacetate (H2DCF-DA,
Invitrogen; Thermo Fisher Scientific, Inc.) for 30 min in the dark.
Levels of reactive oxygen species (ROS) were continuously monitored
for up to 20 min. The fluorescence images were obtained using an
Olympus IX71 fluorescence microscope (Olympus Corp).
Statistical analyses
SigmaPlot version 13.0 (Systat Software, Inc.,
Erkrath, Germany) was used to analyze the significance of all
results. Statistical significance was determined by one-way
analysis of variance using (ANOVA) (P<0.05). A post-hoc test of
ANOVA was conducted by performing a Turkey's test. All experiments
were performed in triplicate, and all data are expressed as the
mean ± standard deviation.
Results
Co-treatment with gefitinib and
vorinostat potentiates apoptotic cell death in lung cancer
First, we examined the inhibitory effect of the
HDACI vorinostat on the viability of NSCLC PC9 and
gefitinib-resistant PC9GR cells using a CellTiter-Glo assay. The
PC9 cell line has a deletional mutation in EGFR exon 19 and the
PC9GR cell line is resistant to gefitinib by having acquired a
secondary T790M mutation in EGFR exon 20 (8,9).
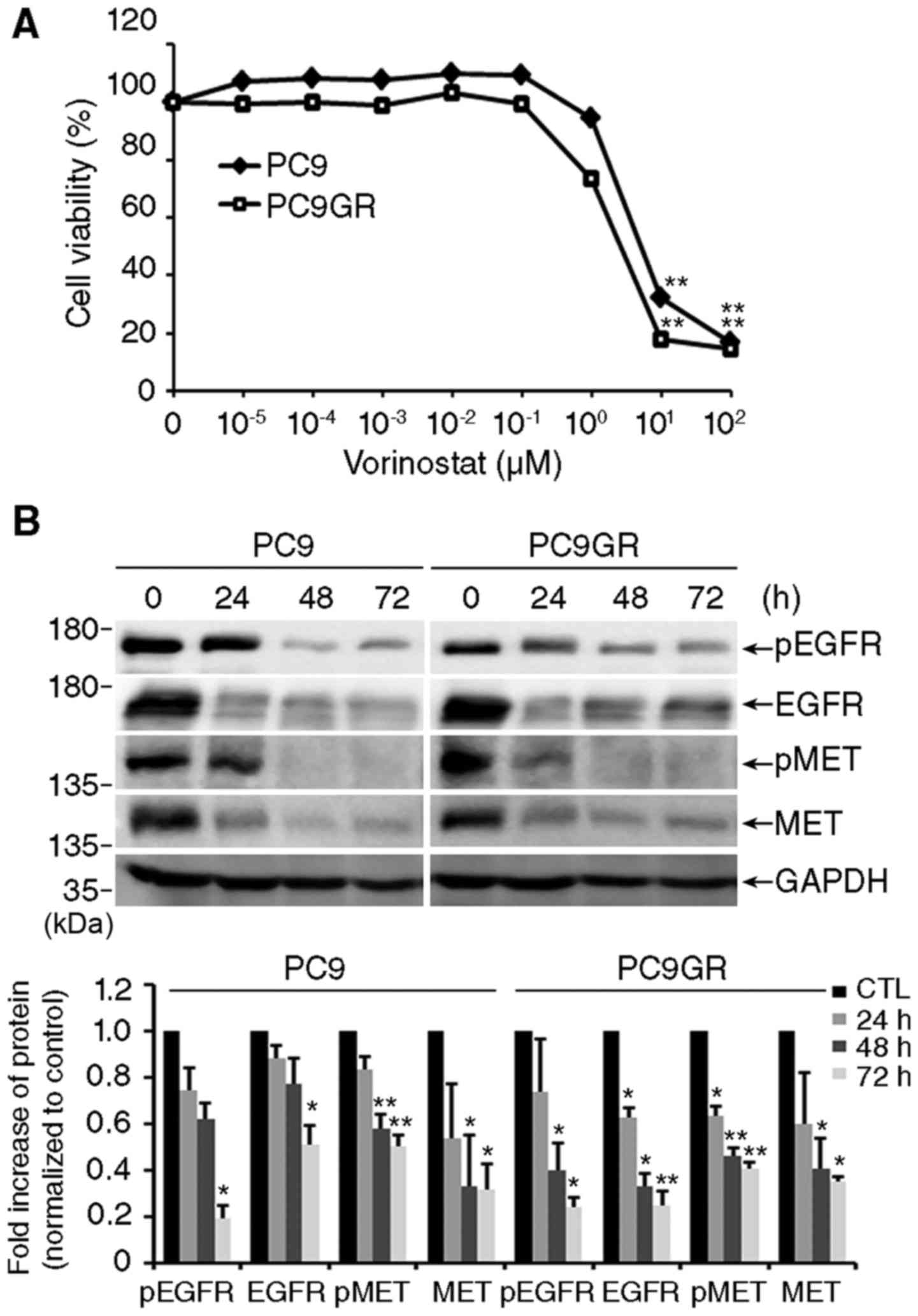
Exposure to vorinostat efficiently reduced the viability of both
cell lines in a concentration-dependent manner (Fig. 1A). The IC50 values of
vorinostat were 2.661 and 6.282 µM for PC9GR and PC9 cells,
respectively. As shown in Fig. 1B,
vorinostat significantly decreased the level of total and
phosphorylated EGFR and MET, which are responsible for the survival
of lung cancer cells (10,11).
Next, we examined the effect of vorinostat on
gefitinib sensitivity in PC9 and PC9GR cells. In accordance with
previous reports (8,9), treatment with 0.01 µM gefitinib alone
significantly decreased the viability of PC9 cells, but it had no
effect on PC9GR cells (Fig. 2A,
upper graphs). Notably, co-treatment with gefitinib and vorinostat
resulted in a synergistic effect in a concentration-dependent
manner not only on PC9 but also on PC9GR cells. The CI values were
<1.0 in all combinations, representing synergistic interaction,
except for that following treatment with 10 µM vorinostat on PC9GR
cells (Fig. 2A, lower tables). To
determine the types of cell death induced by the combined
treatment, we examined the changes in apoptosis-related proteins.
Co-treatment with gefitinib and vorinostat induced cleavage of
caspase-3 and PARP and increased the expression of Bax, a
pro-apoptotic Bcl-2 family protein, in PC9 and PC9GR cells
(Fig. 2B). Mcl-1, an anti-apoptotic
Bcl-2 family protein, decreased in both cell lines after combined
treatment. In accordance with these results, we also observed
nuclear fragmentation and Annexin V/propidium iodide (PI)-positive
cells in both cell lines exposed to the combined treatment and the
reverse of these events by adding the pan-caspase inhibitor Z-VAD
(Fig. 2C and D). These findings
indicate that apoptosis mediates the substantial synergistic
effects of co-treatment with gefitinib and vorinostat in NSCLC
cells with EGFR-activating mutations, including exon 19 deletion
and T790M mutation.
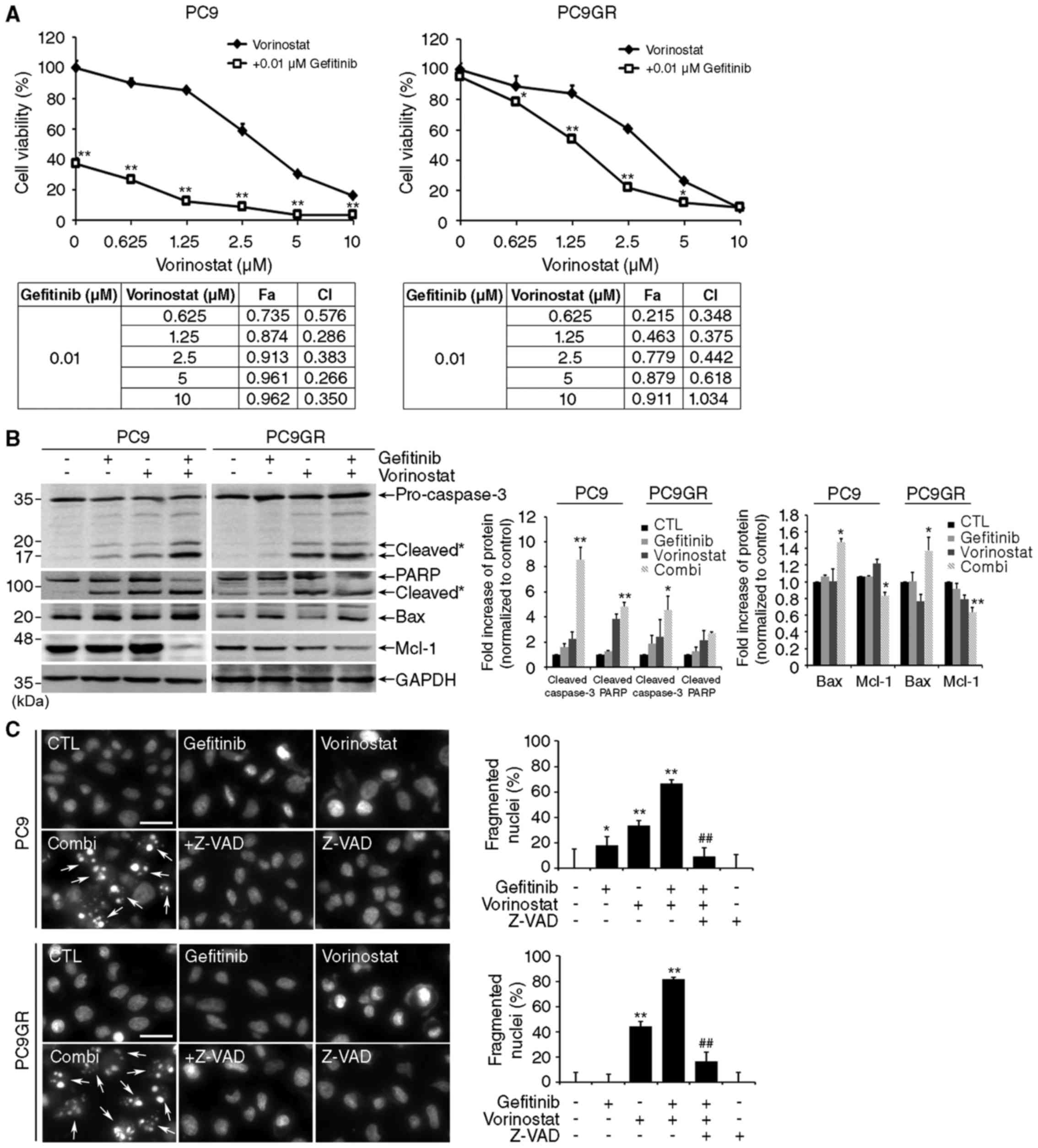 | Figure 2.Co-treatment with gefitinib and
vorinostat increases apoptosis. (A) PC9 and PC9GR cells were
treated with 0.01 µM gefitinib and/or the indicated concentrations
of vorinostat for 48 h, and cell viability was assessed. Data
represent mean ± SD (n=3, *P<0.05, **P<0.001). The fraction
affected value (Fa), indicating the fraction of cells inhibited
after drug exposure, and the combination index (CI) were calculated
using CalcuSyn. (B) Cells were treated with 0.01 µM gefitinib
and/or 5 µM vorinostat for 48 h, and levels of cleaved capsase-3,
PARP, Bax and Mcl-1 were determined by western blotting. GAPDH was
used as a loading control. Cleavage products are labeled with an
asterisk (n=3, *P<0.05, **P<0.001). (C) Nuclear staining with
10 µM Hoechst 33342 in cells treated with 0.01 µM gefitinib and/or
5 µM vorinostat in the presence or absence of 50 M Z-VAD-FMK for 48
h. Left panels: Hoechst staining. Scale bar, 50 µm. Right graphs:
quantification of fragmented nuclei [mean ± SD, n=3; *P<0.05,
**P<0.001 vs. the control, #P<0.05,
##P<0.001 vs. co-treatment (Combi)]. (D)
Representative flow cytometry scatter plots of propidium iodide
(PI) (y-axis) vs Annexin V-FITC (x-axis). Bar charts show
quantitative data of the average of 3 independent flow cytometry
experiments in PC9 and PC9GR cells (n=3; **P<0.001 vs. the
control, #P<0.05, ##P<0.001 vs.
Combi). |
Co-treatment leads to HSP90 cleavage
and degradation of HSP90 client proteins in an ROS-dependent
manner
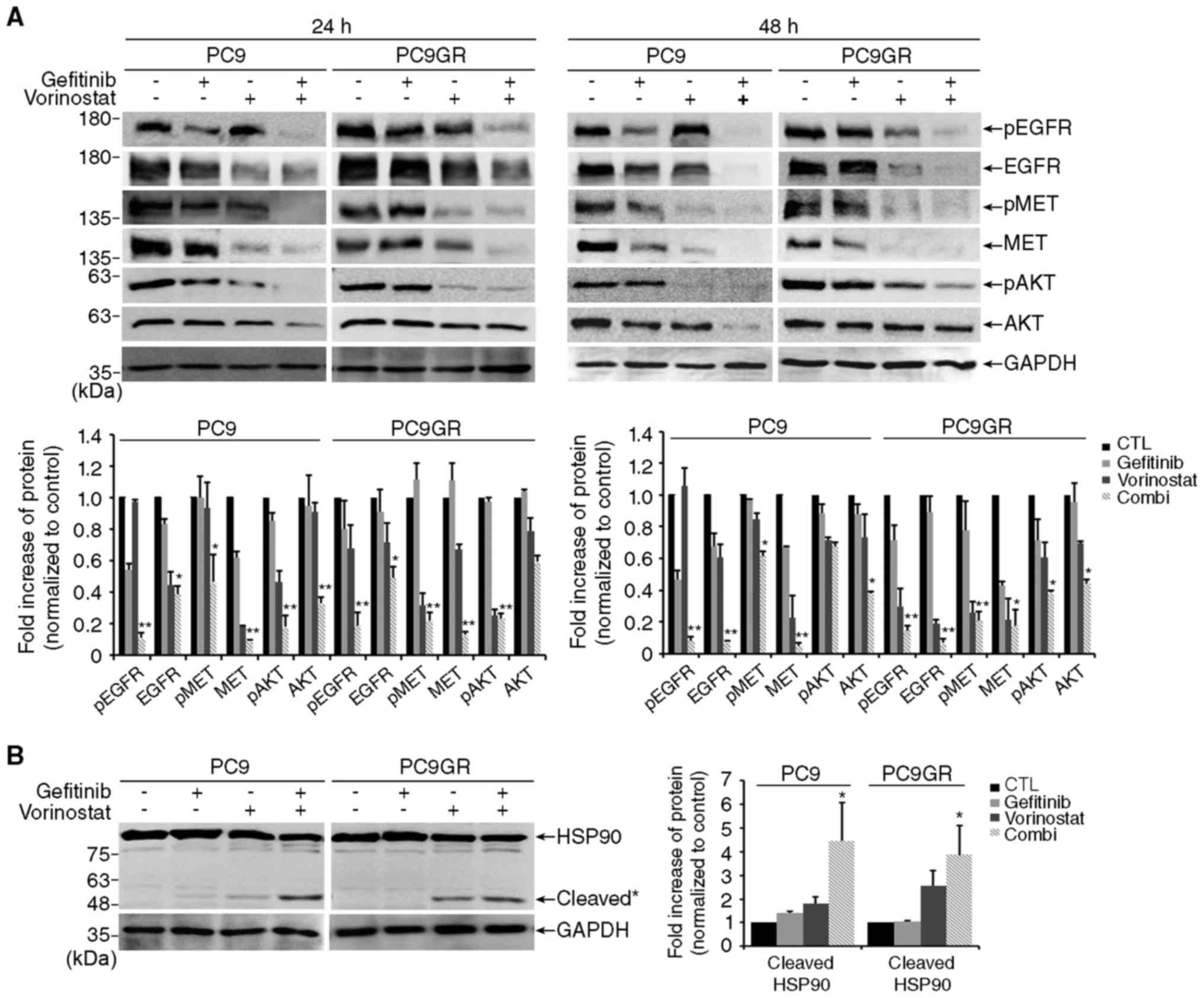
To identify the underlying mechanisms involved in
apoptosis, we examined the levels of EGFR and MET proteins and
their downstream target, AKT. As shown in Fig. 3A, co-treatment with gefitinib and
vorinostat significantly decreased the levels of total EGFR, MET
and AKT proteins and their phosphorylation compared with levels
following treatment with each drug alone. Since EGFR, MET and AKT
are the clients of HSP90 (12), we
examined the level of HSP90 proteins. Treatment with vorinostat
alone increased HSP90 cleavage, and co-treatment significantly
increased HSP90 cleavage in both PC9 and PC9GR cells (Fig. 3B).
It has been reported that oxidative stress caused by
various anticancer drugs induce HSP90 cleavage and degrade its
client proteins in cancer cells (13,14).
In accordance with these reports, we observed a synergistic
increase in ROS by co-treatment with gefitinib and vorinostat in
both PC9 and PC9GR cells compared with that of vorinostat alone
(Fig. 4A). To verify the
contribution of ROS in combined treatment-induced HSP90 cleavage,
we examined the effect of the NADPH oxidase inhibitor, AEBSF. The
addition of AEBSF effectively blocked the decrease in the levels of
phosphorylated and total EGFR and MET (Fig. 4B) and HSP90 cleavage (Fig. 4C) by the combined treatment. These
results indicate that HSP90 cleavage induced by co-treatment with
gefitinib and vorinostat is mediated by ROS production, and that
these events increase the degradation of HSP90 client proteins,
such as EGFR, MET and AKT.
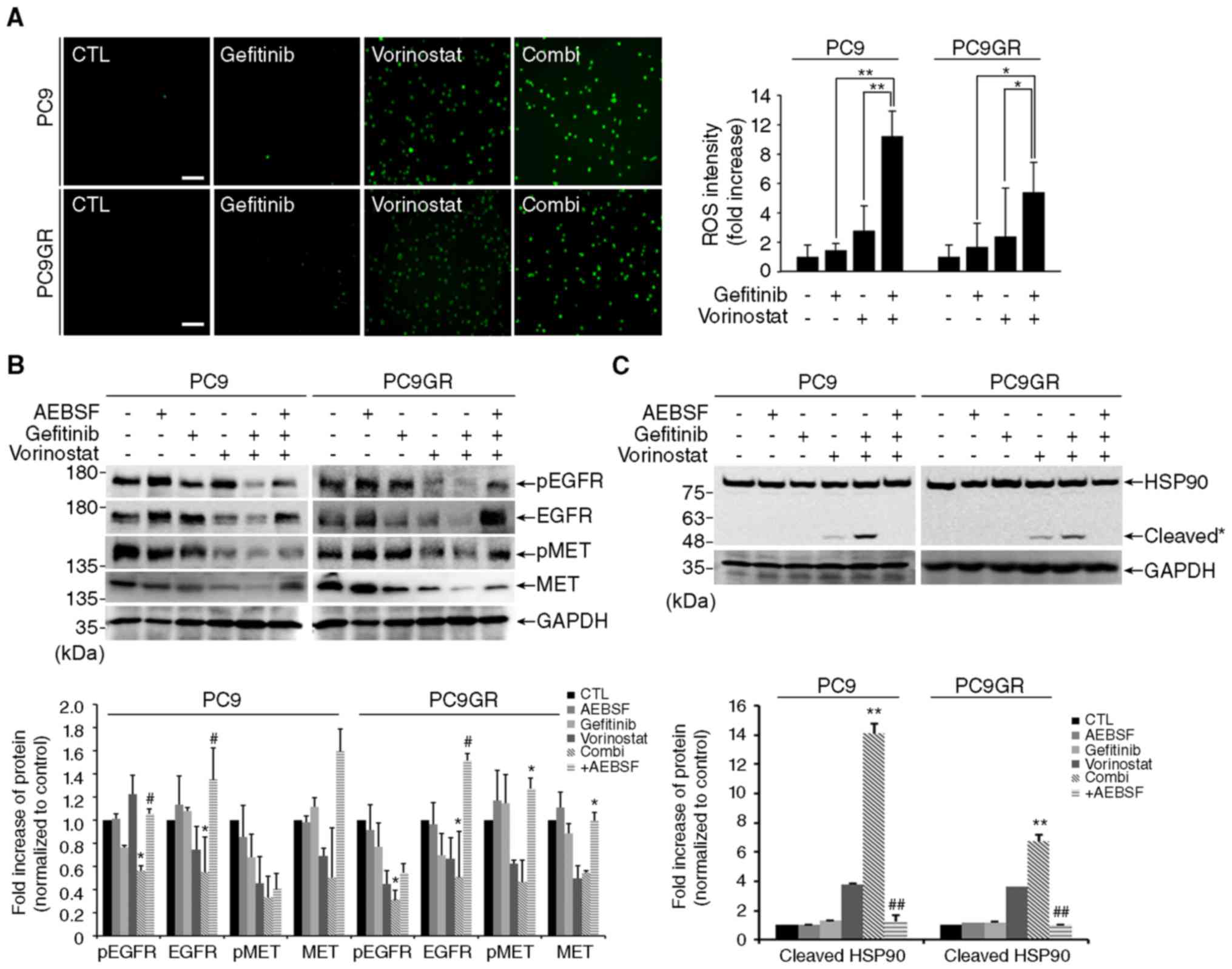 | Figure 4.Co-treatment accumulates reactive
oxygen species (ROS)-dependent HSP90 cleavage and client
degradation. (A) PC9 and PC9GR cells were treated with 0.01 µM
gefitinib and/or 5 µM vorinostat for 16 h, stained with 10 µM
H2DCF-DA, and observed by fluorescence microscopy. Scale
bar, 50 µm. The bar graphs represent the normalized fluorescent
intensity of ROS (mean ± SEM, n=3; *P<0.05, **P<0.001). (B
and C) Cells were treated with 0.01 µM gefitinib and/or 5 µM
vorinostat for 24 h with or without 100 µM
4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride (AEBSF),
and the level of total or phosphorylated EGFR and MET (B) and
cleaved HSP90 (C) were determined by western blotting. GAPDH was
used as a loading control. Cleavage products of HSP90 are labeled
with an asterisk [n=3, *P<0.05, **P<0.001 vs. the control,
#P<0.05, ##P<0.001 vs. co-treatment
(Combi)]. |
ROS-dependent caspase activation is
responsible for HSP90 cleavage by combined treatment
As ROS- or caspase-mediated HSP90 cleavage is
implicated in apoptosis (13,15),
we investigated the types of caspases that are involved in
ROS-dependent cleavage of HSP90 induced by co-treatment with
gefitinib and vorinostat. Addition of AEBSF inhibited the cleavage
of an effector caspase-3 and its activators, caspase-8 and
caspase-9 in PC9 and PC9GR cells exposed to the combined treatment
(Fig. 5A). In accordance with these
results, the caspase-3 inhibitor Z-DEVD completely abolished HSP90
cleavage, and the inhibitors against each caspase, Z-IETD for
caspase-8 and Z-LEHD for caspase-9, were partially effective in
blocking HSP90 cleavage augmented by the combined treatment
(Fig. 5B and C). In addition, the
pan-caspase inhibitor Z-VAD reversed the degradation of HSP90
clients and HSP90 cleavage as well as apoptosis in both cell lines
co-treated with gefitinib and vorinostat (Figs. 5D and 2C). Moreover, the addition of AEBSF
attenuated cell death induced by the combined treatment in the PC9
and PC9GR cells (Fig. 5E). These
results indicate that HSP90 cleavage by co-treatment with gefitinib
and vorinostat is mediated by ROS-dependent caspase activation, and
these events increase apoptosis by degrading HSP90 client proteins,
such as EGFR, MET and AKT.
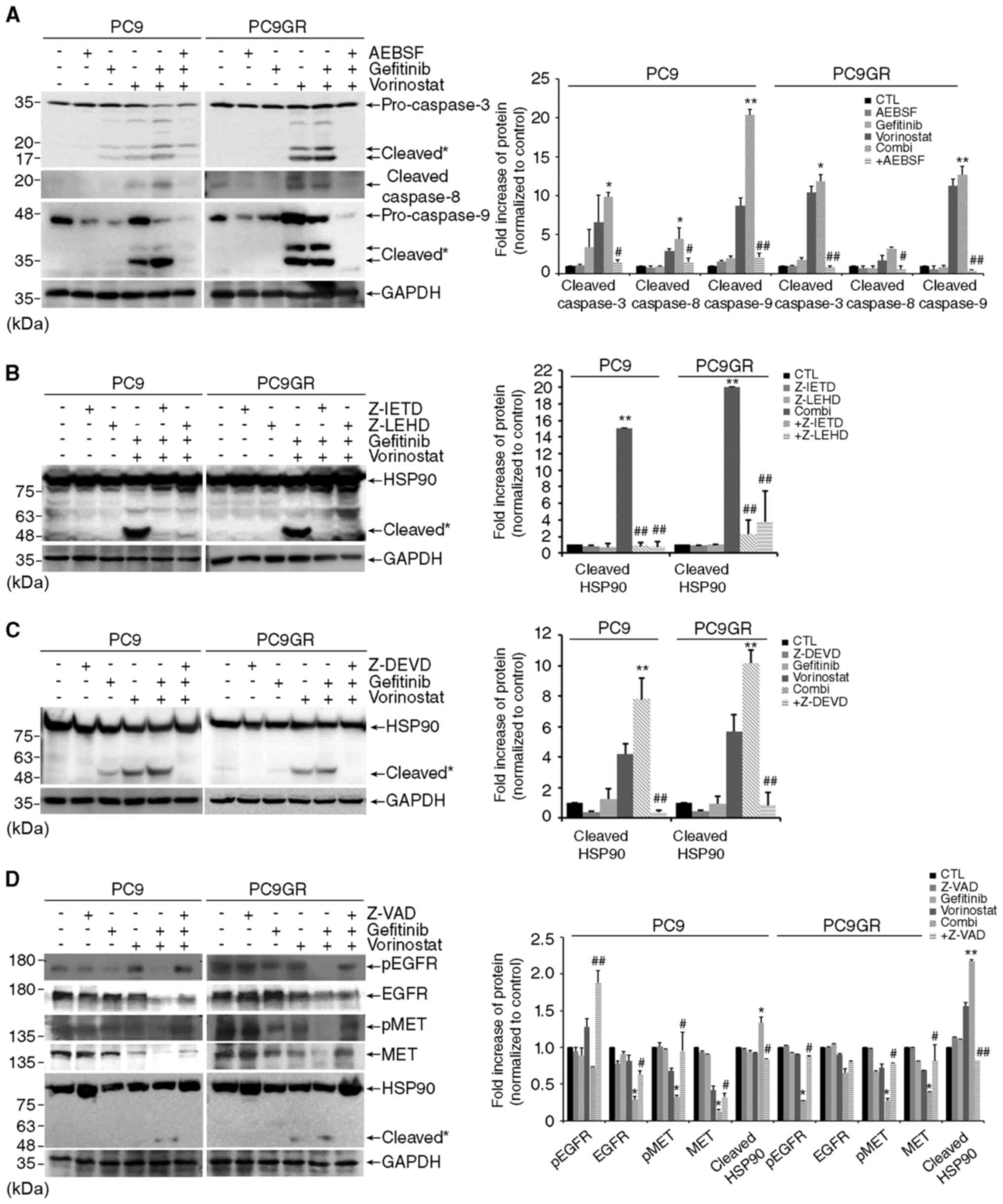 | Figure 5.Co-treatment with gefitinib and
vorinostat induces HSP90 cleavage through reactive oxygen species
(ROS)-dependent caspase activation. (A) PC9 and PC9GR cells were
treated with 0.01 µM gefitinib and/or 5 µM vorinostat for 48 h with
or without 100 µM AEBSF, and caspase-3, −8, and −9 cleavage was
determined by western blotting. Cleavage products of caspase-3, −8,
and −9 are labeled with an asterisk [n=3, *P<0.05, **P<0.001
vs. the control, #P<0.05, ##P<0.001 vs.
co-treatment (Combi)]. (B and C) Cells were treated with 0.01 µM
gefitinib and/or 5 µM vorinostat for 24 h with or without 50 µM
Z-IETD-FMK, Z-LEHD-FMK (B), or Z-DEVD-FMK (C), and HSP90 cleavage
was determined by western blotting (n=3, **P<0.001 vs. the
control, ##P<0.001 vs. Combi). (D) Cells were treated
with 0.01 µM gefitinib and/or 5 µM vorinostat for 24 h with or
without 50 µM Z-VAD-FMK. The levels of total or phosphorylated EGFR
and MET and HSP90 cleavage were detected by western blotting. GAPDH
was used as a loading control. Cleavage products of HSP90 are
labeled with an asterisk (n=3, *P<0.05, **P<0.001 vs. the
control, #P<0.05, ##P<0.001 vs. Combi).
(E) Cells were treated with 0.01 µM gefitinib and/or 5 µM
vorinostat with or without 100 µM AEBSF for 48 h and cell death was
assessed. |
Discussion
The present study revealed that vorinostat in
combination with gefitinib exerted a synergistic anticancer effect
on NSCLC PC9 and PC9GR cells. Our results indicate that
ROS-dependent caspase activation by co-treatment with gefitinib and
vorinostat mediated HSP90 cleavage, and that these events increased
apoptosis by degrading HSP90 client proteins, such as EGFR, MET and
AKT. Accordingly, we suggest fundamental evidence to support that a
combination of HDACI and gefitinib can be used to overcome
gefitinib resistance in NSCLC.
Targeted anticancer therapies have appreciably
advanced since the last decade. In 2015, gefitinib, a non-covalent
inhibitor, was approved by the U.S. Food and Drug Administration
for the first-line treatment of metastatic lung cancer with an
EGFR-activating mutation, such as an EGFR exon 19 deletion and an
EGFR L858R mutation in exon 21 found in 80–90% of all EGFR
mutations in NSCLC (16). However,
progression-free survival of this therapy was reported to be rather
disappointing due to the development of diverse resistance
(17). One of the main mechanisms
by which resistance to gefitinib is acquired is the EGFR T790M
mutation. Another resistance mechanism is activation or
upregulation of bypass RTKs (MET, ERBb3, AXL and IGF1R) and their
downstream signaling molecules (PI3K, AKT, MEK and JAK) (17,18).
Although the second- and third-generation covalent inhibitors
against EGFR T790M have been used for NSCLC, resistance of these
drugs has occurred in patients with new resistant EGFR mutations
and bypass pathways. To overcome resistance against EGFR-TKIs,
therefore, it is useful to downregulate RTKs, for example EGFR
itself or MET, which takes charge of bypassing signaling pathways,
or a downstream signaling molecule, AKT. Our results showed that
HDACI can reduce the level of EGFR, MET and AKT which are
responsible for gefitinib resistance. These results associated with
overcoming gefitinib-resistance will expand the opportunity to
develop combination therapies with EGFR-TKIs as an alternative for
prolonging the control of the disease.
HDACIs, a group of epigenetic drugs, have been
evaluated for their synergistic effects when combined with various
conventional chemotherapeutic or targeted cancer therapies for
different types of tumors (19,20).
It has been reported that HDACIs can increase sensitivity and
reverse resistance to EGFR-TKIs in lung cancers. The suggested
mechanisms of synergy are the induction of E-cadherin, BIM and BAX
(5,21,22,30),
inhibition of the IGF 1R-AKT pathway (23), and accumulation of ROS by
upregulating the major mitochondrial porin voltage-dependent
anion-selective channel protein 1 and modulating the
c-Myc/NRF2/KEAP1 pathway (24) in
NSCLC cell lines. It has also been reported that an HDACI
potentiates apoptosis induced by an EGFR-TKI by HSP90
acetylation-dependent depletion of key survival signaling proteins,
including AKT, EGFR, c-Src and STAT3 (2). Acetylation of K294 (HSP90α)/K287
(HSP90β) reduces its affinity to the co-chaperone machinery,
leading to the degradation of client proteins and apoptosis
(25,26). Therefore, deacetylation of these
proteins by HDAC6 stabilizes HSP90 function, and the HDACI induces
apoptosis through acetylation and activation of HSP90 in cancer
(3,25,27).
One novel finding of our study was that co-treatment with gefitinib
and vorinostat had an additional mechanism of caspase-mediated
degradation of HSP90 with a molecular weight of ~55 kDa in PC9 and
PC9GR NSCLC cells. Moreover, HDACI-induced cleavage of HSP90 was
also detected in other types of cancer cells [renal (SN12C, ACHN
and 786O), prostate (PC3), and breast (MCF-7 and MDA-MB-231) cancer
cell lines (data not shown)]. HSP90 cleavage attenuates the
function of HSP90, results in destabilization of client proteins,
and potentiates apoptosis in cancer cells. There have been reports
showing that HDACI activates ROS-dependent caspase activation,
triggering extrinsic or intrinsic apoptosis in various types of
cancer cells (28,29), and that oxidative stress and
caspase-10 induces HSP90 cleavage (13,15).
Therefore, we predicted that vorinostat would induce HSP90 cleavage
through ROS-dependent caspase activation, and our results have
supported this prediction.
Notably, vorinostat was shown to overcome EGFR-TKI
resistance and synergize with EGFR-TKIs in NSCLCs in vivo.
Busser et al and Jeannot et al reported that the
combination of gefitinib and vorinostat inhibited tumor growth in a
gefitinib-resistant H358 ×enograft model by activating BAX released
from acetylated Ku70 and inhibiting the IGF 1R-AKT pathway,
respectively (22,23). Nakagawa et al showed that the
combined use of vorinostat and EGFR-TKI (gefitinib or osimertinib)
synergistically regressed tumors in xenograft models using PC9 and
PC9GR (T790M) with BIM deletion polymorphism through re-expression
of active BIM (21). In addition, a
phase I/II clinical study of co-treatment with gefitinib and
vorinostat showed a potential improvement in patients with advanced
NSCLC (31). In our study, we found
in vitro that co-treatment with gefitinib and vorinostat
presented a new mechanism by which to induce cell death in NSCLC,
although we did not perform in vivo experiments. We showed
that activation of ROS-dependent caspases led to the downregulation
of HSP90 client proteins through HSP90 cleavage in PC9 and PC9GR
cells. Thus, current research has led to the presumption that
HDACIs are promising drugs that potentiate the therapeutic efficacy
of EGFR-TKIs. Our results, which present the synergistic antitumor
activity of vorinostat in combination with gefitinib, provide the
supportive rationale for the future design of clinical trials based
on these inhibitors in NSCLC patients.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the Korean
Health Technology R&D Project, Ministry of Health and Welfare,
Republic of Korea (HI15C0972), and from the Asan Institute for Life
Sciences, Asan Medical Center (2018–450) Seoul, Republic of
Korea.
Availability of data and materials
The analyzed data sets generated during the study
are available upon reasonable request from the corresponding
author.
Authors' contributions
SEP designed and wrote the manuscript. DEK, JSL and
MJK conducted the research. JKR provided the cell lines and
analyzed the results. SYJ and EKC helped to collect the data and
analyze the results. CSK and JJH were the leading principal
investigators who directed the study, were involved in the
conception of the study, and drafted the manuscript. All authors
read, revised and approved the manuscript and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the study were appropriately
investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Beere HM: ‘The stress of dying’: The role
of heat shock proteins in the regulation of apoptosis. J Cell Sci.
117:2641–2651. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Edwards A, Li J, Atadja P, Bhalla K and
Haura EB: Effect of the histone deacetylase inhibitor LBH589
against epidermal growth factor receptor-dependent human lung
cancer cells. Mol Cancer Ther. 6:2515–2524. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Socinski MA, Goldman J, El-Hariry I,
Koczywas M, Vukovic V, Horn L, Paschold E, Salgia R, West H,
Sequist LV, et al: A multicenter phase II study of ganetespib
monotherapy in patients with genotypically defined advanced
non-small cell lung cancer. Clin Cancer Res. 19:3068–3077. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jhaveri K, Taldone T, Modi S and Chiosis
G: Advances in the clinical development of heat shock protein 90
(Hsp90) inhibitors in cancers. Biochim Biophys Acta. 1823:742–755.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Witta SE, Gemmill RM, Hirsch FR, Coldren
CD, Hedman K, Ravdel L, Helfrich B, Dziadziuszko R, Chan DC, Sugita
M, et al: Restoring E-cadherin expression increases sensitivity to
epidermal growth factor receptor inhibitors in lung cancer cell
lines. Cancer Res. 66:944–950. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Neal JW and Sequist LV: Complex role of
histone deacetylase inhibitors in the treatment of non-small-cell
lung cancer. J Clin Oncol. 30:2280–2282. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hoang T, Campbell TC, Zhang C, Kim K,
Kolesar JM, Oettel KR, Blank JH, Robinson EG, Ahuja HG, Kirschling
RJ, et al: Vorinostat and bortezomib as third-line therapy in
patients with advanced non-small cell lung cancer: A wisconsin
oncology network phase II study. Invest New Drugs. 32:195–199.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rho JK, Choi YJ, Lee JK, Ryoo BY, Na II,
Yang SH, Lee SS, Kim CH, Yoo YD and Lee JC: The role of MET
activation in determining the sensitivity to epidermal growth
factor receptor tyrosine kinase inhibitors. Mol Cancer Res.
7:1736–1743. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rho JK, Lee IY, Choi YJ, Choi CM, Hur JY,
Koh JS, Lee J, Suh BC, Song HJ, Salgaonkar P, et al: Superior
efficacy and selectivity of novel small-molecule kinase inhibitors
of T790M-mutant EGFR in preclinical models of lung cancer. Cancer
Res. 77:1200–1211. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gazdar AF: Epidermal growth factor
receptor inhibition in lung cancer: The evolving role of
individualized therapy. Cancer Metastasis Rev. 29:37–48. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sadiq AA and Salgia R: MET as a possible
target for non-small-cell lung cancer. J Clin Oncol. 31:1089–1096.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mahalingam D, Swords R, Carew JS, Nawrocki
ST, Bhalla K and Giles FJ: Targeting HSP90 for cancer therapy. Br J
Cancer. 100:1523–1529. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Beck R, Verrax J, Gonze T, Zappone M,
Pedrosa RC, Taper H, Feron O and Calderon PB: Hsp90 cleavage by an
oxidative stress leads to its client proteins degradation and
cancer cell death. Biochem Pharmacol. 77:375–383. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pantano C, Shrivastava P, McElhinney B and
Janssen-Heininger Y: Hydrogen peroxide signaling through tumor
necrosis factor receptor 1 leads to selective activation of c-Jun
N-terminal kinase. J Biol Chem. 278:44091–44096. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen H, Xia Y, Fang D, Hawke D and Lu Z:
Caspase-10-mediated heat shock protein 90 beta cleavage promotes
UVB irradiation-induced cell apoptosis. Mol Cell Biol.
29:3657–3664. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jackson SE and Chester JD: Personalised
cancer medicine. Int J Cancer. 137:262–266. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chong CR and Jänne PA: The quest to
overcome resistance to EGFR-targeted therapies in cancer. Nat Med.
19:1389–1400. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wheeler DL, Dunn EF and Harari PM:
Understanding resistance to EGFR inhibitors-impact on future
treatment strategies. Nat Rev Clin Oncol. 7:493–507. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bose P, Dai Y and Grant S: Histone
deacetylase inhibitor (HDACI) mechanisms of action: Emerging
insights. Pharmacol Ther. 143:323–336. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nakagawa T, Takeuchi S, Yamada T, Ebi H,
Sano T, Nanjo S, Ishikawa D, Sato M, Hasegawa Y, Sekido Y and Yano
S: EGFR-TKI resistance due to BIM polymorphism can be circumvented
in combination with HDAC inhibition. Cancer Res. 73:2428–2434.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Busser B, Sancey L, Josserand V, Niang C,
Khochbin S, Favrot MC, Coll JL and Hurbin A: Amphiregulin promotes
resistance to gefitinib in nonsmall cell lung cancer cells by
regulating Ku70 acetylation. Mol Ther. 18:536–543. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jeannot V, Busser B, Vanwonterghem L,
Michallet S, Ferroudj S, Cokol M, Coll JL, Ozturk M and Hurbin A:
Synergistic activity of vorinostat combined with gefitinib but not
with sorafenib in mutant KRAS human non-small cell lung cancers and
hepatocarcinoma. Onco Targets Ther. 9:6843–6855. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Leone A, Roca MS, Ciardiello C,
Terranova-Barberio M, Vitagliano C, Ciliberto G, Mancini R, Di
Gennaro E, Bruzzese F and Budillon A: Vorinostat synergizes with
EGFR inhibitors in NSCLC cells by increasing ROS via up-regulation
of the major mitochondrial porin VDAC1 and modulation of the
c-Myc-NRF2-KEAP1 pathway. Free Radic Biol Med. 89:287–299. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bali P, Pranpat M, Bradner J, Balasis M,
Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, et
al: Inhibition of histone deacetylase 6 acetylates and disrupts the
chaperone function of heat shock protein 90: A novel basis for
antileukemia activity of histone deacetylase inhibitors. J Biol
Chem. 280:26729–26734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Scroggins BT, Robzyk K, Wang D, Marcu MG,
Tsutsumi S, Beebe K, Cotter RJ, Felts S, Toft D, Karnitz L, et al:
An acetylation site in the middle domain of Hsp90 regulates
chaperone function. Mol Cell. 25:151–159. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nishioka C, Ikezoe T, Yang J, Takeuchi S,
Koeffler HP and Yokoyama A: M S-275, a novel histone deacetylase
inhibitor with selectivity against HDAC1, induces degradation of
FLT3 via inhibition of chaperone function of heat shock protein 90
in AML cells. Leuk Res. 32:1382–1392. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ruefli AA, Ausserlechner MJ, Bernhard D,
Sutton VR, Tainton KM, Kofler R, Smyth MJ and Johnstone RW: The
histone deacetylase inhibitor and chemotherapeutic agent
suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway
characterized by cleavage of Bid and production of reactive oxygen
species. Proc Natl Acad Sci USA. 98:10833–10838. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim DH, Lee J, Kim KN, Kim HJ, Jeung HC,
Chung HC and Kwon HJ: Anti-tumor activity of
N-hydroxy-7-(2-naphthylthio) heptanomide, a novel histone
deacetylase inhibitor. Biochem Biophys Res Commun. 356:233–238.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ng KP, Hillmer AM, Chuah CT, Juan WC, Ko
TK, Teo AS, Ariyaratne PN, Takahashi N, Sawada K, Fei Y, et al: A
common BIM deletion polymorphism mediates intrinsic resistance and
inferior responses to tyrosine kinase inhibitors in cancer. Nat
Med. 18:521–528. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Han JY, Lee SH, Lee GK, Yun T, Lee YJ,
Hwang KH, Kim JY and Kim HT: Phase I/II study of gefitinib
(Iressa(®)) and vorinostat (IVORI) in previously treated
patients with advanced non-small cell lung cancer. Cancer Chemother
Pharmacol. 75:475–483. 2015. View Article : Google Scholar : PubMed/NCBI
|