Introduction
In our previous study, 5 epithelial-derived
carcinoma cell lines, including human non-small cell lung cancer
(NSCLC) NCI-H1299, human nasopharyngeal carcinoma CNE1, human
gastric cancer MGC, human breast cancer MCF-7 and human hepatoma
Hep3b-2, were used to screen Taxol-resistant cells. The results
confirmed that NCI-H1299 cells highly expressing oncoprotein 18
(Op18)/stathmin exhibited resistance to Taxol, and strong
capabilities of cell proliferation, migration and invasion,
compared with the other cell lines (1). Silencing of Op18/stathmin by RNA
interference (RNAi) resulted in inhibition of cell proliferation
and motility, and strengthened the sensitivity of NCI-H1299 cells
to Taxol. In vivo experiments confirmed that RNAi of
Op18/stathmin combined with Taxol co-operatively decreased the
tumorigenesis of transplanted NCI-H1299 cells and tumor growth, and
promoted high-grade differentiation of xenografts (2). By tracing the genetic background of
NCI-H1299 cells and retrieving relative literatures, it was
determined that NCI-H1299 cells are innately p53 deficient, and
lack p53 protein expression (3,4). We
hypothesized that p53 deficiency is associated with the high
malignancy levels of NCI-H1299 cells exhibiting high expression of
Op18/stathmin.
Wild-type p53 (p53wt)
prevents cell cycle progression and the repair of damaged and
mutant genes, and induces apoptosis and inhibition of proliferation
in tumor development. p53wt is activated and
highly expressed in instances of DNA damage caused by stress,
including ultraviolet radiation, hypoxia and drugs, promotes the
sensitivity of tumors to treatment and increases cell apoptosis
(5,6). The majority of antitumor drugs induce
p53 expression and activation; for example, in colon cancer HCT-116
cells with p53wt introduced, Adriamycin enhanced
p53 expression and inhibition of cell proliferation (7). Similarly, high concentrations of
Nutlin-3 induced high expression of p53 resulting in apoptosis in
colon cancer RKO and prostate cancer LNCaP cells with
p53wt, and the status of cell apoptosis was
positively associated with the levels of p53 expression (8).
The microtubule regulator Op18/stathmin, a small
molecule of phosphoprotein that is highly expressed in solid
tumors, serves a crucial role in integrating and transducing
various signals from intra- and extra-cellular stimuli (9). It directly regulates the dynamics
equilibrium of the microtubule cytoskeleton, and controls cellular
biological behavior, including cell cycle progression, metastasis
and invasion, through phosphorylated inactivation and
dephosphorylated activation (9–11).
Co-transfection of Op18/stathmin luciferase reporter and
p53wt carrier confirmed that
p53wt inhibited the promoter of Op18/stathmin,
which resulted in decreased Op18/stathmin mRNA transcription and
protein expression, and arrested the cell cycle at the
G2/M phases in HT1080 fibrosarcoma cells (12).
The present study is a continuation of our previous
research which primarily focused on investigating whether
p53wt deficiency was associated with Taxol
resistance mediated by Op18/stathmin signaling in NCI-H1299 cells.
Additionally, the changes in Taxol resistance following the
introduction of exogenous p53wt and the molecular
mechanism regulating Op18/stathmin signaling was investigated to
determine the effects of exogenous p53wt on
Op18/stathmin signaling and the underlying molecular mechanisms,
and the association between p53 deficiency and the malignancy of
NCI-H1299 cells was confirmed.
Materials and methods
Cell lines and cell culture
The NSCLC NCI-H1299 cell line [American Type Culture
Collection (ATCC), Manassas, VA, USA; ATCC number, CRL-5803™] was
grown in RPMI-1640 medium (cat. no. 01-100-1ACS; Biological
Industries, Kibbutz Beit Haemek, Israel) supplemented with 10%
fetal bovine serum (FBS; cat. no. 04-001-1ACS; Biological
Industries), 100 IU/ml penicillin and 100 µg/ml streptomycin (cat.
no. SV30010; HyCone; GE Healthcare Life Sciences, Logan, UT, USA)
at 37°C in humidified atmosphere containing 5% CO2.
Antibodies and chemical reagents
The primary antibodies comprised of rabbit
polyclonal anti-stathmin (1:3,000; cat. no. 569391; EMD Millipore,
Billerica, MA, USA), anti-extracellular signal-regulated kinase 1
(ERK1; 1:1,000; cat. no. sc-93), anti-cyclin-dependent 2 (CDC2;
1:1,000; cat. no. sc-954), anti-nuclear factor-κB (NF-κB; 1:1,000;
cat. no. SC-109), anti-B-cell lymphoma-2 (Bcl-2; 1:1,000; cat. no.
sc-492), mouse monoclonal anti-β-actin (1:2,000; cat. no.
sc-47778), anti-phospho(p)-ERK1 (1:1,000; cat. no. sc-7383),
anti-caspase-3 (1:1,000; cat. no. sc-7272), anti-p53 (1:1,000; cat.
no. sc-126), anti-Bcl-2-associated X protein (1:1,000; Bax; cat.
no. sc-7480) (Santa Cruz Biotechnology, Inc., Dallas, TX, USA),
mouse monoclonal anti-caspase-8 (1:1,000; cat. no. 9746), rabbit
monoclonal anti-caspase-9 (1:1,000; cat. no. 9502), rabbit
polyclonal anti-p-thr161-CDC2 (1:1,000; cat. no. 9114) (Cell
Signaling Technology, Inc., Danvers, MA, USA), anti-p-stathmin
Ser25 (1:1,000; cat. no. ab194752), anti-p-stathmin Ser63 (1:1,000;
cat. no. ab76583), anti-interleukin-10 (IL-10; 1:1,000; cat. no.
ab34843) and anti-IL-6 (1:1,000; cat. no. ab6672) (Abcam,
Cambridge, MA, USA).
The secondary antibodies used were horseradish
peroxidase (HRP)-conjugated goat anti-rabbit IgG (which was diluted
at 1:3,000 for the detection of stathmin, NF-κB, IL-6 and IL-10,
and at 1:2,500 for detecting caspase-9, Bcl-2, p-stathmin-Ser25,
-Ser63, phospho-Thr161-CDC2, CDC2 and ERK; cat. no. sc-2004) and
rabbit anti-mouse IgG (which was diluted at 1:3,000 for the
detection of β-actin and p53, and at 1:2,500 for analyzing
caspases-3, −8, Bcl-2-associated X and phosphor-ERK; cat. no.
sc-358914) (Santa Cruz Biotechnology, Inc.).
The NF-κB inhibitor ammonium pyrrolidine
dithiocarbamate (PDTC; cat. no. 5108-96-3; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) (13) and
Taxol (cat. no. sc-201439; Santa Cruz Biotechnology, Inc.) were
separately dissolved in dimethyl sulfoxide (DMSO), stored at −20°C
and diluted to appropriate concentrations (Taxol, 100 nM; or PDTC,
5 or 10 µM) prior to use.
Plasmid construction and cell
transfection
Nearly confluent cells at the logarithmic growth
phase were divided into three groups (blank, C3 and
p53wt) for transfection as follows: Blank control
without any plasmid (blank); empty vector pEGFP-C3 (C3); and
pEGFP-C3-p53 (p53wt). The plasmid pEGFP-C3-p53 was
constructed by inserting the p53wt gene into the
pEGFP-C3 vector between the two cleavage sites of XholI and
KpnI, and PEGFP-C3 was labeled with neomycin and kanamycin
resistance genes. G418 (400 µg/ml; Sigma-Aldrich; Merck KGaA) was
used to screen cells following transfection for 5 h. The PEGFP-C3
and pEGFP-C3-p53 plasmids were provided by Professor Cao Ya (Cancer
Research Institute, Central South University, Changsha, China).
Cell transfection was performed with
Lipofectamine® 2000 (cat. no. 11668-019; Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocols. Additionally 4 µg plasmids in 10 µl Lipofectamine 2000
were transiently transfected into cells at a transfection
efficiency of >80% in 250 µl incomplete RPMI-1640 medium.
Western blot analysis
Protein was extracted with lysis buffer consisting
of 50 mM pH 8.0 Tris-HCl, 1 mM ethylenediaminetetraacetic acid, 2%
SDS, 5 mM dithiothreitol and 10 mM phenylmethylsulfonyl fluoride.
The protein concentration was determined using BCA Protein Assay
Reagent (Pierce; Thermo Fisher Scientific, Inc.). Total proteins
(50 µg) were then separated by 10% SDS-PAGE and electro-transferred
onto nitrocellulose membranes. The membranes were blocked with
phosphate-buffered saline (PBS) containing 5% non-fat milk
overnight at 4°C, incubated with the aforementioned primary
antibodies overnight at 4°C, and then with the aforementioned
HRP-conjugated secondary antibodies for 2 h at room temperature. An
enhanced chemiluminescence detection kit (Thermo Fisher Scientific,
Inc.) was used for immunoblotting. Protein belts were exposed on
the films in a dark room, then scanned and saved for the edition by
Photoshop 7.0 software (Adobe Systems Inc., San Jose, CA, USA).
MTT assay
Cells (5×103 cells/well) were seeded in a
96-well plate and each sample was placed in 6 parallel wells. A
total of 10 µl 5 mg/ml MTT (Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China) was added to each well for 4
h for 24, 48 or 72 h. The supernatant was then discarded, 100 µl
DMSO was added and the plate was shaken with a Transference
Decoloring Shaker (ZD-2008; Haimen Kylin-Bell Instrument
Manufacturing Co., Ltd., Shanghai, China) at a fixed low speed at
room temperature for 10 min. The optical density (OD) value was
measured using a microplate reader (BioTek Instruments, Inc.,
Winooski, VT, USA) at a wavelength of 490 nm. The relative
proliferative ratio of the cells per well was calculated according
to the following formula: Relative proliferation (%) =
(ODtransfection/ODcontrol) × 100%. The
relative proliferative ratio of the blank control was set as 100%
at all time points.
Colony formation analysis
Cells were seeded in media at a density of
2×103 cells/well in 6-well plates (2 parallel
wells/sample) for ~2 weeks at 37°C in an incubator with an
atmosphere containing 5% CO2. When clear colonies were
observed by the naked eye, the plates were washed three times with
PBS and fixed with 100% methanol for 15 min at room temperature.
The cells were then stained with crystal violet for 15 min at room
temperature and washed with water to remove excess dye. Colonies
containing >50 cells were counted under a light inverted
microscope (Leica Microsystems GmbH, Wetzlar, Germany) at ×100
magnification. Colony formation rate (%) = (means number of
colonies/2000) × 100%. The experiment was repeated three times.
Wound healing assays
Cells were cultured overnight at 37°C in an
atmosphere containing 5% CO2 to yield a monolayer in
6-well plates, and then the monolayer was scratched using 200-µl
pipette tips, washed twice with PBS and RPMI-1640 medium was added.
Wound healing status was monitored at 0, 12, 24 and 36 h, and
images were captured with an inverted light microscope at ×4
magnification in order to analyze the capability of cell migration
in the 2-dimensional plane.
Transwell assays
The upper chambers of a Transwell plate with 8.0 µm
pore size of polycarbonate membrane of polystyrene plates (cat. no.
3422; Costar, Corning Incorporated, Corning, NY, USA) were
pretreated with heated serum-free RPMI-1640 medium for 30 min at
37°C and then the medium was removed. Cells were adjusted to a
concentration of 2×105 cells/ml in RPMI-1640 medium with
0.2% FBS. A total of 200 µl sample was added to the upper chamber,
and the lower chamber was filled 800 µl RPMI-1640 medium with 10%
FBS. The plates were incubated at 37°C for 24 h.
The upper chamber was removed, the medium was
discarded and a cotton swab was used to remove cells on the bottom
of the upper chamber. The remaining migrated cells on the back of
the membrane in the bottom of upper chamber were fixed with 100%
methanol for 10 min at room temperature and stained with 0.1%
crystal violet for 10 min at room temperature for Transwell
analysis in 3-dimensional space. Images were captured in 5 random
visual fields under an inverted light microscope at ×40
magnification. Experiments were repeated three times.
ELISA assays of autocrine IL-10 and
IL-6
A total of 1×106 cells/well were
incubated in 24-well plates containing serum-free incomplete
RPMI-1640 medium without the special indicator phenol red. After 24
h at 37°C, the supernatant was collected for detection of autocrine
IL-10 and IL-6 levels from the tumor cells using ELISA.
The absorbance value was measured at a wavelength of
450 nm with the Human IL-10 (cat. no. EK1102) and Human IL-6 (cat.
no. EK1062) ELISA kits [Hangzhou Multi Sciences (Lianke) Biotech
Co., Ltd., Hangzhou, China], according to the manufacturer's
protocols.
Fluorescence-activated cell sorting
(FACS) analysis
Cells were washed with ice-cold PBS and digested
with 0.25% trypsin. Cells were collected for centrifugation at 697
× g for 5 min at room temperature, and the pellet was suspended
with PBS as a single-cell solution, which was centrifuged at 697 ×
g at room temperature and washed twice with PBS. Cells were
resuspended with 300 µl PBS and fixed with 700 µl 70% ethanol at
4°C overnight for the detection of cell cycle distribution and
apoptosis.
Cell cycle distribution and apoptosis were detected
using FACS, which was performed by a specialized institute (Beijing
Dingguo Changsheng Biotechnology Co., Ltd., Beijing, China).
Statistics
Statistical analysis was performed using the
statistical software SPSS, version 17.0 (SPSS, Inc., Chicago, IL,
USA). Data are presented as the mean ± standard deviation. Analysis
of variance and least significant difference method were applied to
perform multiple comparisons between the groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
Introduction of p53wt
decreases Op18/stathmin expression and phosphorylation
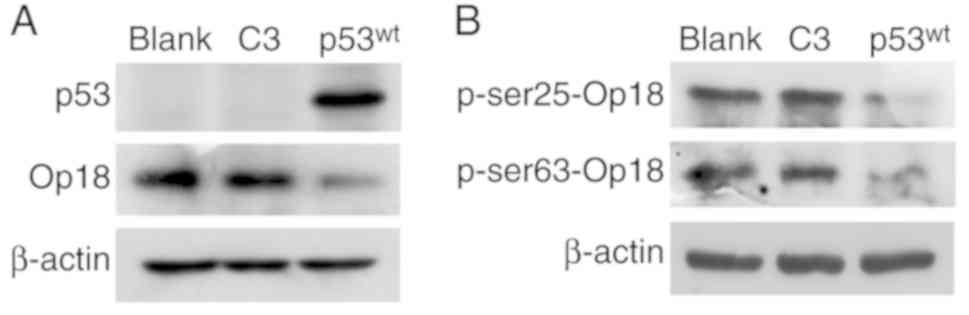
Western blotting for p53 demonstrated that the
p53wt lane exhibited a band while the blank and the C3
lanes did not exhibit any trace of p53, which demonstrated that the
NCI-H1299 cells were originally p53-deficient, and that exogenous
p53wt was successfully introduced and expressed
in the cells. There were no differences in the expression between
the blank and the C3 controls, but the expression of Op18/stathmin
was notably impaired in the p53wt group, compared with
the two control groups (Fig.
1A).
Similarly, the levels of Op18/stathmin
phosphorylation at the Ser25 and Ser63 sites were notably decreased
in the p53wt group, compared with the two control
groups, and there were no evident changes in the expression of
Op18/stathmin at these two sites of phosphorylation between the
blank and C3 control groups (Fig.
1B).
p53 impairs the capabilities of cell
proliferation and colony formation
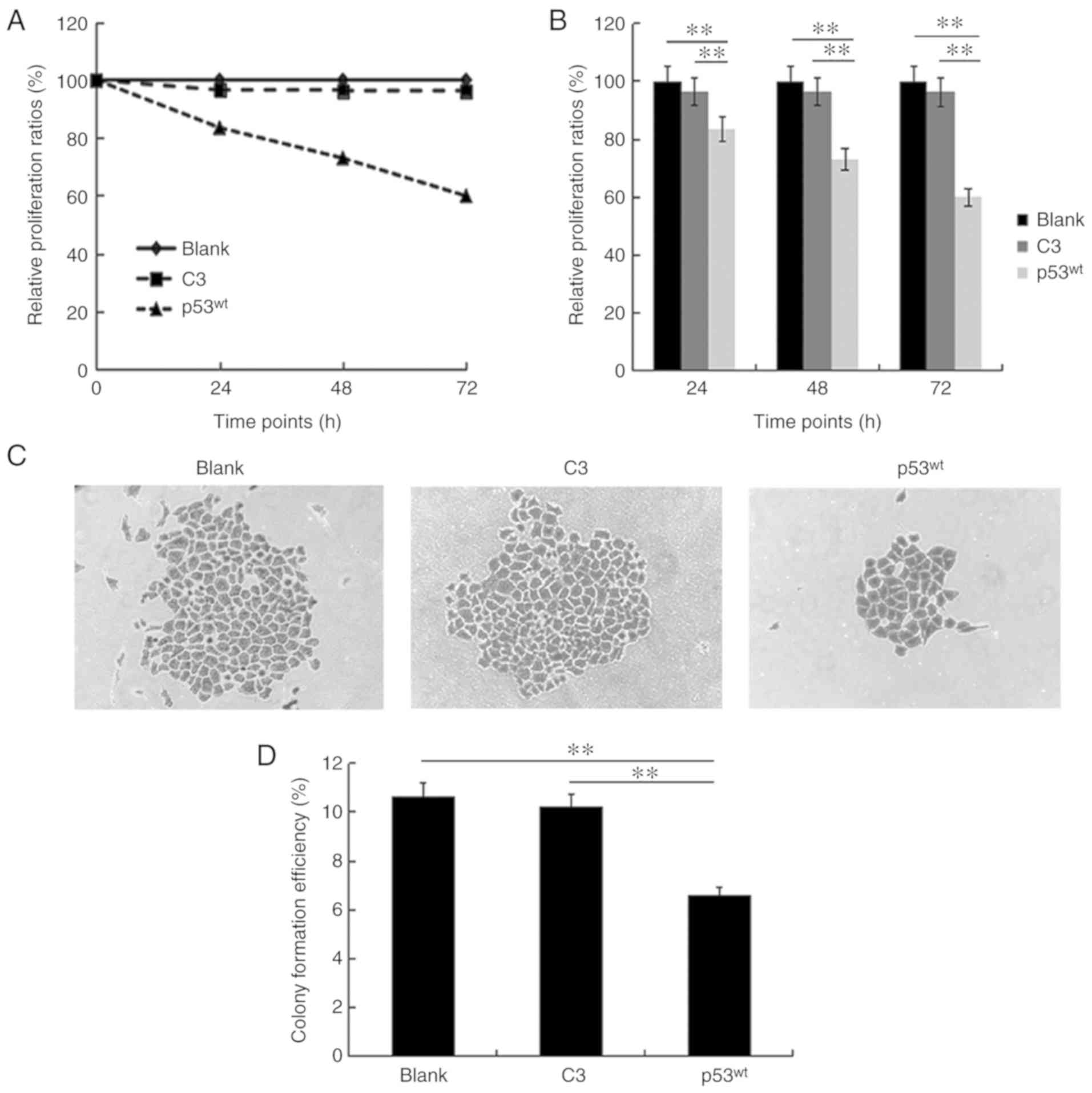
Cell proliferation curves demonstrated that the
relative proliferation ratios were 96.5, 96.3 and 96.2% in the C3
control group at the 24, 48 and 72 h time points, respectively,
which were similar to the blank group, that was set as 100% at all
time points. Compared with the two approximately parallel curves
for the blank and C3 control groups, the representative curve for
the p53wt group descended steeply with proliferation
ratios of 83.5, 72.9 and 59.9% at the three time points,
respectively (Fig. 2A). The
histograms demonstrated that no statistical difference existed
between the blank and C3 control groups at the three time points,
but the relative proliferation ratio of the p53wt group
was significantly reduced, compared with the two control groups at
all time points (P<0.01; Fig.
2B).
Colony formation analysis demonstrated that a number
of large colonies composed of several small colonies with merging
borders, as depicted in the blank and C3 control groups. By
contrast, there were only a few sparsely distributed small colonies
in the p53wt group (Fig.
2C). The mean colony formation ratios, which implied the mean
percentage of forming colonies (>50 cells) in 2,000 cells, were
10.65 and 10.23% in the two control groups, respectively, and 6.62%
in the p53wt group, which was significantly reduced,
compared with the other groups (P<0.01). The difference between
the ratios of the two control groups was not significant (Fig. 2D).
p53 suppresses cell migration in
multi-dimensional spaces
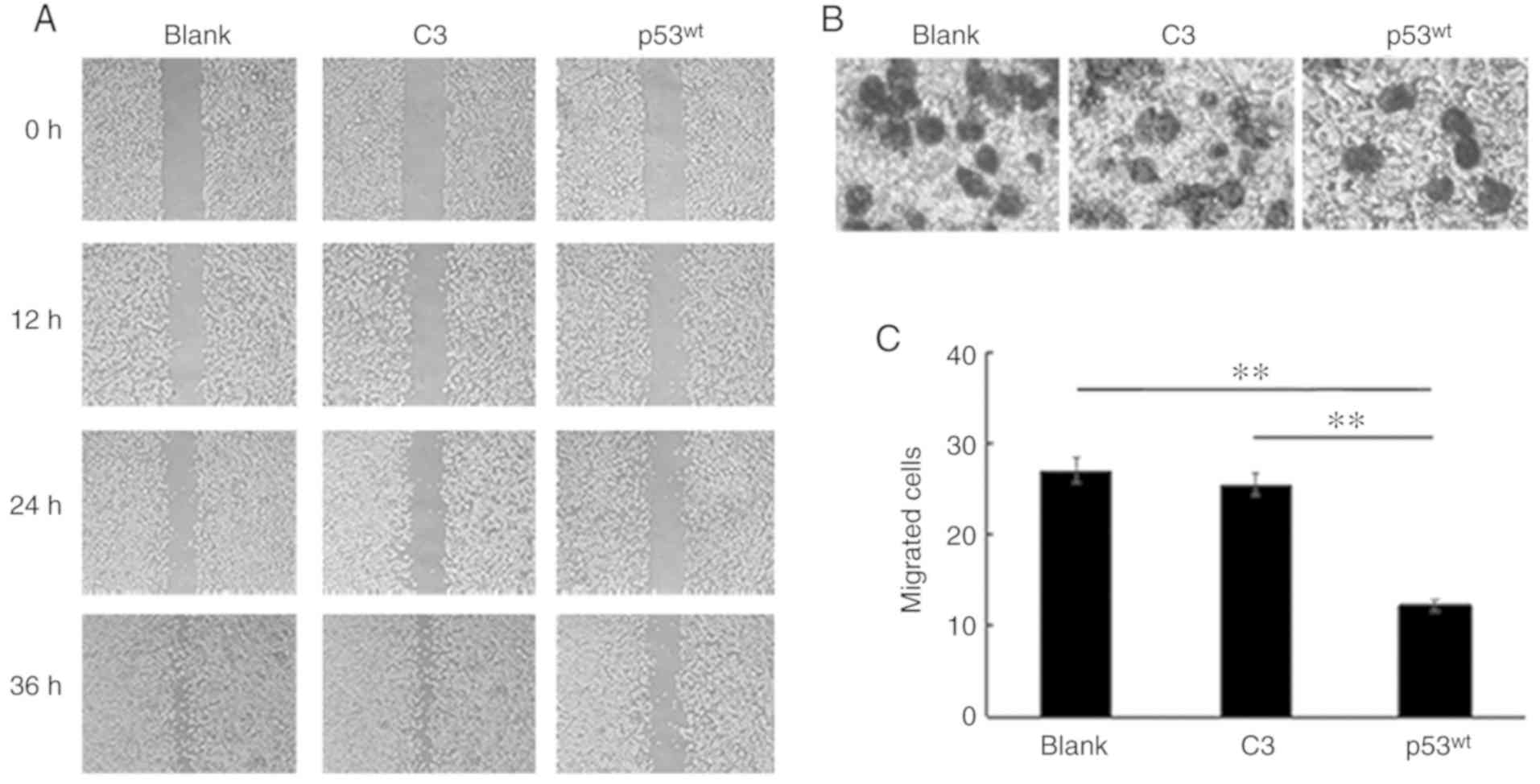
The wound healing assays demonstrated that a large
proportion of the scratched area remained empty among all three
groups at the 12 h time point. After 24 h, the wound width of the
C3 group was reduced, compared with the blank group, but the
difference was not notable by the naked eye. The wounds had almost
healed at 36 h in the blank and C3 control groups; however, a large
area of the scratched region remained empty in the p53wt
group. p53wt introduction inhibited the migration
of NCI-H1299 cells in the 2-dimensional plane (Fig. 3A).
The Transwell assays identified that the mean number
of migrated cells was 28, 26 and 14 among the blank, C3 and
p53wt groups, respectively. Therefore, the number of
migrated cells was notably decreased following
p53wt introduction (Fig. 3B). Histograms indicated that there
was no apparent difference in the number of infiltrating cells
between the blank and C3 control groups, but the number of migrated
cells was significantly reduced in the p53wt group,
compared with the two control groups; therefore,
p53wt significantly reduced the motility of cells
in 3-dimensional space (P<0.01; Fig.
3C).
p53 negatively modulates the
activities of CDC2 and ERK by inhibiting NF-κB expression
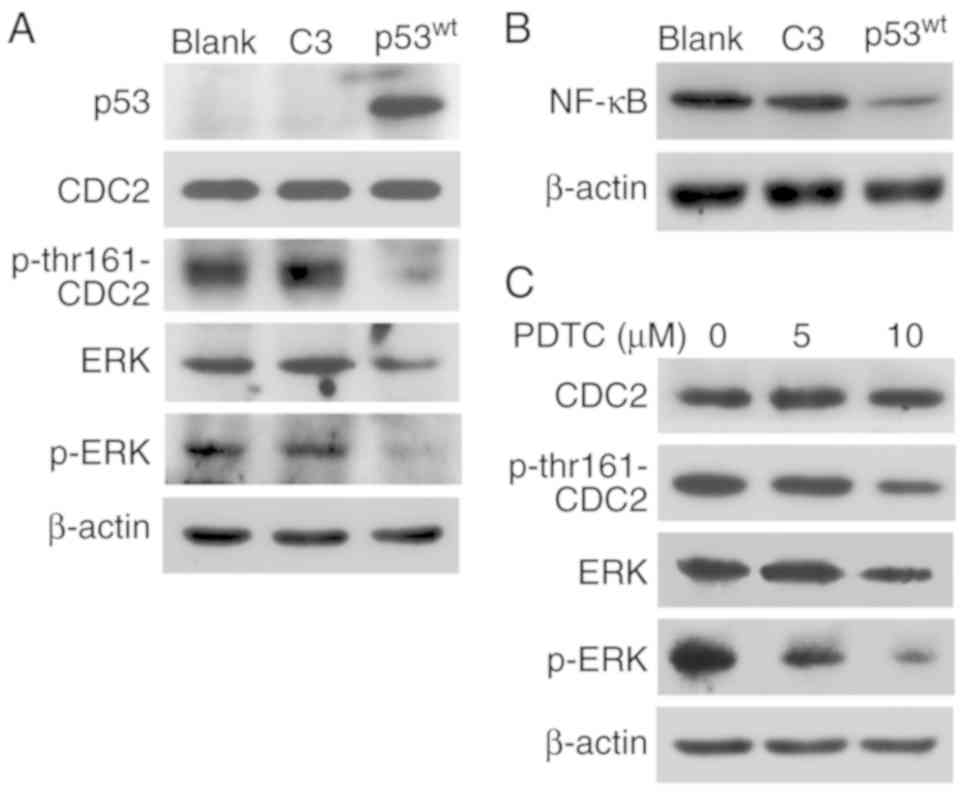
Western blotting indicated that p53 expression
successfully attenuated CDC2 phosphorylation at the thr161 site.
The levels of ERK and p-ERK were also downregulated in the
p53wt group, but those of CDC2 were similar among the
three groups (Fig. 4A).
Additionally, introduction of exogenous p53wt
reduced the expression of NF-κB (Fig.
4B).
The NF-κB inhibitor PDTC was employed to block NF-κB
signaling in p53-deficient NCI-H1299 cells. Western blot analysis
demonstrated that the levels of p-thr161-CDC2, ERK and p-ERK, with
ERK being slightly decreased at 5 µM, compared with at 0 µM and
notably reduced at 10 µM, while p-ERK gradually decreased in a PDTC
concentration-dependent manner. Blockage of NF-κB signaling also
downregulated the activities of p-thr161-CDC2 and ERK, which is in
agreement with the results of p53 introduction, and the
concentration gradient of PDTC did not exert any effects on CDC2
expression (Fig. 4C).
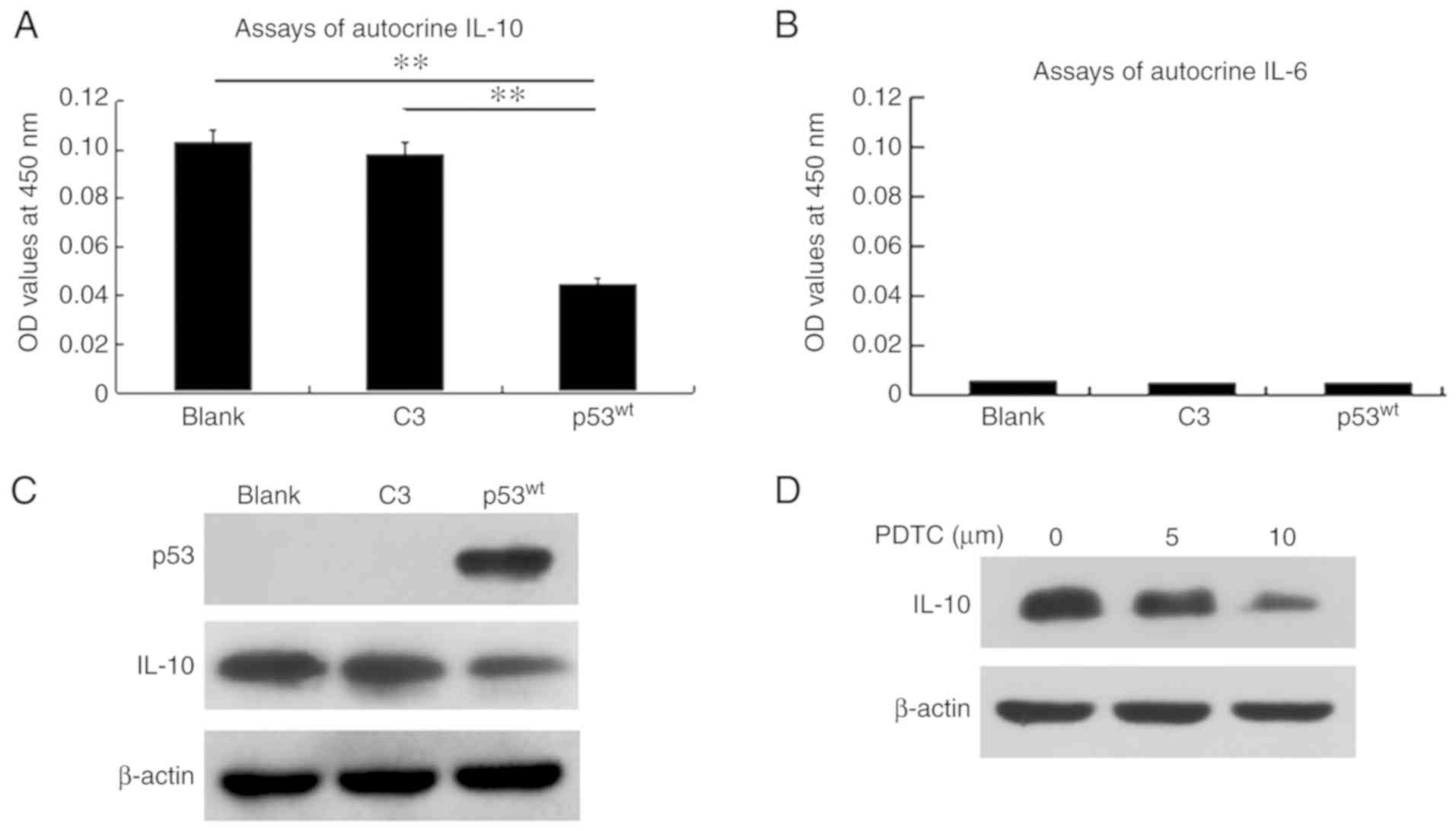
p53 inhibits IL-10 autocrine and
protein expression
ELISA confirmed that the mean autocrine IL-10 OD
level in the culture supernatant was 0.109, 0.093 and 0.055 in the
blank, C3 and p53wt groups, respectively. The mean level
of the p53wt group was significantly decreased, compared
with the two control groups (P<0.01), and there was no
significant difference between those of the blank and C3 control
groups (Fig. 5A). ELISA
demonstrated that IL-6 autocrine OD levels were low in the three
groups and p53 did not affect IL-6 (Fig. 5B).
Western blot analysis demonstrated that IL-10 was
expressed normally in the blank and C3 control groups, which was
similar between them, but was notably attenuated in the
p53wt group. Additionally, IL-6 was not expressed in the
three groups (Fig. 5C).
Concentration gradients of the inhibitor PDTC
downregulated the expression of IL-10 in a concentration-dependent
manner in NCI-H1299 cells, which implied that blockage of NF-κB
signaling induced the identical inhibitory effects on IL-10
expression as exogenous p53wt induction (Fig. 5D).
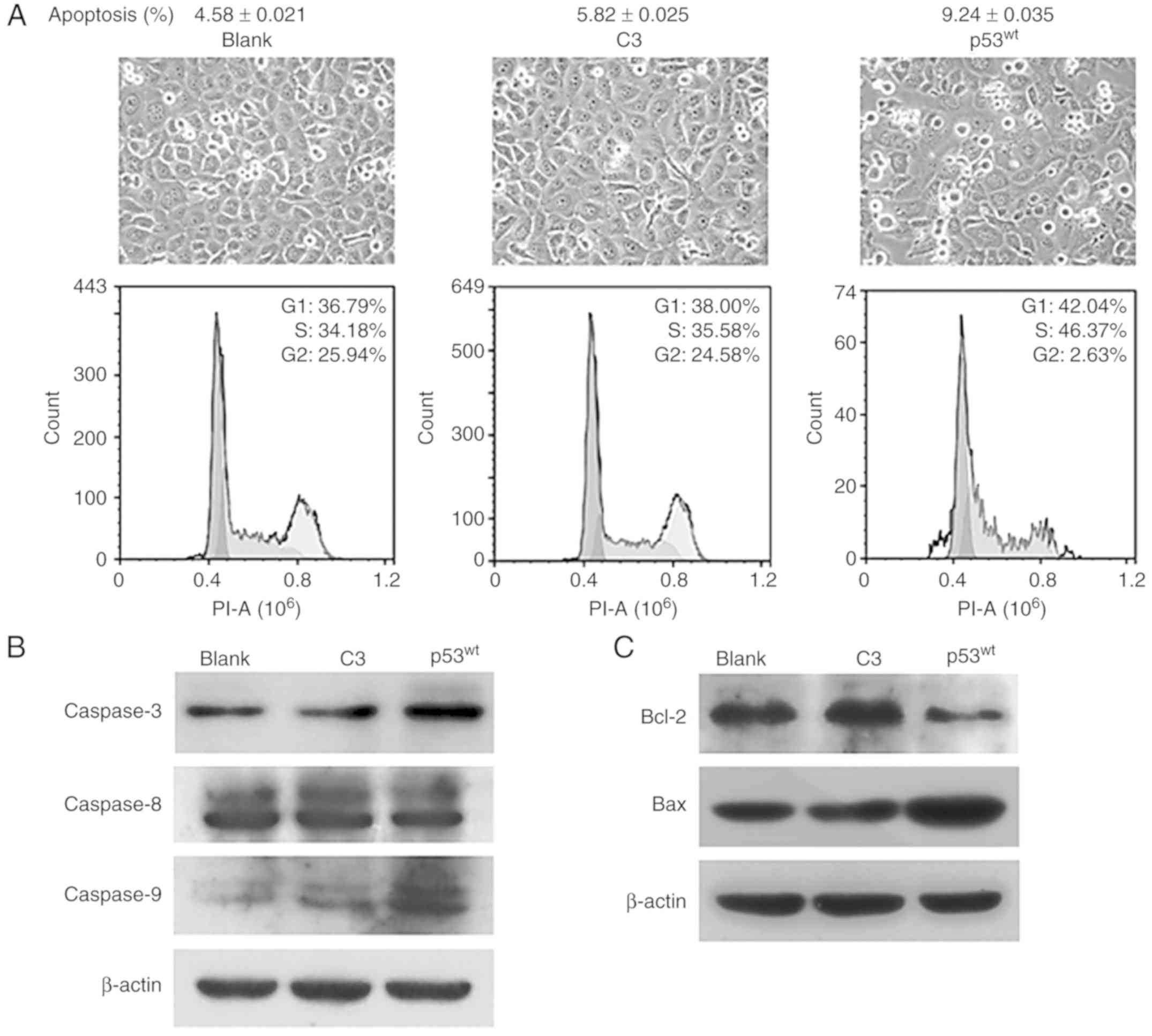
p53 induces cell cycle arrest at the
G1/S phases and cellular apoptosis
The images of cell growth demonstrated that cells
were nearly confluent in the blank and C3 control groups, but there
was reduced quantity of cells in the p53wt group. FACS
analysis validated that the proportion of cell that entered the
G2 phase was 25.94, 24.58 and 2.63% among the blank, C3
and p53wt groups, respectively, while the cellular
apoptosis ratios were 4.58, 5.82 and 9.24%, respectively. By
contrast, the majority of NCI-H1299 cells were arrested at
G1/S phases when transfected with exogenous
p53wt (Fig.
6A).
The expression levels of caspase-3 and −9 were
increased in the p53wt group, compared with the two
control groups, while no evident changes were observed in caspase-8
expression levels in the three groups (Fig. 6B). Introduction of p53 attenuated
the expression of the anti-apoptotic protein Bcl-2, and upregulated
the levels of its counterpart Bax (Fig.
6C). The expression levels of all of these molecules in the
blank control group were similar to those in the C3 group (Fig. 6B and C).
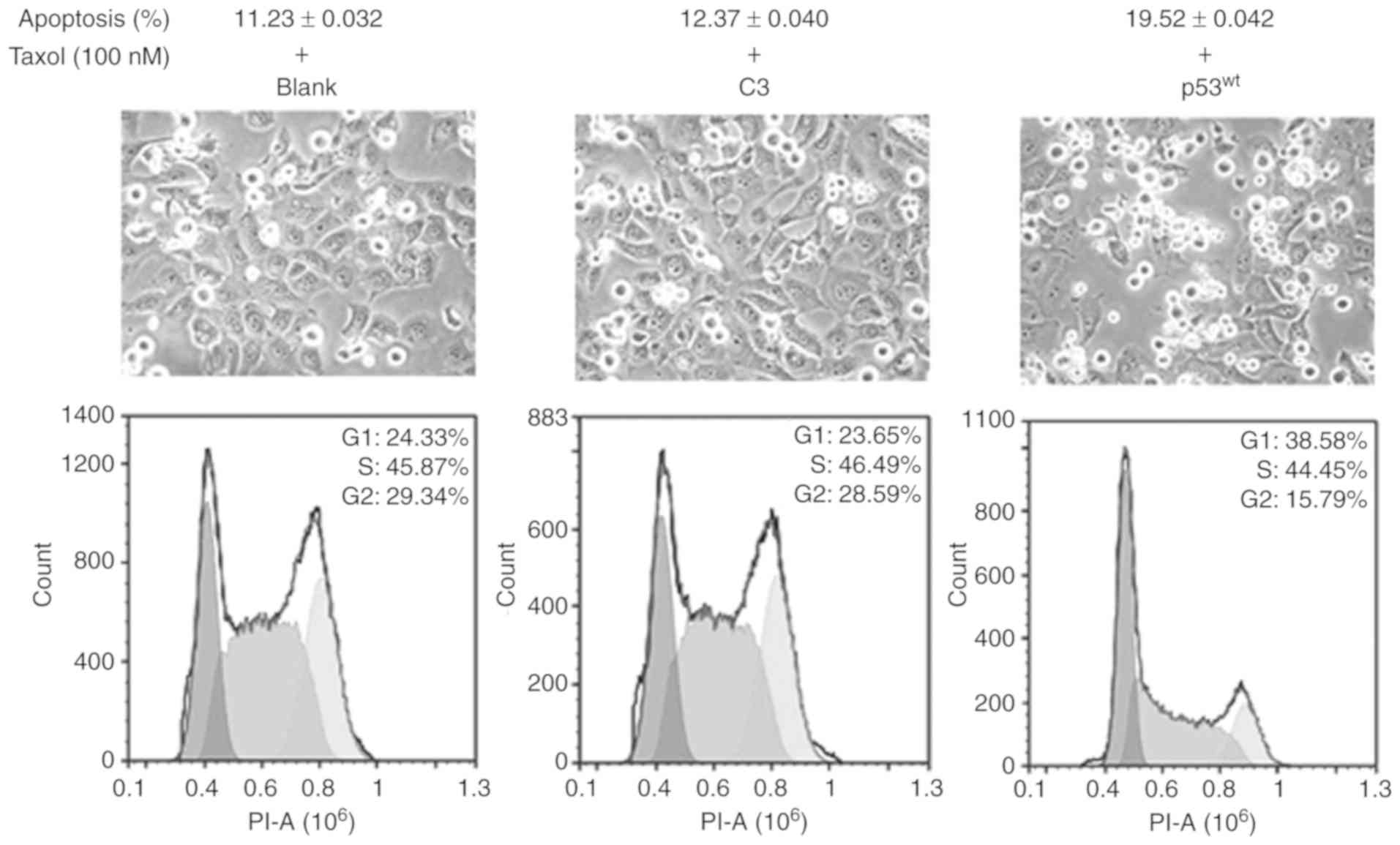
p53 promotes the sensitivity of
NCI-H1299 cells to Taxol
The images of cell growth demonstrated that there
was a large amount of cells in the suspension in the three groups
after 24 h treatment with 100 nM Taxol, and the adherent cells
became significantly sparse in the p53wt group, compared
with the two control groups. FACS analysis indicated that the
ratios of cell apoptosis were 11.23, 12.37 and 19.52% among the
blank, C3 and p53wt groups, respectively. The ratios of
G2 phase cells were 29.34, 28.59 and 15.79% among the
blank, C3 and p53wt groups, respectively. These results
indicated that p53wt introduction resulted in an
increase in the cytotoxicity of Taxol in NCI-H1299 cells and a
decrease in the ratios of cells at the G2 phases
(Fig. 7).
Discussion
p53 is a tumor suppressor gene that primarily
arrests aberrant cells at the G1/S phase checkpoint for
repair of damaged DNA, which results in apoptosis when the degree
of destroyed DNA is beyond the threshold of repair (5). Mutation of p53 that occurs in
the majority of tumors results in failure to control cell growth,
apoptosis and DNA repair, and ultimately becomes a tumor-promoting
gene that induces carcinogenesis and drug resistance (14). There are >50% of tumors that
exhibit p53 gene deficiency or mutation; therefore,
utilization of p53wt against tumors is an
attractive treatment strategy (15,16).
Our previous studies confirmed that silencing
Op18/stathmin would result in the inhibition of cell migration and
invasion, which implied that the downregulation of Op18/stathmin
expression was involved in the inhibition of cell migration and
invasion in NCI-H1299 cells following the introduction of exogenous
p53wt (1,2). The present study confirmed that the
introduction of exogenous p53wt successfully
inhibits Op18/stathmin expression and phosphorylation, and
decreases the capacities of proliferation and colony formation, as
well as migration in multi-dimensional spaces in NCI-H1299 cells.
In melanoma A375, Hs294T and G361 cells, a previous study also
determined that increased p53 expression resulted in inhibition of
cellular growth and viability following treatment with the histone
deacetylase sirtuin type 1 inhibitor Tenovin-1 (17).
The present study specifically demonstrated the
status of Op18/stathmin expression and its phosphorylation
following the introduction of exogenous p53wt;
however, further experiments primarily indicated the changes of
regulatory molecules CDC2, ERK and NF-κB on Op18/stathmin signaling
for the intervention of exogenous p53wt which
negatively modulated the activities of CDC2 and ERK by inhibiting
NF-κB expression. CDC2 is a positive regulator of the cell cycle
process, in which aberrant activation is associated with
acceleration of cell proliferation and malignant transformation in
the development of tumors (18–20).
ERK is a major member of the mitogen-activated protein kinases
family, and is also involved in the positive regulation of cell
proliferation and differentiation (21,22).
In Epstein-Barr virus (EBV)-infected nasopharyngeal carcinoma CNE1
cells, Op18/stathmin was identified as the downstream target of
CDC2 and ERK, and EBV-encoding latent membrane protein 1 promotes
cell cycle progression through the regulation of CDC2- and
ERK-mediated Op18/stathmin signaling (23,24).
Previous studies indicated that Op18/stathmin was dephosphorylated
in NCI-H1299 cells treated with the CDC2 blocker purvalanol A or
ERK blocker PD98059, both of which enhance the sensitivity of cells
to Taxol (25,26). Due to the activity of Op18/stathmin
regulating microtubules being regulated by the balance of
phosphorylation and dephosphorylation, the phosphorylation levels
of Op18/stathmin at the Ser25 and 63 sites following the
introduction of p53wt were detected. The present
study demonstrated that exogenous p53wt
expression inhibits the phosphorylation of CDC2-thr161 and ERK,
downregulates the activities of CDC2 and ERK kinases, and
negatively regulates Op18/stathmin phosphorylation at the Ser25 and
Ser63 sites. The decreased phosphorylation of Op18/stathmin was
associated with the downregulation of Op18/stathmin expression and
the loss of the activities of its upstream kinases, ERK and CDC2,
following exogenous p53wt introduction.
The transcription factor NF-κB is associated with
cell proliferation, and blocking of NF-κB signaling results in
inhibition of tumor growth (27,28).
In the present study, introduction of exogenous
p53wt decreased the expression levels of NF-κB
and the activities of CDC2 and ERK, while PDTC blocking of NF-κB
signaling also resulted in decreased levels of CDC2 and ERK
activity in the NCI-H1299 cells. These results demonstrated that
p53 negatively regulates the activation of CDC2 and ERK via NF-κB,
and indirectly influences Op18/stathmin expression and
phosphorylation. Therefore, p53 regulates Op18/stathmin signaling
through the p53-NF-κB-CDC2/ERK-Op18/stathmin pathway.
IL-10 is a pleiotropic cytokine and is considered as
an immunosuppressor that mediates tumor immune evasion through
inhibition of the host immune response, therefore serving a vital
role in tumor survival and metastasis (29). IL-6 is also a multi-functional
cytokine secreted by a variety of cell types and is frequently
involved in a series of physiological and pathological effects,
including inflammation and fibrosis (30). Aberrant expression of IL-6 is
associated with the poor prognosis of patients with glioma
(31), and gallbladder and colon
cancer types (32,33). The effects of
p53wt on the release of autocrine cytokines IL-10
and IL-6 and their protein expression levels in NCI-H1299 cells
were also investigated in the present study, which indicated that
p53 inhibits IL-10 in vitro autocrine and protein
expression, but does not exert any influence on IL-6. We
hypothesized that p53-mediated Op18/stathmin signaling is
implicated in the immune evasion of tumors via autocrine IL-10.
However, IL-6 autocrine and expression remained at low levels in
the NCI-H1299 cells with or without exogenous p53. A previous study
reported that autocrine IL-10 suppressed the production of
pro-inflammatory cytokines, including tumor necrosis factor-α, IL-1
and IL-6, and further mediated immune evasion by inhibiting the
ability of antigen-presenting cells to present antigens to T cells
(33).
Caspase-3 and −9 are the executor and promoter of
the endogenous cell death pathway, respectively (25). Bcl-2 is an anti-apoptotic protein
and Bax primarily acts against the anti-apoptotic activity of Bcl-2
(25,35,36).
In the present study, p53 upregulated the expression levels of
caspase-3 and −9, and Bax, and decreased the levels of Bcl-2
expression in NCI-H1299 cells. p53 resulted in cell cycle arrest at
the G1/S phases and an increase in cell apoptosis,
enhancing the sensitivity of NCI-H1299 cells to Taxol, which
indicates that the lack of p53 is associated with the development
of Taxol resistance in NCI-H1299 cells with high expression of
Op18/stathmin. Introduction of exogenous p53wt
resulted in a majority of cells arresting at the G1/S
phases, but the ratios of NCI-H1299 cells halted at the
G1/S phases was only slightly reduced following the
co-treatment of p53wt and Taxol, which may be
associated with Taxol primarily inducing cells arresting at the
G2/M phase, eventually causing a decrease of
G1 stage cells following the synergistic induction of
p53wt and Taxol, compared with the treatment of
p53wt alone (37).
Acknowledgements
The authors would like to thank Professor Cao Ya
from the Cancer Research Institute of Central South University
(Changsha, China) for providing the plasmids.
Funding
This study was financed by the Key Scientific
Research Project of Colleges and Universities of Hunan Province
(grant no. 12A018), the National Natural Science Foundation of
China (grant no. 81272274) and the Natural Science Foundation of
Hunan Province (grant no. 12JJ3104).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
XL was primarily responsible for the design of the
study and the revision of the manuscript. The experiments, analysis
and interpretation of the data were primarily conducted by SC.
Members of the research team also included YZ, FS, DL and TY, who
participated in part of the experiments, analysis and
interpretation of the data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lin X, Liao Y, Yang J, Su L, Zou H and Zuo
Y: Regulation of the drug-resistance of carcinoma cells mediated by
Op18/stathmin. Chem Life. 33:265–268. 2013.
|
|
2
|
Long D, Yu T, Chen X, Liao Y and Lin X:
RNAi targeting STMN alleviates the resistance to taxol and
collectively contributes to down regulate the malignancy of NSCLC
cells in vitro and in vivo. Cell Biol Toxicol. 34:7–21. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Guntur VP, Waldrep JC, Guo JJ, Selting K
and Dhand R: Increasing p53 protein sensitizes non-small cell lung
cancer to paclitaxel and cisplatin in vitro. Anticancer Res.
30:3557–3564. 2010.PubMed/NCBI
|
|
4
|
Lv Y, Huo Y, Yu X, Liu R, Zhang S, Zheng X
and Zhang X: TopBP1 contributes to the chemoresistance in non-small
cell lung cancer through upregulation of p53. Drug Des Devel Ther.
10:3053–3064. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Riley T, Sontag E, Chen P and Levine A:
Transcriptional control of human p53-regulated genes. Nat Rev Mol
Cell Biol. 9:402–412. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Matt S and Hofmann TG: The DNA
damage-induced cell death response: A roadmap to kill cancer cells.
Cell Mol Life Sci. 73:2829–2850. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang EB, Yin DD, Sun M, Kong R, Liu XH,
You LH, Han L, Xia R, Wang KM, Yang JS, et al: P53-regulated long
non-coding RNA TUG1 affects cell proliferation in human non-small
cell lung cancer, partly through epigenetically regulating HOXB7
expression. Cell Death Dis. 5:e12432014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kracikova M, Akiri G, George A,
Sachidanandam R and Aaronson SA: A threshold mechanism mediates p53
cell fate decision between growth arrest and apoptosis. Cell Death
Differ. 20:576–588. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin X and Cao Y: Advances in the
regulation of the signals on Op18/stathmin. Life Sci Res.
11:195–199. 2007.
|
|
10
|
Cassimeris L: The oncoprotein 18/stathmin
family of microtubule destabilizers. Curr Opin Cell Biol. 14:18–24.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Belletti B and Baldassarre G: Stathmin: A
protein with many tasks. New biomarker and potential target in
cancer. Expert Opin Ther Targets. 15:1249–1266. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Johnsen JI, Aurelio ON, Kwaja Z, Jörgensen
GE, Pellegata NS, Plattner R, Stanbridge EJ and Cajot JF:
p53-mediated negative regulation of stathmin/Op18 expression is
associated with G2/M cell-cycle arrest. Int J Cancer.
88:685–691. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bessho R, Matsubara K, Kubota M, Kuwakado
K, Hirota H, Wakazono Y, Lin YW, Okuda A, Kawai M and Nishikomori
R: Pyrrolidine dithiocarbamate, a potent inhibitor of nuclear
factor kappa B (NF-kappa B) activation, prevents apoptosis in human
promyelocytic leukemia HL-60 cells and thymocytes. Biochem
Pharmacol. 48:1883–1889. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shen F, Wu Y and Lin X: The pleiotropic
function of P53. Basic Clin Med. 35:1672–1676. 2015.(In
Chinese).
|
|
15
|
Olive KP, Tuveson DA, Ruhe ZC, Yin B,
Willis NA, Bronson RT, Crowley D and Jacks T: Mutant p53 gain of
function in two mouse models of Li-Fraumeni syndrome. Cell.
119:847–860. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fuster JJ, Sanz-Gonzalez SM, Moll UM and
Andres V: Classic and novel roles of p53: Prospects for anticancer
therapy. Trends Mol Med. 13:192–199. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wilking MJ, Singh C, Nihal M, Zhong W and
Ahmad N: SIRT1 deacetylase is overexpressed in human melanoma and
its small molecule inhibition imparts anti-proliferative response
via p53 activation. Arch Biochem Biophys. 563:94–100. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu P, Kao TP and Huang H: CDK1 promotes
cell proliferation and survival via phosphorylation and inhibition
of FOXO1 transcription factor. Oncogene. 27:4733–4744. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Perez de Castro I, de Carcer G and
Malumbres M: A census of mitotic cancer genes: New insights into
tumor cell biology and cancer therapy. Carcinogenesis. 28:899–912.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dhillon AS, Hagan S, Rath O and Kolch W:
MAP kinase signalling pathways in cancer. Oncogene. 26:3279–3290.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang J and Wu GS: Role of autophagy in
cisplatin resistance in ovarian cancer cells. J Biol Chem.
289:17163–17173. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lin X, Liu S, Luo X, Ma X, Guo L, Li L, Li
Z, Tao Y and Cao Y: EBV-encoded LMP1 regulates Op18/stathmin
signaling pathway by cdc2 mediation in nasopharyngeal carcinoma
cells. Int J Cancer. 124:1020–1027. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin X, Tang M, Tao Y, Li L, Liu S, Guo L,
Li Z, Ma X, Xu J and Cao Y: Epstein-Barr virus-encoded LMP1
triggers regulation of the ERK-mediated Op18/stathmin signaling
pathway in association with cell cycle. Cancer Sci. 103:993–999.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen X, Liao Y, Long D, Yu T, Shen F and
Lin X: The Cdc2/Cdk1 inhibitor, purvalanol A, enhances the
cytotoxic effects of taxol through Op18/stathmin in non-small cell
lung cancer cells in vitro. Int J Mol Med. 40:235–242. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin X, Liao Y, Chen X, Long D, Yu T and
Shen F: Regulation of oncoprotein 18/stathmin signaling by ERK
concerns the resistance to taxol in nonsmall cell lung cancer
cells. Cancer Biother Radiopharm. 31:37–43. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huan L, Bao C, Chen D, Li Y, Lian J, Ding
J, Huang S, Liang L and He X: MicroRNA-127-5p targets the
biliverdin reductase B/nuclear factor-κB pathway to suppress cell
growth in hepatocellular carcinoma cells. Cancer Sci. 107:258–266.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lu Y, Liu C, Cheng H, Xu Y, Jiang J, Xu J,
Long J, Liu L and Yu X: Stathmin, interacting with Nf-κB, promotes
tumor growth and predicts poor prognosis of pancreatic cancer. Curr
Mol Med. 14:328–339. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Incrocci R, Barse L, Stone A, Vagvala S,
Montesano M, Subramaniam V and Swanson-Mungerson M: Epstein-barr
virus latent membrane protein 2A (LMP2A) enhances IL-10 production
through the activation of Bruton's tyrosine kinase and STAT3.
Virology. 500:96–112. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kawaratani H, Moriya K, Namisaki T, Uejima
M, Kitade M, Takeda K, Okura Y, Kaji K, Takaya H, Nishimura N, et
al: Therapeutic strategies for alcoholic liver disease: Focusing on
inflammation and fibrosis (Review). Int J Mol Med. 40:263–270.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shan Y, He X, Song W, Han D, Niu J and
Wang J: Role of IL-6 in the invasiveness and prognosis of glioma.
Int J Clin Exp Med. 8:9114–9120. 2015.PubMed/NCBI
|
|
32
|
Zhang M, Gong W, Zhang Y, Yang Y, Zhou D,
Weng M, Qin Y, Jiang A, Ma F and Quan Z: Expression of
interleukin-6 is associated with epithelial-mesenchymal transition
and survival rates in gallbladder cancer. Mol Med Rep.
11:3539–3546. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Olsen J, Kirkeby LT, Olsen J, Eiholm S,
Jess P, Gögenur I and Troelsen JT: High interleukin-6 mRNA
expression is a predictor of relapse in colon cancer. Anticancer
Res. 35:2235–2240. 2015.PubMed/NCBI
|
|
34
|
Mittal SK and Roche PA: Suppression of
antigen presentation by IL-10. Curr Opin Immunol. 34:22–27. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Klumpp D, Misovic M, Szteyn K, Shumilina
E, Rudner J and Huber SM: Targeting TRPM2 channels impairs
radiation-induced cell cycle arrest and fosters cell death of T
cell leukemia cells in a Bcl-2-dependent manner. Oxid Med Cell
Longev 2016. 80267022016.
|
|
36
|
Veena VK, Kennedy K, Lakshmi P, Krishna R
and Sakthivel N: Anti-leukemic, anti-lung, and anti-breast cancer
potential of the microbial polyketide 2,4-diacetylphloroglucinol
(DAPG) and its interaction with the metastatic proteins than the
antiapoptotic Bcl-2 proteins. Mol Cell Biochem. 414:47–56. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Oyaizu H, Adachi Y, Okumura T, Okigaki M,
Oyaizu N, Taketani S, Ikebukuro K, Fukuhara S and Ikehara S:
Proteasome inhibitor 1 enhances paclitaxel-induced apoptosis in
human lung adenocarcinoma cell line. Oncol Rep. 8:825–829.
2001.PubMed/NCBI
|