Introduction
It has been reported that tescalcin (TESC) is
upregulated by treatment with class I histone deacetylase (HDAC)
inhibitors. In addition, TESC plays important roles related to
chromatin remodeling, transcriptional regulation, and epigenetic
modification (1) by interacting
with the cytosolic tail domain of Na+/H+
exchanger isoform type-1 (NHE1), implicating novel gene functions
in the regulation of NHE1 in cardiac tissues (2). This finding was further supported by a
study that revealed that Na+/H+ exchanger was
regulated by both post-translational modifications and a number of
regulatory binding proteins, including calmodulin, moesin and TESC
(3). Notably, it was demonstrated
that NHE1 was directly associated with cellular transformation,
invasion and metastasis in cancer, suggesting that NHE1 is a
potential novel drug target for anticancer therapeutics (4), and this was further supported by the
observation of TESC overexpression in melanoma and colorectal and
gastric cancers (5–7). However, the regulation of TESC
expression in cancer and its underlying mechanisms are largely
unknown.
TESC overexpression in K562 cells has been
demonstrated to regulate cell proliferation via activation of ERK
signaling, while the expression of Ets transcription factors,
including Ets-1, Ets-2, and Fli-1, was blocked at the mRNA level
following TESC knockdown (8). The
Ets gene family belongs to one of the largest family of
transcriptional targets in cancer (9–12).
Recent research further indicated that high levels of DNA
methylation were frequently observed in the binding sites of Ets
transcription factors, including PDEF, GABPA, ELF1, FLI1 and ETS2
(13).
Epigenetic alterations caused by the methylation of
cytosine guanine dinucleotide (CpG) islands are reported to play
pivotal regulatory roles in gene expression, differentiation,
apoptosis and tumorigenesis (14,15).
In addition, promoter methylation is considered an important
regulatory mechanism for the expression of gastric cancer-related
genes, such as NDRG2, MGMT, hMLH1, RASSF1A, p16 and RUNX3, which
have been revealed to be involved in gastric carcinogenesis
(16,17). Furthermore, tumor suppressor genes
and oncogenes are mainly detected in repetitive sequences within
the genome as well as in regions of hypermethylation where the gene
promoters are located with CpG islands. Notably, promoter
hypomethylation has been revealed to be associated with oncogene
activation, whereas hypermethylation of CpG sites was revealed to
be associated with inactivation of tumor-suppressor genes (18–21).
In addition, DNA methylation may inhibit gene expression by either
blocking transcription factors or recruiting methyl-CpG binding
proteins (MBDs) via histone methylation (22).
Methyl-CpG-binding domain (MBD) proteins, including
MBD1/2/3/4/5/6 and MeCP2, are classified as epigenetic regulators
(23). Recent studies have
demonstrated the crucial functions of methyl-CpG binding proteins
in epigenetic events in cancer (24), and MBD proteins were revealed to be
involved in tumorigenesis as the initiators of DNA methylation
(25). Among these proteins, MBD1
is the largest, and it has been demonstrated to be associated with
cancer manifestation (26,27). MBD1 plays a role in the drug
resistance of cancers and immune cells (28). MBD1 evoked epigenetic changes via
DNA methylation and the repressive H3K9me3 histone mark (29). Methylation of the lysine residue of
histone H3 (H3K4) is highly associated with the transcriptional
activation of genes, whereas methylation of H3K9 (H3K9me3) results
in the recruiting of factors that inactivate transcription
(30). Thus, these studies
indicated that MBDs may act as a link between DNA methylation and
histone modification (31).
Although the roles of the TESC gene have been
studied in cancer, the detailed molecular mechanism remains
unclear. In addition, the involvement of TESC expression in the
connection of DNA methylation and histone methylation via MBD1 is
largely unknown. Therefore, the aim of the present study was to
investigate the role of TESC expression in epigenetic regulation.
Our data revealed that DNA methylation was negatively associated
with TESC gene expression in gastric cancer cells. Moreover, both
DNA methylation and histone modification via MBD1 may play
potential roles in the regulation of TESC expression.
Materials and methods
Cell lines and culture conditions
The human gastric cancer cell lines, AGS, SNU-216,
NCI-N87, SNU-620, SNU-638, NUGC-3, and MKN-74; human colon cancer
cell lines, COLO205, DLD-1, HCT116, HT-29, KM12C, KM12SM, LS174T
and SW480; human liver cancer cell lines, SK-Hep-1 and Huh-7; human
cervical cancer cell lines, CaSki and SiHa; human lung cancer cell
line, A549; and human breast cancer cell line, MCF-7, were
purchased from the American Type Culture Collection (ATCC;
Manassas, VA, USA) and grown in Dulbecco's modified Eagle's medium
(DMEM) supplemented with 10% fetal bovine serum (FBS). Cells were
grown at 37°C in a humidified, 5% CO2/air atmosphere. To
identify regulation by methyltransferase, 5-aza-2′-deoxycytidine (1
µM; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), a
methyltransferase inhibitor, was added to the culture medium for 72
h to induce demethylation of the cytosine residues.
Sodium bisulfite modification and
methylation-specific PCR (MSP)
Chromosomal DNA was isolated from the cell cultures
grown in 100 mm cell culture dishes (Corning Inc., Corning, NY,
USA) using a genomic DNA purification kit (Promega Corp., Madison,
WI, USA) according to the manufacturer's protocol. The extracted
DNA was eluted with 250 µl of distilled water. Sodium bisulfite
modification of genomic DNA was carried out using an EZ DNA
Methylation kit (Zymo Research Corp., Irvine, CA, USA) according to
the manufacturer's protocol using 0.1 mg of purified DNA. PCR was
carried out as described previously using primers (Table SI) and a Power SYBR-Green kit
(Applied Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) (20). The methylation index
was calculated for each sample using the following formula:
Methylation index = [1/(1 + 2 - (CTu - CTme)] × 100%, where CTu was
the average cycle threshold (CT) obtained from duplicate
quantitative PCR analyses using the unmethylated primer pair and
CTme was the average CT obtained using the methylated primer
pair.
Cell viability assay
Cell viability was assessed with WST-1 assays (Roche
Diagnostics GmbH, Mannheim, Germany) according to the
manufacturer's protocol. Briefly, 10 µl of WST-1 reagent was added
to each well of a 96-well plate (1×103 cells/well).
After incubation for 1 h, the conversion of the WST-1 reagent into
chromogenic formazan was evaluated with a spectrophotometer
(Molecular Devices, LLC, Sunnyvale, CA, USA).
RT-PCR and real-time PCR
Total RNA from cells cultured in a 100-mm cell
culture dish (Corning Inc.) was prepared using TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols. Reverse transcription was conducted using
10 µg of total RNA with a reverse transcription kit (Promega
Corp.). The expression levels of the studied genes were measured by
RT-PCR and real-time PCR analysis. One microliter of cDNA was used
for the PCR, and triplicate reactions were performed for each
sample using an AccuPower PCR premix (Bioneer Corp., Daejeon,
Korea) and a Power SYBR-Green kit (Applied Biosystems; Thermo
Fisher Scientific, Inc.) with gene-specific primers on an ABI
ProFlex PCR system (Applied Biosystems; Thermo Fisher Scientific,
Inc.) and an ABI StepOnePlus instrument (Applied Biosystems; Thermo
Fisher Scientific, Inc.). The primers used for this selected gene
are listed in Table SI. RNA
quantity was normalized to that of β-actin or GAPDH, and gene
expression was quantified according to the 2-ΔCq method (32).
Western blot analysis
Cells cultured in a 100-mm cell culture dish
(Corning Inc.) were resuspended in RIPA lysis buffer [50 mmol/l
Tris-HCl, pH 7.4, 150 mmol/l NaCl, 1% NP40, 0.25% Na-deoxycholate,
1 mmol/l phenylmethylsulfonyl fluoride (PMSF), 1 mmol/l sodium
orthovanadate, 1X protease inhibitor cocktail] (Sigma-Aldrich;
Merck KGaA). Protein was assessed using a BCA Protein Assay Kit
(Thermo Fisher Scientific, Inc.). Proteins were size-fractionated
by 12% SDS-PAGE and transferred to a polyvinylidene difluoride
(PVDF) membrane (Millipore Corp., Billerica, MA, USA). Non-specific
binding was blocked by incubation with phosphate-buffered saline
with Tween-20 (PBST) with 5% powdered milk and 1% Triton X-100.
Membranes were incubated overnight at 4°C with primary antibodies
(TESC; dilution 1:1,000; cat. no. 11125-1-AP; ProteinTech Group,
Inc., Chicago, IL, USA), (MBD1, Sp-1 and MeCP2; dilution 1:1,000;
cat nos. SC-55473, SC-420 and SC-20700; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA), (H3K4me1, H3K4me2, H3K4me3, H3K9me3,
H3K27me3 and H3K36me3; dilution 1:1,000; cat. nos. ab8895, ab7766,
ab8580, ab8898, ab6147 and ab9050, respectively; Abcam, Cambridge,
MA, USA) followed by incubation with polyclonal HRP-conjugated
secondary antibodies (dilution 1:2,000; cat. nos. a6667 and a9917;
Sigma-Aldrich; Merck KGaA) for 1 h at room temperature. The
membranes were incubated in Clarity Western ECL Substrate (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and the protein bands were
visualized by exposure to X-ray film. Protein loading was
visualized by incubation of stripped membranes with a monoclonal
antibody to β-actin (dilution 1:1,000; cat. no. SC-47778; Santa
Cruz Biotechnology, Inc.).
Cell transfection
SNU-638 and NCI-N87 cells (1×105
cell/well) grown in 24-well plates (Corning Inc.) were transfected
with specific-siRNAs (30 nmol/ml) targeting TESC (Bioneer Corp.)
and the pCMV-sports6-TESC vector (2 µg/µl) for 24 h using
Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocols. The medium was replaced
with DMEM containing 10% FBS for 24 h. After recovery, cell
viability was evaluated with the WST-1 assay. The pCMV-sports6
control and pCMV-sports6 TESC vectors were obtained from the Korea
Human Gene Bank (Medical Genomics Research Center, Korea Institute
of Bioscience and Biotechnology, Daejeon, Korea).
Nuclear fractionation
Fractionation of nuclear extracts was carried out
using a Nuclear Extract kit (Active Motif, Carlsbad, CA, USA)
according to the manufacturer's instructions. Pellets were
resuspended in 50 ml of complete lysis buffer, and supernatants
were used as the nuclear fractions after centrifugation at 14,000 ×
g for 10 min at 4°C.
Chromatin immunoprecipitation (ChIP)
assay
ChIP assays were carried out using an EZ ChIP
Chromatin Immunoprecipitation kit (EMD Millipore, Billerica, MA,
USA) as described in the supplier's protocol. Briefly, the
cross-linked chromatin was sonicated after cell lysis and then
incubated with antibodies against MBD1, HDAC2, Oct-1 and Sp-1
(dilution 1:1,000; cat. nos. SC-55473, SC-7899, SC-232 and SC-420,
respectively; Santa Cruz Biotechnology, Inc.) at 4°C overnight. The
immunocomplex was precipitated with Protein A-agarose (EMD
Millipore), and the beads were washed, sequentially treated with 10
µl of RNase A (37°C for 30 min) and 75 µl of Proteinase K (45°C for
4 h), and incubated at 65°C overnight to reverse cross-link the
chromatin. The DNA was recovered by phenol-chloroform extraction
and co-precipitation with glycogen and dissolved in 50 µl of
Tris-EDTA (TE) buffer. Extracted DNA was amplified by PCR using 1
µl of the precipitated DNA. PCR primers (sequences are presented in
Table SI) were designed to amplify
the expected Oct-1 or Sp-1 binding sites at the TESC gene promoter.
The real-time PCR conditions were 40 cycles at 94°C for 40 sec,
60°C for 1 min, and 72°C for 40 sec.
Statistical analysis
The one-way analysis of variance (ANOVA) with the
Tukey's post hoc test and the Student's t-test were used to detect
differences in the methylation and expression levels in low- and
high-TESC-expressing gastric cancer cell lines using SPSS for
Windows, release 17.0 (SPSS, Inc., Chicago, IL, USA). P-values
<0.05 were considered to indicate a statistically significant
difference.
Results
Differential expression of TESC mRNA
and protein in gastric cancer cell lines
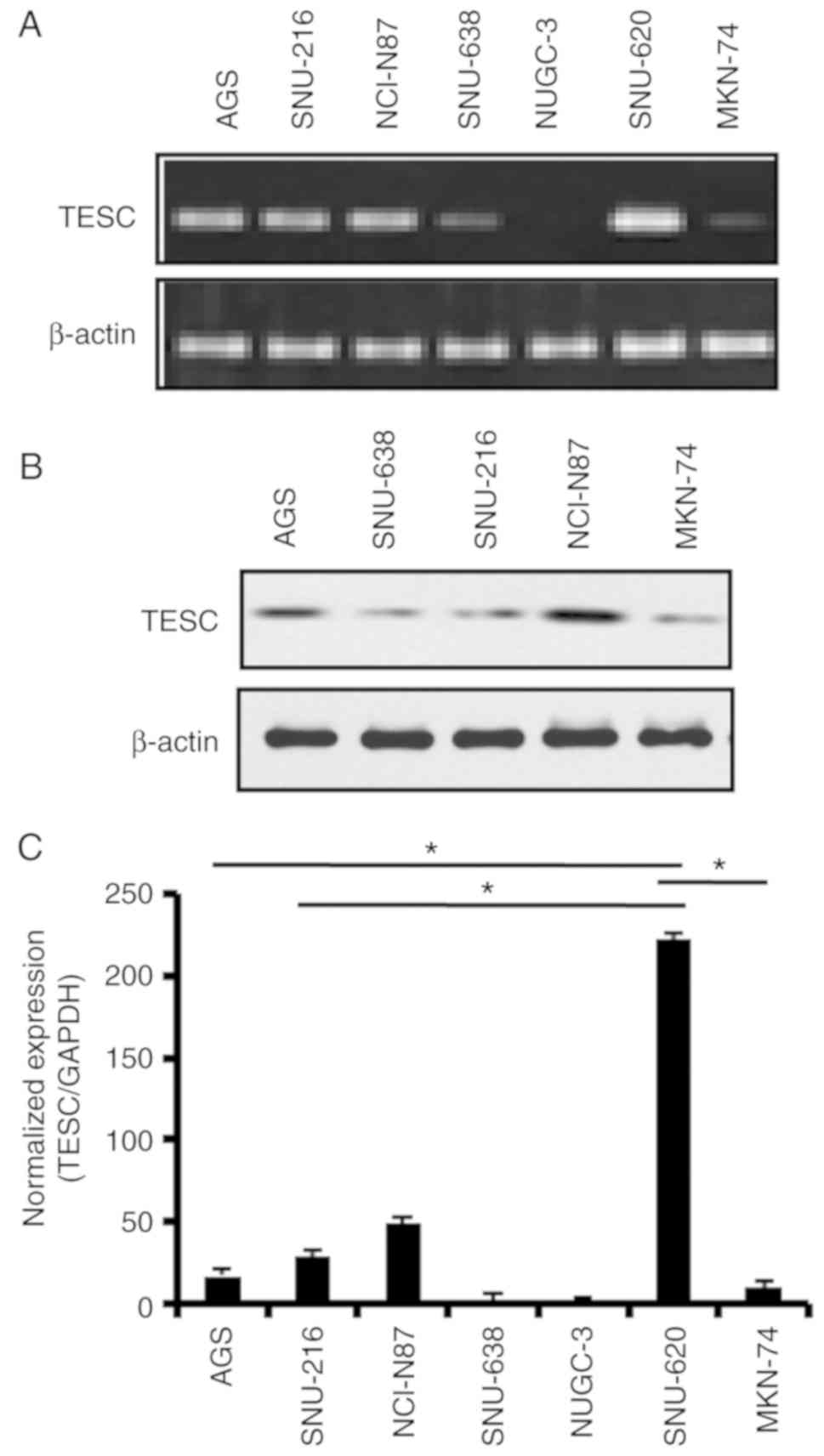
To profile TESC gene expression in gastric cancer,
we examined TESC mRNA and protein expression status in gastric
cancer cell lines (Fig. 1A and B).
RT-PCR revealed the upregulation of TESC mRNA expression in the
SNU-620 cell line and a moderate level of TESC mRNA in the AGS,
SNU-216, and NCI-N87 cell lines. Conversely, the TESC level was
significantly downregulated in the SNU-638, NUGC-3 and MKN-74 cell
lines. The results of quantitative real-time PCR also supported
this observation (Fig. 1C).
Moreover, the protein level of TESC also exhibited differential
expression in the gastric cancer cell lines (Fig. 1B). Collectively, our results
indicated that the TESC gene was differentially expressed in cancer
cell lines. Given the recent studies indicating that epigenetic
regulatory mechanisms such as promoter methylation are one of the
vital mechanisms for the regulation of gene expression (33,34),
these results may indicate that the hypermethylation of the
promoter inactivates the expression of the TESC gene.
Aberrant hypermethylation of the TESC
gene promoter in gastric cancer cell lines
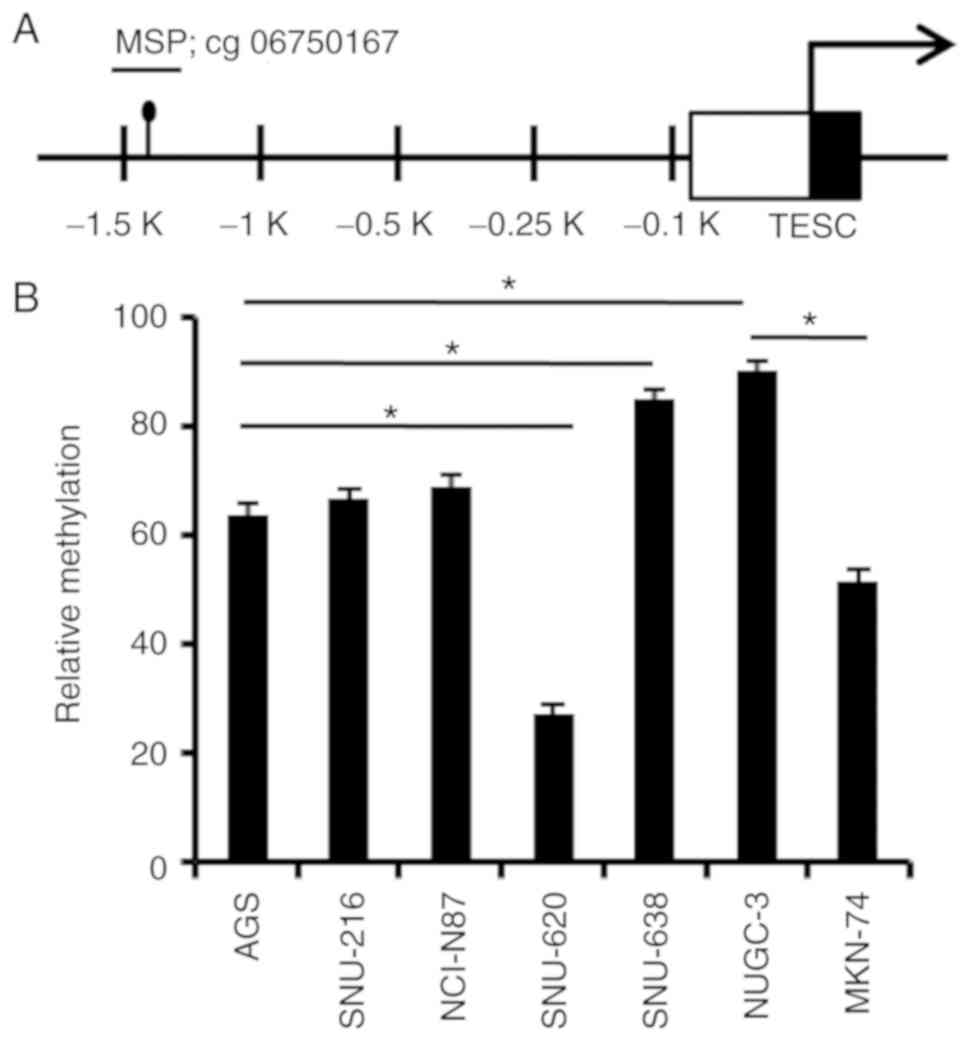
The methylation level of the TESC gene in various
CpG sites of the proximal promoter using the NCBI GEO database
(www.ncbi.nlm.nih.gov/geo, datasets for
GSE25869) was then analyzed and the significant hypermethylation of
the TESC promoter was revealed in various normal tissues compared
with that in cancer tissues, including colon, breast, brain, lung
and gastric cancers (data not shown). It was thus hypothesized that
promoter methylation of the TESC gene was a key mechanism for
regulating TESC expression. cg06750167 (probe ID) of CpG sites on
the TESC gene promoter revealed very different methylation levels
in gastric cancer compared with those in normal tissues (Fig. 2A). To confirm the differential
methylation and its expression level, real-time MSP
(methylation-specific PCR) was performed in gastric cancer cell
lines to analyze the specific CpG site methylation status in
gastric cancer cell lines. As revealed in Fig. 2B, a significant amplification was
detected in gastric cancer cell lines, including AGS, SNU-216,
NCI-N87, SNU-638 and NUGC-3 cells, by comparing the methylation
levels of the methylation-specific signal with the
unmethylation-specific signal (Fig.
2B). Thus, these results revealed that the CpG site of the TESC
promoter was likely to be hypermethylated in gastric cancer cell
lines, which may suggest that the increased methylation level of
the cg06750167 site was critical in determining the expression
pattern of the TESC gene in gastric cancer.
Demethylation of the CpG site on the
TESC promoter following treatment with 5-aza-dC in gastric cancer
cell lines
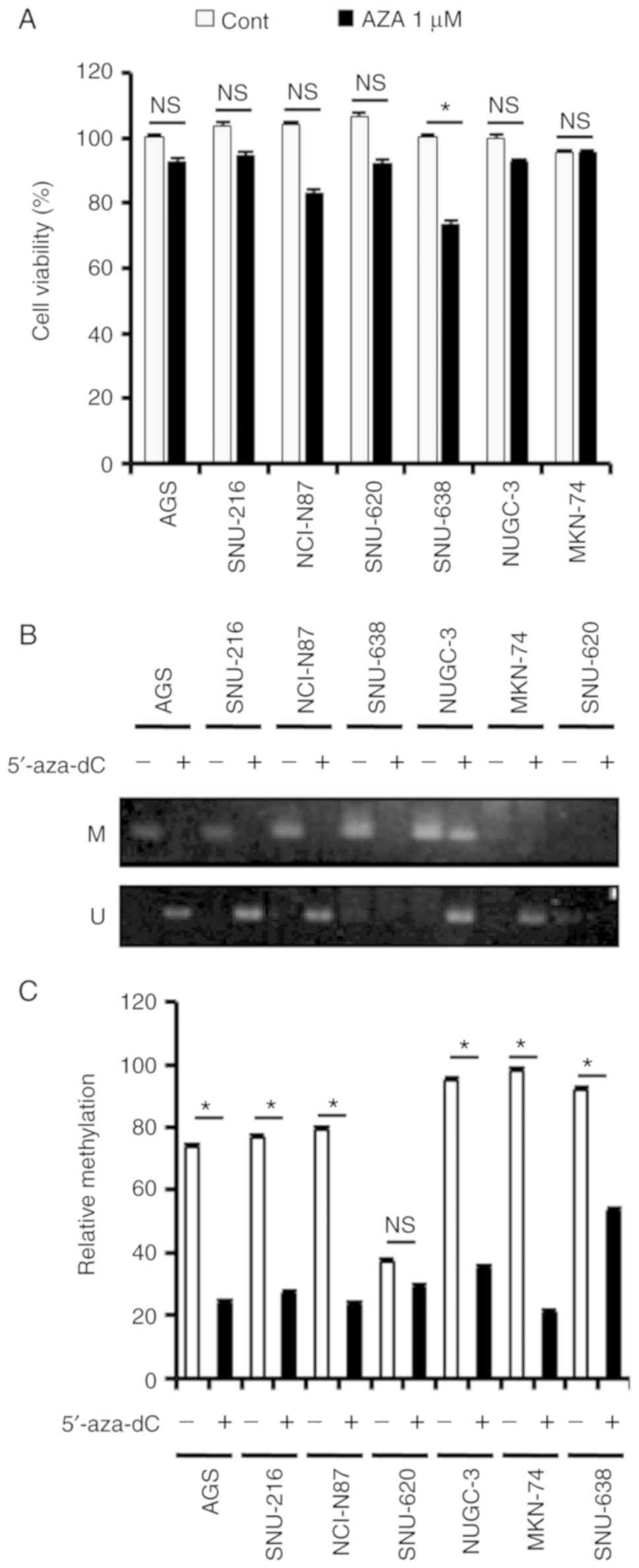
Given the epigenetic regulation mechanism via the
relationship between DNA methylation and opposite gene expression
(33), and the finding that the
TESC promoter was highly methylated in gastric cancer cell lines
with the exception of SNU-620 cells, cell viability following
5-aza-2c-deoxycytidine (5′-aza-dC) treatment was examined, however
no obvious changes were observed, except in SNU-638 cells (Fig. 3A). Since the DNA methyltransferase
(DNMT) inhibitor 5′-aza is able to cause DNA demethylation in the
genome and induce the expression of silenced genes, additively, 1
µM of aza is not an effective concentration to induce cell death in
gastric cancer cell lines. Thus, the cell viability was not
significantly altered in gastric cancer cell lines after AZA
treatment. It is known that the epigenetic change in the
methylation status of the specific CpG site is involved in
transcription regulation (35).
Thus, the promoter status after 5′-aza-dC treatment was examined.
To this end, 5-aza-dC, a methyltransferase inhibitor, was added to
the gastric cancer cell lines, and methylation levels were
evaluated by MSP and real-time MSP (Fig. 3B and C). As revealed in Fig. 3B, there was significant
amplification with unmethylated primers in AGS, SNU-216, NCI-N87,
SNU-638, NUGC-3 and MKN-74 cells following treatment with
5′-aza-dC. This was further supported by the significant
hypomethylation on the TESC promoter in gastric cancer cell lines
treated with 5′-aza-dC (Fig. 3C).
Numerical evaluation revealed 2.5-, 2.5-, 2.5-, 2.5-, 4- and 1-fold
decreases in methylation on the TESC promoter following treatment
with 5′-aza-dC in AGS, SNU-216, NCI-N87, NUGC-3, MKN-74 and SNU-638
cells, respectively. Therefore, these findings indicated that
5′-aza-dC-induced demethylation of the CpG site on the TESC
promoter may be crucial in determining the expression of the TESC
gene.
Demethylation of the TESC promoter by
5′-aza-dC leads to the expression of TESC in gastric cancer cell
lines
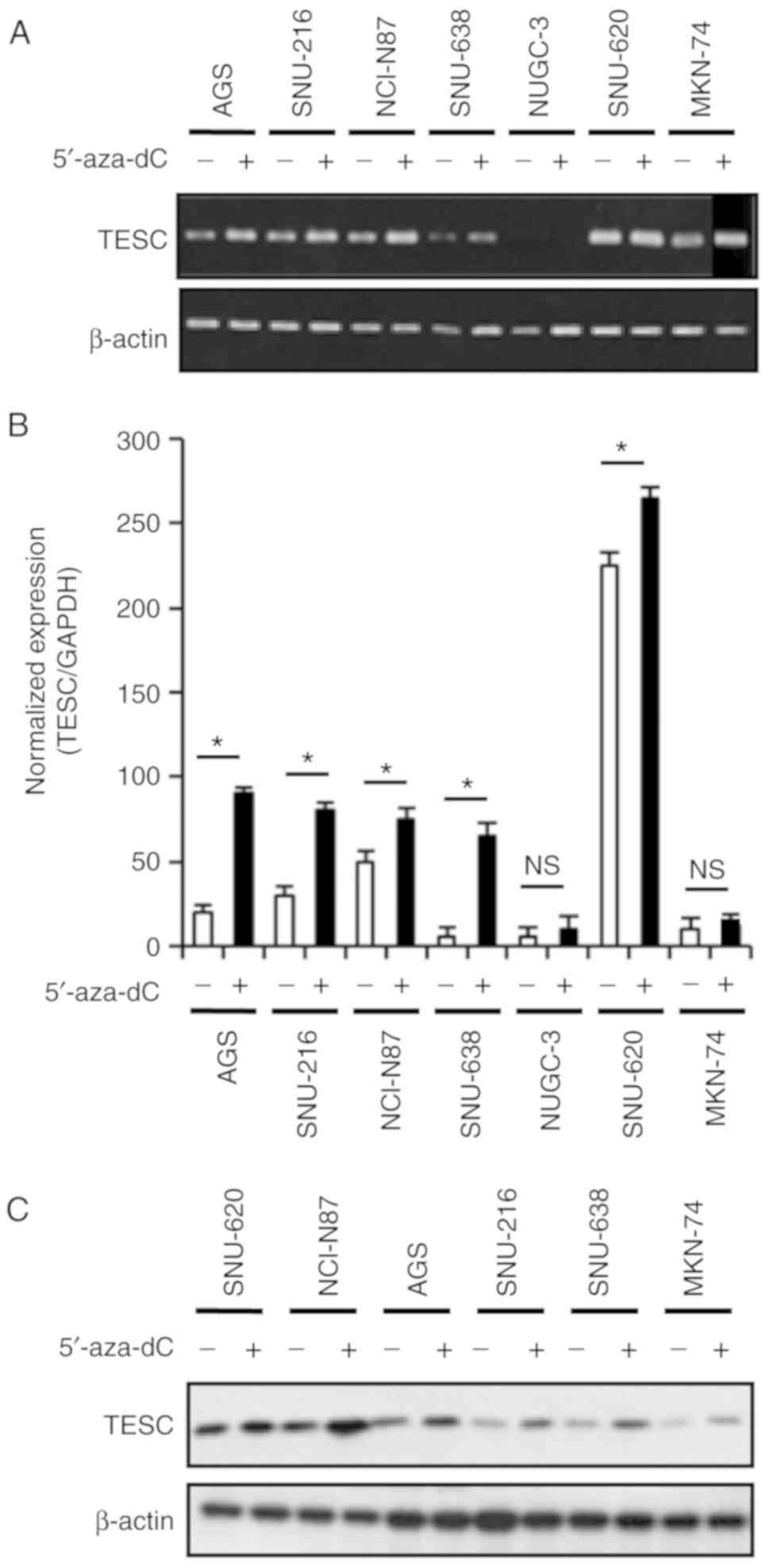
Given that the treatment with 5′-aza-dC depleted
promoter methylation, it was examined whether promoter
hypomethylation induced by 5′-aza-dC (1 µM) treatment for 3 days
led to the expression of the TESC gene. The RT-PCR and real-time
PCR results indicated the upregulation of TESC mRNA following
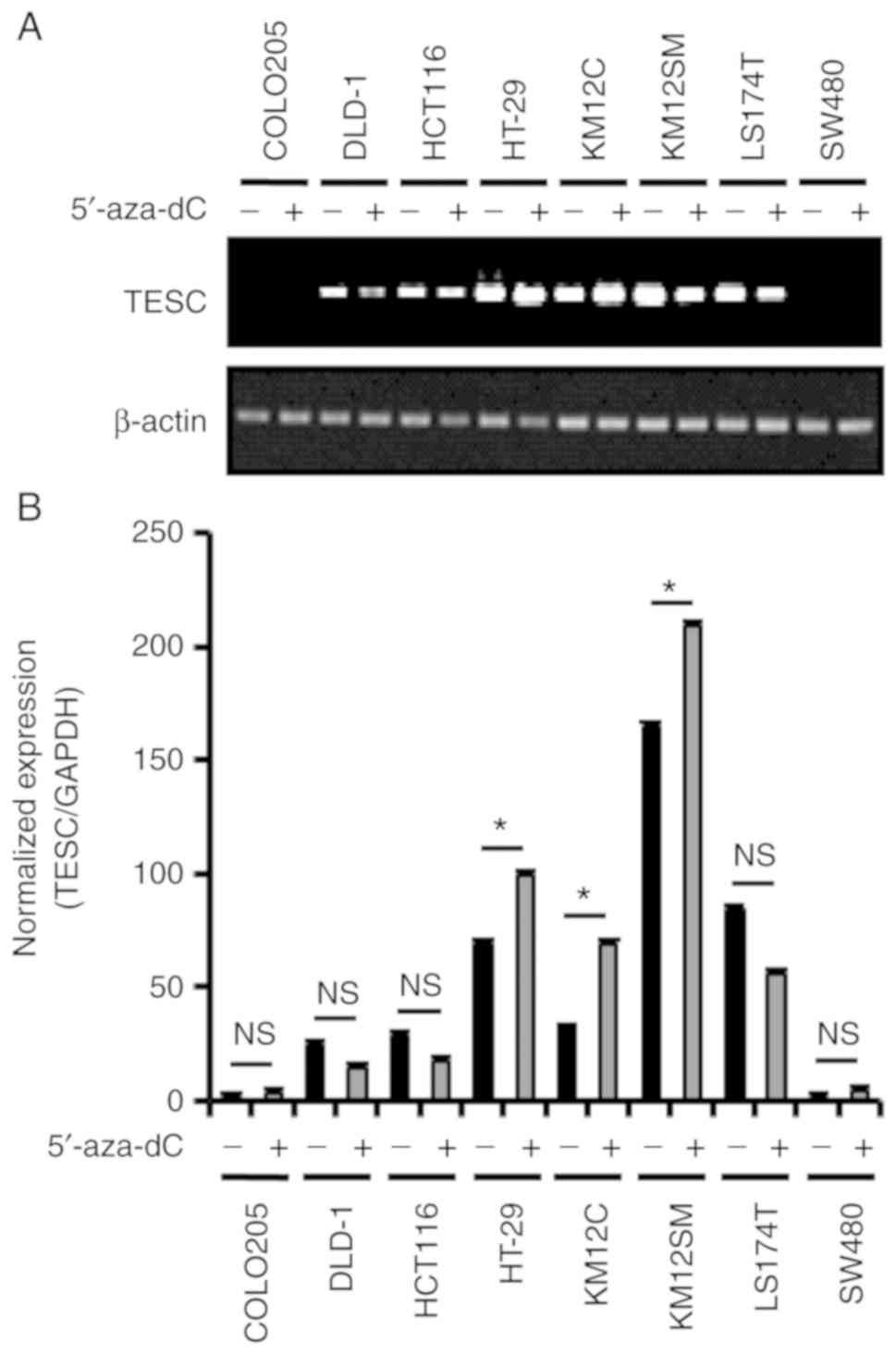
5′-aza-dC treatment in some gastric and colorectal cancer cell
lines (Figs. 4A and B, and 5A and B). Quantitative real-time PCR data
also revealed that 5′-aza-dC treatment increased TESC expression by
5-, 3-, 0.5-, 6- and 0.7-fold in the AGS, SNU-216, NCI-N87, SNU-638
and SNU-620 gastric cancer cell lines, respectively, and there were
0.5-, 2- and 0.8-fold increases in the HT-29, KM12C, and KM12SM
(Figs. 4B and 5B) colorectal cancer cell lines,
respectively. In agreement with the TESC expression results, the
protein levels in 5-aza-dC-treated gastric cancer cell lines were
upregulated after 5′-aza-dC treatment (Fig. 4C). In the MKN-74 cell line, the
expression of TESC was weakly increased after 5′-aza-dC treatment
as shown in Fig. 4A and C. The
results revealed that the response of MKN-74 cells after 5′-aza-dC
treatment was not different compared to the responses of other
gastric cancer cell lines. Overall, these data indicated that the
5′-aza-dC-induced hypomethylation in the TESC promoter restored
TESC expression. These results strongly indicated that TESC gene
expression was associated with the methylation status of a specific
CpG site, and the methylation level of this CpG site was one of the
key factors for TESC mRNA and protein expression in gastric cancer
cell lines. Therefore, the transcriptional and translational
regulation mechanism that regulates TESC gene expression via DNA
methylation was identified.
Epigenetic modification of the TESC
gene is mediated by MBD1 and Oct-1 via HDAC2 in gastric cancer cell
lines
It has been reported that the accumulation of the
MBD protein associated with the 5′-methylated cytosine residue is
involved in the transcriptional regulation of promoter methylation
(36). MBD proteins form a complex
with corepressors, including HDACs and Sin3A, and facilitate the
assembly of a repressive chromatin structure (37,38).
Another silencing mechanism via DNA methylation is the binding of
transcription factors such as Sp-1 and Oct-1 at gene promoters
(39–41). A recent study revealed that the
mouse TESC gene contained CpG islands on the promoter region and
putative transcription factor binding sites for Sp-1, EGFR1,
ZBP-89, AP-2 and CDF-1 (42).
To determine whether methylation of the CpG site at
the TESC promoter influences the binding of candidate transcription
factors, the promoter region was divided into four parts (R1-R4),
ranging from −0.1 to −1.5K, to identify the localization of the
transcription factors (Fig. 6A). To
assess the chromatin localization of the putative regulators such
as MBD1, HDAC2, Oct-1 and Sp-1, ChIP was performed with relevant
antibodies in hypermethylated AGS and SNU-638 cells to assess the
chromatin localization of the regulators (Fig. 6B and C). The binding of the
epigenetic transcription repressor complex MBD1 and HDAC2 was only
observed in the R4 region, which contains the differential CpG
methylation site. The binding of MBD1 and HDAC2 was observed in
TESC downregulated AGS and SNU-638 cells, but not in
5′-aza-dC-treated AGS and SNU-638 cells (Fig. 6B and C). Furthermore, the ChIP
analysis of the transcription factors Oct-1 and Sp-1 at the R4
region revealed the significant localization of Oct-1 in AGS and
SNU-638 cells with a hypermethylated TESC promoter. The Oct-1
localization was reduced following treatment with 5′-aza-dC
(Fig. 6B and C). Collectively,
these findings indicated that in AGS and SNU-638 cells, the binding
of MBD1 to the methylated CpG site of the TESC promoter resulted in
the recruitment of corepressors, such as HDAC2, and that the
binding of Oct-1 was also critical for the transcriptional
inhibition of TESC by binding to MBD1 and HDAC2.
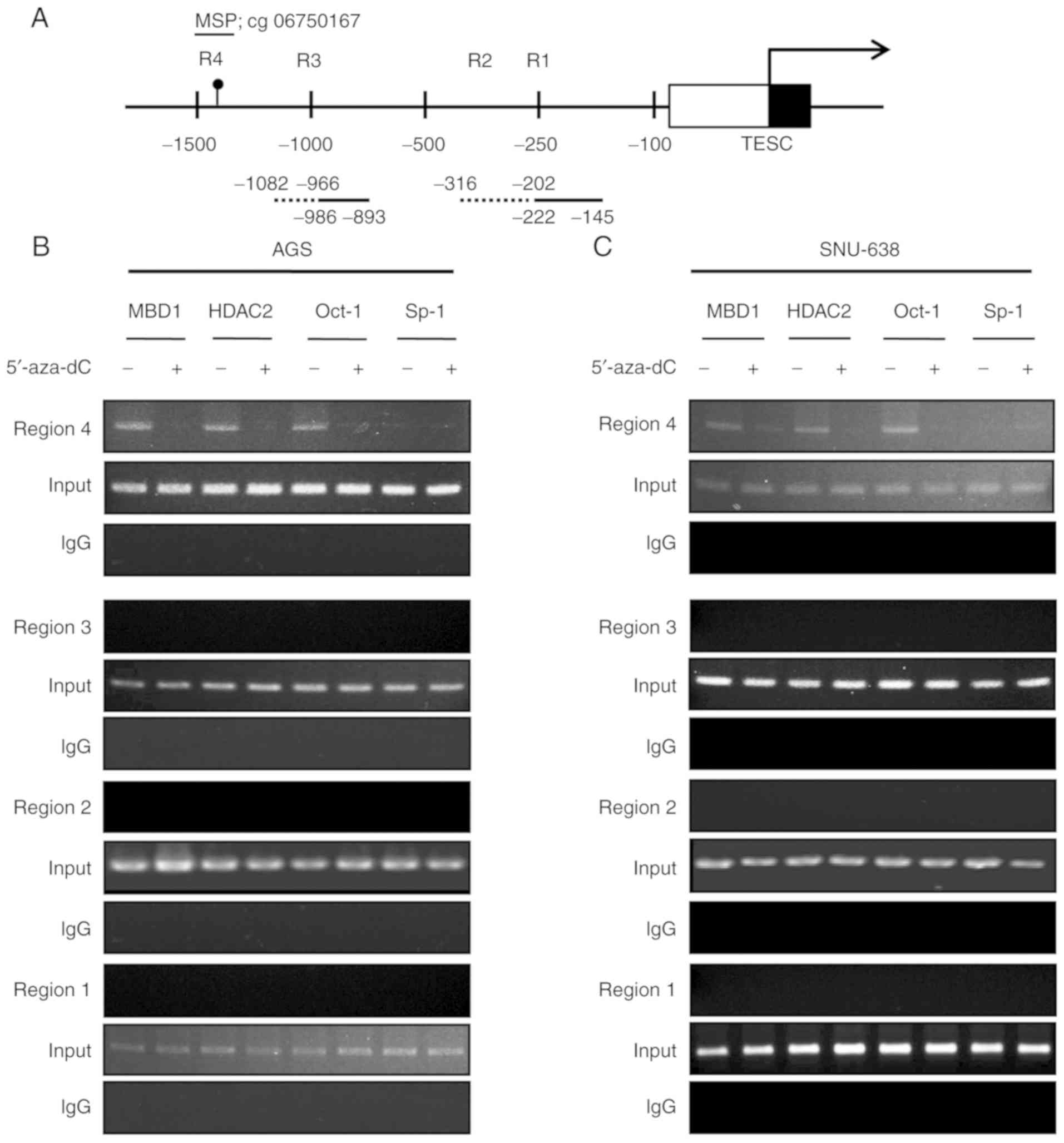 | Figure 6.ChIP analysis of the TESC promoter
against the candidates MBD1, HDAC2, Oct-1, and Sp-1. ChIP assays
were conducted on the TESC promoter using anti-MBD1, anti-HDAC2,
anti-Oct-1, and anti-Sp-1 by PCR to amplify the CpG site. (A)
Schematic diagram of the TESC promoter indicating four candidate
regions (R1-R4) for putative transcription factor binding. PCR
analysis of the IP derived from DMSO- or 5′-aza-dC-mediated (B) AGS
and (C) SNU-638 cells was performed using primers for the putative
regions (R1-R4). Each sample was examined in three independent
reactions. ChIP, chromatin immunoprecipitation; TESC, tescalcin;
MBD, methyl-CpG binding domain protein; HDAC, histone deacetylase;
IP, immunoprecipitated DNA; DMSO, dimethyl sulphoxide. |
TESC negatively regulates MBD1 and
histone methylation and enhances cell survival in gastric
cancer
Given the aforementioned results, whether TESC loss
negatively regulated MBD1 expression was determined, and for this,
a small interfering RNA against TESC (siTESC) and was constructed
and used to treat NCI-N87 cells, which exhibited high TESC
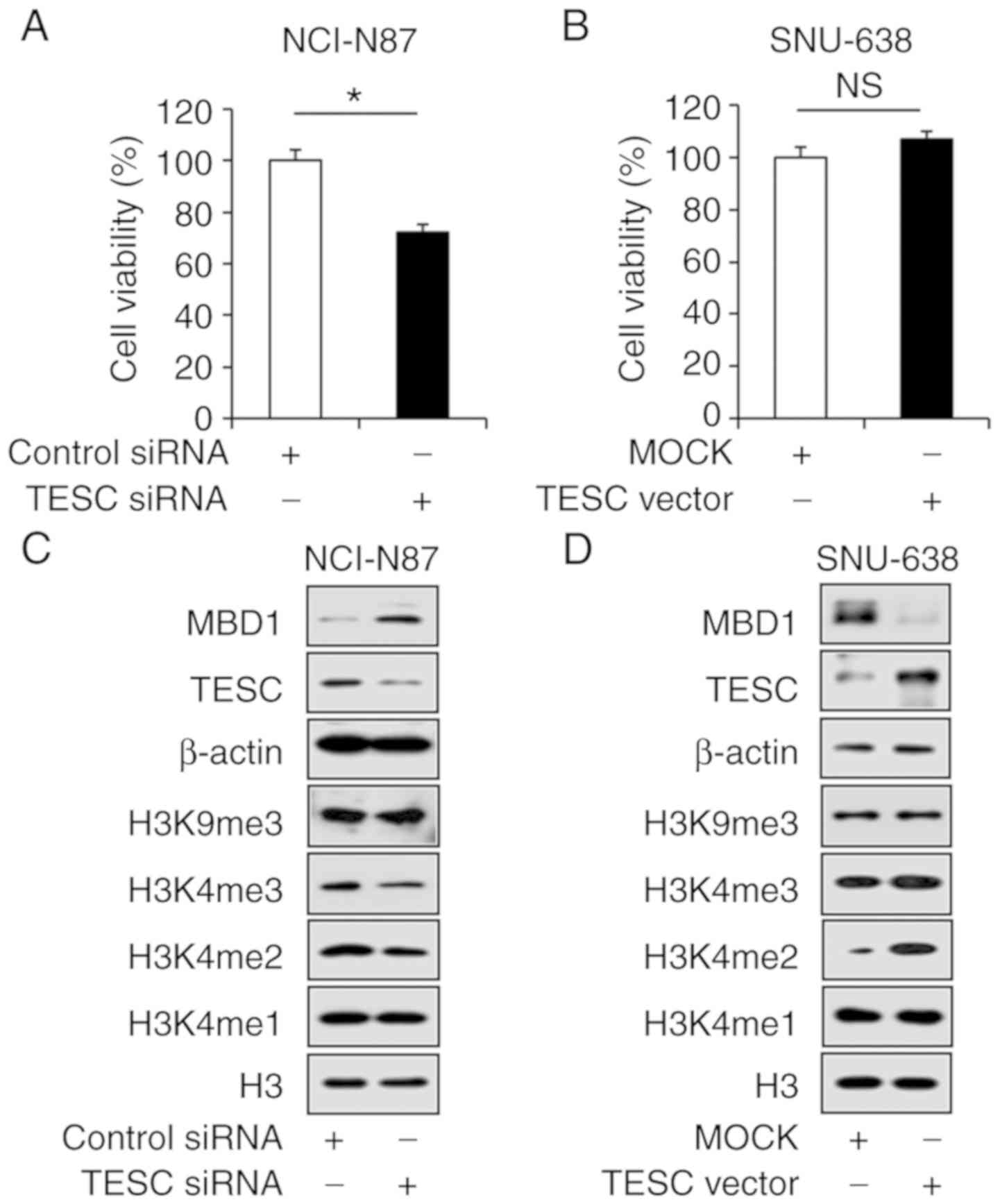
expression levels. Fig. 7C revealed
the significant downregulation of TESC expression in NCI-N87 cells
that were treated with siTESC compared with that in cells
transfected with siCont (Fig. 7C).
In addition, the WST-1 assay results indicated that siTESC reduced
the viability of NCI-N87 cells by 30% compared to that in the
controls (Fig. 7A). Notably, the
upregulation of MBD1 following siTESC treatment and the
downregulation of H3K4me2 and H3K4me3, which are active
transcription markers was detected. However, no obvious change was
observed for other histone methylations such as H3K4me1 and H3K9me3
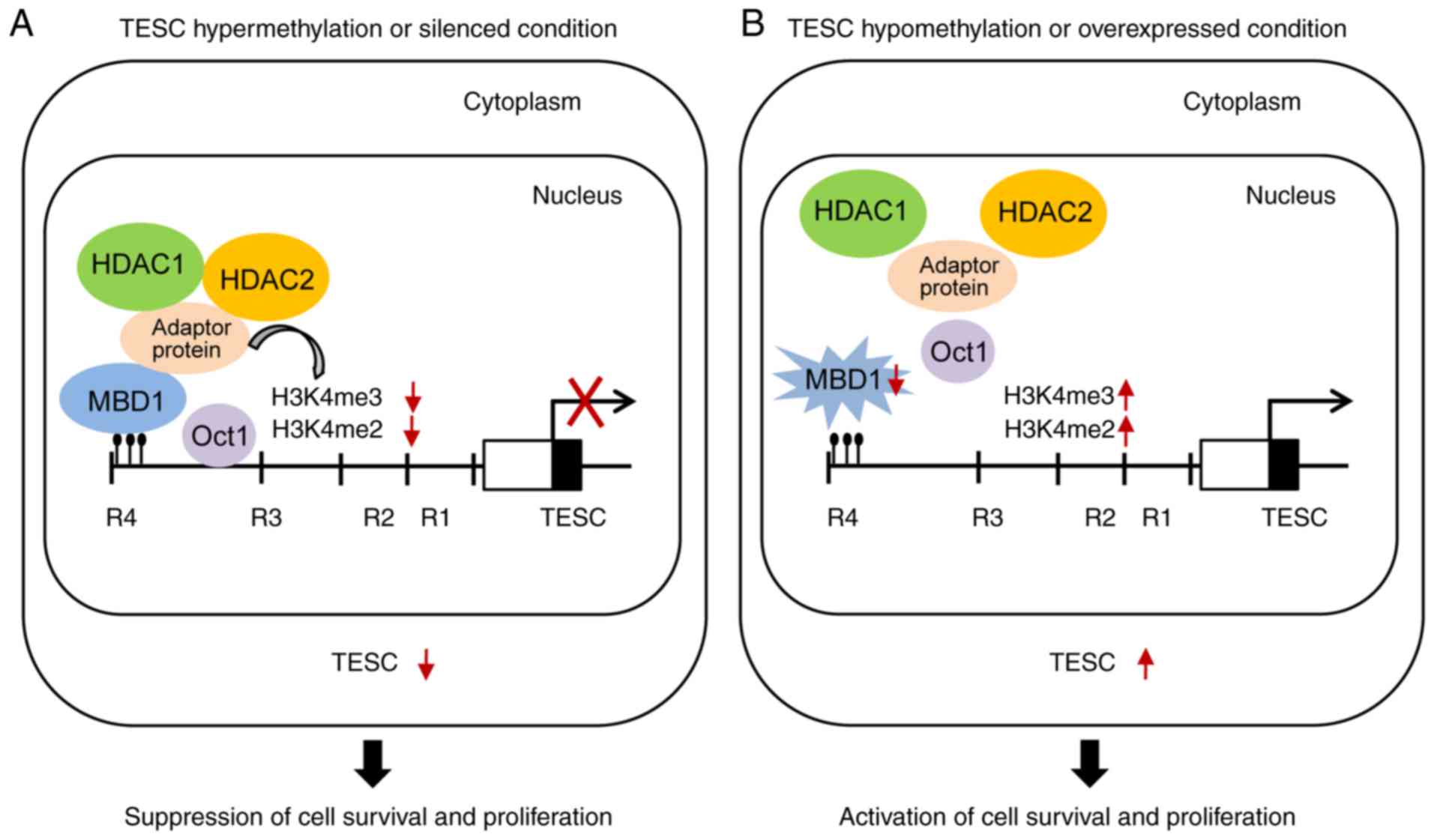
(Fig. 7C). Thus, our results
indicated that the silencing of TESC by promoter hypermethylation
suppressed the survival of gastric cancer cells (Fig. 8A).
Considering the result that TESC loss suppresses the
survival of gastric cancer cells, TESC was overexpressed using the
p-CMV-sports6-TESC vector in SNU-638 cells, which express a low
level of TESC. Fig. 7B revealed
that overexpression of TESC increased cell proliferation by 10%
(Fig. 7B). In addition, western
blot analysis demonstrated that TESC overexpression significantly
inhibited MBD1 expression, while the levels of H3K4me2 and H3K4me3
increased (Fig. 7D). Collectively,
our findings provided evidence that TESC upregulated H3K4me2 and
H3K4me3 for transcriptional activation and thus indicated that
overexpression of TESC by promoter hypomethylation promoted cell
survival in gastric cancer (Fig.
8B).
Discussion
The present study demonstrated that TESC expression
was negatively regulated by promoter methylation via MBD1 and that
its overexpression promoted the survival of gastric cancer cells,
but not colorectal cancer cells. It has been reported the
upregulation of TESC expression in radiation-induced thyroid cancer
as well as acute myeloid leukemia resulted in acquired resistance
against sorafenib via an interaction with NHE1 (43,44).
Previously, our research revealed that TESC expression was
upregulated in colorectal cancer tissues compared with that in
normal tissues and that the inhibition of TESC blocked NF-kB
signaling and decreased cell survival in vitro and in
vivo (6). Furthermore, the
upregulation of TESC expression promoted migration and invasion via
the activation of epithelial-mesenchymal transition (EMT) in
colorectal cancer cells (7).
Overall, these studies suggested that the identification of the
regulatory mechanism of TESC expression could provide effective
therapeutic strategies for diverse types of cancer.
In the present study, the differential expression of
TESC in gastric cancer cell lines was observed; TESC was
upregulated in AGS, SNU-216, NCI-N87, and SNU-620 cells but was
downregulated in SNU-638, NUGC-3 and MKN-74 cells. However,
comparison to a normal gastric epithelial cell line could not be
performed since we could not obtain a normal gastric cell line and
the ATCC does not carry such a cell line. No association between
methylation and malignant status was discovered. Notably, TESC
expression was negatively regulated via epigenetic regulation such
as DNA methylation and histone modification (33). DNA methylation is considered one of
the most powerful epigenetic modifications (34) and plays important roles in silencing
of tumor suppressor genes via hypermethylation and activating of
oncogenes via hypomethylation in cancer (45). To identify specific demethylated
genes in the NCBI Dataset, 32 pairs of human normal gastric and
cancer tissues were analyzed to discover specific methylation
signatures using microarray with the Illumina HumanMethylation27
BeadChip (HumanMethylation27_270596_v.1.2). Furthermore, it was
revealed that TESC expression was negatively regulated by promoter
methylation in gastric cancer cells, not colorectal cancer cells.
Real-time MSP revealed the hypermethylation of the TESC promoter in
gastric cancer cells, except for SNU-620 cells. In addition, its
hypermethylation was confirmed only in HT-29 and KM12C colorectal
cancer cells. Consistent with these results, when cells were
treated with 5′-aza-dC, a methyltransferase inhibitor, real-time
MSP/RT-PCR and western blot analyses indicated the critical role
that the methylation level of the promoter cg06750167 site in the
TESC gene plays in its expression. Accumulating evidence has
indicated that promoter methylation was involved in the
transcription regulatory mechanism of cancer-related genes, such as
MTO1, MRPL41, TCF21 and ZNF331 (46–48).
Moreover, additional evidence has suggested that CpG sites in the
TESC promoter caused different methylation patterns in cold blood
from 336 Mexican-American newborns after prenatal phthalate
exposure (49). Additionally,
patients with major depressive disorder were revealed to have
hypermethylation of CpG sites on a TESC gene-regulating genetic
variant (rs7294919) (50). In
addition, histone deacetylases (HDACs) play crucial roles in
epigenetic regulation of protein expression (51). HDAC inhibition also regulates
processes such as DNA histone methylation and gene expression via
DNMT1 regulation (52). This was
supported by the finding that treatment with Class I HDAC
inhibitors upregulated the TESC gene in neurons and had a
neuroprotective effect via an epigenetic mechanism (1).
Several researchers have reported the epigenetic
mechanisms through the regulation of genes by DNA methylation via
transcription factors such as E2F, Sp-1, Oct-1 and CREB, or via
methyl-CpG-binding proteins such as MBD1, MBD2, MBD3 and MeCp2
(22,53–57).
Notably, the present results demonstrated that TESC expression was
suppressed by the binding of MBD1, HDAC2, and Oct-1 to its
promoter; conversely, 5′-aza-dC treatment restored TESC expression
by blocking MBD1, HDAC2, and Oct-1 binding on putative methylation
sites. In support of these data, putative Sp-1 and Sp3 binding on
the promoter of the mouse TESC gene was reported by Perera et
al (41). However, Sp-1 binding
was not revealed on the TESC promoter in DMSO- or 5′-aza-dC-induced
gastric cancer cells. MBD1, a transcriptional regulator, has been
shown to repress the transcription of tumor suppressor genes, such
as p16 and VHL, by binding their CpG sites (58). It has been reported that the binding
of MBDs and HDACs (HDAC1 and HDAC2) to promoters plays a crucial
role in the suppression of protein expression (59). In line with this, the present study
demonstrated that vital regulators, including MBD1, HDAC2 and
Oct-1, via CpG methylation were involved in the regulation of TESC
expression and that 5′-aza-dC blocked these repressors by
demethylation.
MBD proteins are considered critical regulators of
gene regulation via chromatin modification (60). Moreover, the MBD proteins have been
demonstrated to bind DNAs containing methylated CpG sites (61). To investigate the effects of the
binding of mono-, di-, and trimethylated histone H3K4 and
trimethylated histone H3K9 on the regulation of TESC expression,
histone methylation status was examined in siTESC-transfected
NCI-N87 cells and in TESC vector-transfected SNU-638 cells. Histone
methylation analysis revealed that differential TESC expression was
associated with MBD1 and histone methylation. Suppression of TESC
upregulated MBD1 expression and conversely downregulated H3K4me3
and decreased cell viability. In addition, loss of H3K4me2/3 caused
cell death, whereas high expression of H3K4me2/3 induced cell
survival, resulting in increased migration and invasion (62–64).
In these findings, loss of TESC mediated the increase in MBD1 and
decrease in H3K4me2/3 and then induced cell death. In contrast,
overexpression of TESC using a TESC expression vector decreased
MBD1 levels and enhanced the expression of H3K4me2/3. Previously,
our team of researchers reported that TESC expression was involved
in cell migration, invasion and EMT in colorectal cancer, but that
depletion of TESC induced cell death (6,7). Based
on our findings, we propose a detailed mechanism for the regulation
of TESC expression in which TESC expression promotes cell survival
for invasion and migration via DNA hypomethylation, decreases MBD1,
and increases H3K4me2/3, whereas loss of TESC may induce cell death
via promoter hypermethylation, accumulation of MBD1, and inhibition
of H3K4me2/3. In a future study, we may analyze the methylation
status of TESC promoter using tissues of patients with gastric
cancer compared to normal tissues and define the relationships
between TESC methylation and gastric cancer.
In conclusion, TESC expression was regulated by the
methylation status of the promoter in gastric cancer cell lines,
indicating a relationship between TESC expression and its
epigenetic modification, such as promoter methylation and histone
methylation. Furthermore, 5′-aza-dC prevented the binding of MBD1,
HDAC2, and Oct-1 to CpG sites in the TESC promoter. Moreover,
knockdown of TESC inhibited H3K4me2/3, which is a transcription
active code, and decreased cell survival, whereas overexpression of
TESC enhanced H3K4me2 for transcriptional activation and increased
cell survival. Collectively, our findings provided novel evidence
for the epigenetic regulation of the CpG site of TESC, suggesting a
mechanistic link between TESC expression and epigenetic regulation.
These findings also indicated novel cancer therapeutic strategies
based on this identified epigenetic regulatory mechanism in gastric
cancer cells.
Supplementary Material
Supporting Data
Acknowledgements
We would like to thank Dr Hogyu David Seo
(Department of Biological Sciences, Korea Advanced Institute of
Science and Technology), and Dr Soo Young Jun (Genome Research
Center, Korea Research Institute of Bioscience and Biotechnology)
for editing help of this manuscript.
Funding
The present study was supported by the Basic Science
Research Program and Mid-Career Researcher Program through the
National Research Foundation of Korea (NRF) funded by the Korea
government (2017R1D1A1B03031502, 2016R1A2B4007413 and
2017R1A2B2005629).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
TWK, SRH, YHK and HGL designed the experiments and
analyzed the data. TWK, SRH, JTK, SMY, MSL, SHL and YHK performed
the experiments. TWK, SRH, YHK and HGL wrote the manuscript. YHK
and HGL supervised the overall project. All authors read and
approved the manuscript and agree to be accountable for all aspects
of the research in ensuring that the accuracy or integrity of any
part of the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patients consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
TESC
|
tescalcin
|
|
MBD
|
methyl-CpG binding domain protein
|
|
HDAC
|
histone deacetylase
|
|
CpG
|
cytosine guanine dinucleotide
|
|
MSP
|
methylation-specific PCR
|
|
RT-PCR
|
reverse transcription-polymerase chain
reaction
|
References
|
1
|
Takamatsu G, Katagiri C, Tomoyuki T,
Shimizu-Okabe C, Nakamura W, Nakamura-Higa M, Hayakawa T,
Wakabayashi S, Kondo T, Takayama C, et al: Tescalcin is a potential
target of class Ihistone deacetylase inhibitors in neurons. Biochem
Biophys Res Commun. 482:1327–1333. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mailänder J, Müller-Esterl W and Dedio J:
Human homolog of mouse tescalcin associates with
Na+/H+ exchanger type-1. FEBS Lett.
507:331–335. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Malo ME and Fliegel L: Physiological role
and regulation of the Na+/H+ exchanger. Can J
Physiol Pharmacol. 84:1081–1095. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Loo SY, Chang MK, Chua CS, Kumar AP,
Pervaiz S and Clement MV: NHE-1: A promising target for novel
anti-cancer therapeutics. Curr Pharm Des. 18:1372–1382. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fan J, Xing Y, Wen X, Jia R, Ni H, He J,
Ding X, Pan H, Qian G, Ge S, et al: Long non-coding RNA ROR decoys
gene-specific histone methylation to promote tumorigenesis. Genome
Biol. 16:1392015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kang YH, Han SR, Kim JT, Lee SJ, Yeom YI,
Min JK, Lee CH, Kim JW, Yoon SR, Yoon DY, et al: The EF-hand
calcium-binding protein tescalcin is a potential oncotarget in
colorectal cancer. Oncotarget. 5:2149–2160. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kang J, Kang YH, Oh BM, Uhm TG, Park SY,
Kim TW, Han SR, Lee SJ, Lee Y and Lee HG: Tescalcin expression
contributes to invasive and metastatic activity in colorectal
cancer. Tumour Biol. 37:13843–13853. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Levay K and Slepak VZ: Tescalcin is an
essential factor in megakaryocytic differentiation associated with
Ets family gene expression. J Clin Invest. 117:2672–2683. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Seth A, Ascione R, Fisher RJ,
Mavrothalassitis GJ, Bhat NK and Papas TS: The ets gene
family. Cell Growth Differ. 3:327–334. 1992.PubMed/NCBI
|
|
10
|
Wasylyk B, Hahn SL and Giovane A: The Ets
family of transcription factors. Eur J Biochem. 211:7–18. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Oikawa T and Yamada T: Molecular biology
of the Ets family of transcription factors. Gene. 303:11–34. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Seth A and Watson DK: ETS transcription
factors and their emerging roles in human cancer. Eur J Cancer.
41:2462–2478. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hogart A, Lichtenberg J, Ajay SS, Anderson
S; NIH Intramural Sequencing Center, ; Margulies EH and Bodine DM:
Genome-wide DNA methylation profiles in hematopoietic stem and
progenitor cells reveal overrepresentation of ETS transcription
factor binding sites. Genome Res. 22:1407–1418. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Schones DE and Zhao K: Genome-wide
approaches to studying chromatin modifications. Nat Rev Genet.
9:179–191. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu B, Tahk S, Yee KM, Fan G and Shuai K:
The ligase PIAS1 restricts natural regulatory T cell
differentiation by epigenetic repression. Science. 330:521–525.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ling ZQ, Tanaka A, Li P, Nakayama T,
Fujiyama Y, Hattori T and Sugihara H: Microsatellite instability
with promoter methylation and silencing of hMLH1 can regionally
occur during progression of gastric carcinoma. Cancer Lett.
297:244–251. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qu Y, Dang S and Hou P: Gene methylation
in gastric cancer. Clin Chim Acta. 424:53–65. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yamamoto E, Suzuki H, Takamaru H, Yamamoto
H, Toyota M and Shinomura Y: Role of DNA methylation in the
development of diffuse-type gastric cancer. Digestion. 83:241–249.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu JS, Koujak S, Nagase S, Li CM, Su T,
Wang X, Keniry M, Memeo L, Rojtman A, Mansukhani M, et al:
PCDH8, the human homolog of PAPC, is a candidate
tumor suppressor of breast cancer. Oncogene. 27:4657–4665. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ai L, Kim WJ, Kim TY, Fields CR, Massoll
NA, Robertson KD and Brown KD: Epigenetic silencing of the tumor
suppressor cystatin M occurs during breast cancer progression.
Cancer Res. 66:7899–7909. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wolf I, O'Kelly J, Rubinek T, Tong M,
Nguyen A, Lin BT, Tai HH, Karlan BY and Koeffler HP:
15-hydroxyprostaglandin dehydrogenase is a tumor suppressor of
human breast cancer. Cancer Res. 66:7818–7823. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hendrich B and Tweedie S: The methyl-CpG
binding domain and the evolving role of DNA methylation in animals.
Trends Genet. 19:269–277. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hendrich B and Bird A: Identification and
characterization of a family of mammalian methyl-CpG binding
proteins. Mol Cell Biol. 18:6538–6547. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ballestar E and Esteller M:
Methyl-CpG-binding proteins in cancer: Blaming the DNA methylation
messenger. Biochem Cell Biol. 83:374–384. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sansom OJ, Maddison K and Clarke AR:
Mechanisms of disease: Methyl-binding domain proteins as potential
therapeutic targets in cancer. Nat Clin Pract Oncol. 4:305–315.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jørgensen HF, Ben-Porath I and Bird AP:
Mbd1 is recruited to both methylated and nonmethylated CpGs via
distinct DNA binding domains. Mol Cell Biol. 24:3387–3395. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu H, Jin G, Wang H, Wu W, Liu Y, Qian J,
Fan W, Ma H, Miao R, Hu Z, et al: Methyl-CpG binding domain 1 gene
polymorphisms and lung cancer risk in a Chinese population.
Biomarkers. 13:607–617. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang J, Tai LS, Tzang CH, Fong WF, Guan XY
and Yang M: 1p31, 7q21 and 18q21 chromosomal aberrations and
candidate genes in acquired vinblastine resistance of human
cervical carcinoma KB cells. Oncol Rep. 19:1155–1164.
2008.PubMed/NCBI
|
|
29
|
Monnier P, Martinet C, Pontis J, Stancheva
I, Ait-Si-Ali S and Dandolo L: H19 lncRNA controls gene expression
of the Imprinted Gene Network by recruiting MBD1. Proc Natl Acad
Sci USA. 110:20693–20698. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang P, Lin C, Smith ER, Guo H, Sanderson
BW, Wu M, Gogol M, Alexander T, Seidel C, Wiedemann LM, et al:
Global analysis of H3K4 methylation defines MLL family member
targets and points to a role for MLL1-mediated H3K4 methylation in
the regulation of transcriptional initiation by RNA polymerase II.
Mol Cell Biol. 29:6074–6085. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zheng YG, Wu J, Chen Z and Goodman M:
Chemical regulation of epigenetic modifications: Opportunities for
new cancer therapy. Med Res Rev. 28:645–687. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gibney ER and Nolan CM: Epigenetics and
gene expression. Heredity. 105:4–13. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kulis M and Esteller M: DNA methylation
and cancer. Adv Genet. 70:27–56. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nile CJ, Read RC, Akil M, Duff GW and
Wilson AG: Methylation status of a single CpG site in the IL6
promoter is related to IL6 messenger RNA levels and rheumatoid
arthritis. Arthritis Rheum. 58:2686–2693. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Prokhortchouk E and Hendrich B: Methyl-CpG
binding proteins and cancer: Are MeCpGs more important than MBDs?
Oncogene. 21:5394–5399. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jones PL, Veenstra GJ, Wade PA, Vermaak D,
Kass SU, Landsberger N, Strouboulis J and Wolffe AP: Methylated DNA
and MeCP2 recruit histone deacetylase to repress transcription. Nat
Genet. 19:187–191. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ng HH, Zhang Y, Hendrich B, Johnson CA,
Turner BM, Erdjument-Bromage H, Tempst P, Reinberg D and Bird A:
MBD2 is a transcriptional repressor belonging to the MeCP1 histone
deacetylase complex. Nat Genet. 23:58–61. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Clark SJ, Harrison J and Molloy PL: Sp1
binding is inhibited by mCpmCpG methylation.
Gene. 195:67–71. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim TW, Lee SJ, Oh BM, Lee H, Uhm TG, Min
JK, Park YJ, Yoon SR, Kim BY, Kim JW, et al: Epigenetic
modification of TLR4 promotes activation of NF-κB by regulating
methyl-CpG-binding domain protein 2 and Sp1 in gastric cancer.
Oncotarget. 7:4195–4209. 2016.PubMed/NCBI
|
|
41
|
Murayama A, Sakura K, Nakama M,
Yasuzawa-Tanaka K, Fujita E, Tateishi Y, Wang Y, Ushijima T, Baba
T, Shibuya K, et al: A specific CpG site demethylation in the human
interleukin 2 gene promoter is an epigenetic memory. EMBO J.
25:1081–1092. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Perera EM, Bao Y, Kos L and Berkovitz G:
Structural and functional characterization of the mouse tescalcin
promoter. Gene. 464:50–62. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Stein L, Rothschild J, Luce J, Cowell JK,
Thomas G, Bogdanova TI, Tronko MD and Hawthorn L: Copy number and
gene expression alterations in radiation-induced papillary thyroid
carcinoma from chernobyl pediatric patients. Thyroid. 20:475–487.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Man CH, Lam SS, Sun MK, Chow HC, Gill H,
Kwong YL and Leung AY: A novel tescalcin-sodium/hydrogen exchange
axis underlying sorafenib resistance in FLT3-ITD+ AML.
Blood. 123:2530–2539. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qureshi SA, Bashir MU and Yaqinuddin A:
Utility of DNA methylation markers for diagnosing cancer. Int J
Surg. 8:194–198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kim TW, Kim B, Kim JH, Kang S, Park SB,
Jeong G, Kang HS and Kim SJ: Nuclear-encoded mitochondrial MTO1 and
MRPL41 are regulated in an opposite epigenetic mode based on
estrogen receptor status in breast cancer. BMC Cancer. 13:5022013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen B, Zeng C, Ye Y, Wu D, Mu Z, Liu J,
Xie Y and Wu H: Promoter methylation of TCF21 may repress autophagy
in the progression of lung cancer. J Cell Commun Signal.
12:423–432. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang Y, He T, Herman JG, Linghu E, Yang Y,
Fuks F, Zhou F, Song L and Guo M: Methylation of ZNF331 is an
independent prognostic marker of colorectal cancer and promotes
colorectal cancer growth. Clin Epigenetics. 9:1152017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Solomon O, Yousefi P, Huen K, Gunier RB,
Escudero-Fung M, Barcellos LF, Eskenazi B and Holland N: Prenatal
phthalate exposure and altered patterns of DNA methylation in cord
blood. Environ Mol Mutagen. 58:398–410. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Han KM, Won E, Kang J, Choi S, Kim A, Lee
MS, Tae WS and Ham BJ: TESC gene-regulating genetic variant
(rs7294919) affects hippocampal subfield volumes and
parahippocampal cingulum white matter integrity in major depressive
disorder. J Psychiatr Res. 93:20–29. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Delcuve GP, Khan DH and Davie JR: Roles of
histone deacetylases in epigenetic regulation: emerging paradigms
from studies with inhibitors. Clin Epigenetics. 4:52012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sarkar S, Abujamra AL, Loew JE, Forman LW,
Perrine SP and Faller DV: Histone deacetylase inhibitors reverse
CpG methylation by regulating DNMT1 through ERK signaling.
Anticancer Res. 31:2723–2732. 2011.PubMed/NCBI
|
|
53
|
Campanero MR, Armstrong MI and Flemington
EK: CpG methylation as a mechanism for the regulation of E2F
activity. Proc Natl Acad Sci USA. 97:6481–6486. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tian HP, Lun SM, Huang HJ, He R, Kong PZ,
Wang QS, Li XQ and Feng YM: DNA methylation affects the
SP1-regulated transcription of FOXF2 in breast cancer cells.
J Biol Chem. 290:19173–19183. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Garrity PA, Chen D, Rothenberg EV and Wold
BJ: Interleukin-2 transcription is regulated in vivo at the level
of coordinated binding of both constitutive and regulated factors.
Mol Cell Biol. 14:2159–2169. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Iguchi-Ariga SM and Schaffner W: CpG
methylation of the cAMP-responsive enhancer/promoter sequence
TGACGTCA abolishes specific factor binding as well as
transcriptional activation. Genes Dev. 3:612–619. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Nair SS, Coolen MW, Stirzaker C, Song JZ,
Statham AL, Strbenac D, Robinson MD and Clark SJ: Comparison of
methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding
domain (MBD) protein capture for genome-wide DNA methylation
analysis reveal CpG sequence coverage bias. Epigenetics. 6:34–44.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Fujita N, Takebayashi S, Okumura K, Kudo
S, Chiba T, Saya H and Nakao M: Methylation-mediated
transcriptional silencing in euchromatin by methyl-CpG binding
protein MBD1 isoforms. Mol Cell Biol. 19:6415–6426. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Patra SK, Patra A, Zhao H, Carroll P and
Dahiya R: Methyl-CpG-DNA binding proteins in human prostate cancer:
expression of CXXC sequence containing MBD1 and repression of MBD2
and MeCP2. Biochem Biophys Res Commun. 302:759–766. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bird AP and Wolffe AP: Methylation-induced
repression-belts, braces, and chromatin. Cell. 99:451–454. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Dhasarathy A and Wade PA: The MBD protein
family - reading an epigenetic mark? Mutat Res. 647:39–43. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Ng HH, Xu RM, Zhang Y and Struhl K:
Ubiquitination of histone H2B by Rad6 is required for efficient
Dot1-mediated methylation of histone H3 lysine 79. J Biol Chem.
277:34655–34657. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Fernandez-Capetillo O, Allis CD and
Nussenzweig A: Phosphorylation of histone H2B at DNA double-strand
breaks. J Exp Med. 199:1671–1677. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Tamagawa H, Oshima T, Shiozawa M, Morinaga
S, Nakamura Y, Yoshihara M, Sakuma Y, Kameda Y, Akaike M, Masuda M,
et al: The global histone modification pattern correlates with
overall survival in metachronous liver metastasis of colorectal
cancer. Oncol Rep. 27:637–642. 2012.PubMed/NCBI
|