Introduction
Melanoma derived from melanocytes that exhibit the
capacity to be more aggressive and a marked resistance to treatment
is leading to an increased morbidity (5%) and mortality rates (15%)
globally (1). Advanced melanoma has
historically been associated with a poor prognosis and a 5-year
survival rate of <10% (2,3). The most important clinical features of
melanoma in the initial stages (I/II) are the local migration and
long distance metastases through the lymph nodes (4,5). In
addition, previous studies have demonstrated that melanoma
frequently metastasized to other organs, which is another factor
that contributes to the low survival rate (6,7).
Although surgery, conventional and comprehensive treatments may
produce modest benefits to patients and improve patients' survival;
clinical trials have indicated that the short survival time of
patients with melanoma remained and <15% of patients survived
(8,9). Therefore, investigating a more
efficient approach to inhibit the local and long distance migration
of melanoma metastases may improve the survival rate of
patients.
Programmed death-1 (PD-1) is a type of
immunosuppressive molecule that is activated by programmed death
receptor 1 (10,11). PD-1 is a member of the cluster of
differentiation 28 (CD28) superfamily, which originated and was
cloned through the apoptosis of T cells in mice hybridoma (12). Targeting PD-1 immunity against
tumors, anti-infection, autoimmune disease and organ
transplantation survival have demonstrated marked outcomes in
previous studies (13,14). PD-1 ligand L1 and the antibody of
PD-1 are able to produce the same effects via binding with PD-1
(15). Notably, a previous study has
indicated that PD-1 is expressed on the surface of various
malignancies, including hepatoma, non-small cancer lung cell and
colon cancer (16). The antibody
target for PD-1 (anti-PD-1) was able to inhibit PD-1 expression
(17). However, the function of
anti-PD-1 and the survival of patients with cancer remain to be
elucidated. Furthermore, the role of PD-1 expression on melanoma
cells remains unknown. The present study investigated the
therapeutic effects of anti-PD-1 in a mouse model of melanoma.
Tumor-bearing mice were used to evaluate the efficacy of anti-PD-1
and the overall survival according to PD-L1 status in mice with
melanoma was assessed by meta-analysis.
Previous studies have indicated that a recombinant
adenovirus constructed based on the Adeno-X expression system,
resulted in a gene therapy vehicle to treat human cancer (18,19).
Adenovirus vectors are one of the most widely used vectors in gene
therapy applications for the treatment of diverse human diseases,
including cancer (20). The
immunotherapy accompanied with other therapeutic methods has
revealed therapeutic effects in animal models and different
clinical trials (21–23). The use of antineoplastic drugs
combined with immunotherapy has been demonstrated to effectively
target tumor cells with specific recognition molecules or domains
of antigens or receptors (24–27). A
number of different agents for the treatment of cancer, including
chemoradiotherapy and immunotherapy, are being assessed in
preclinical trials (28,29). Immunotherapy agents, in which an
antibody or interleukin is inserted into a gene expression vector,
have led to positive outcomes regarding tumor apoptosis in patients
with advanced tumors pre-clinically and clinically (30). The most crucial area of gene therapy
is to establish an effective gene delivery system. In the present
study, the oncolytic adenovirus-mediated gene therapy system
expressing anti-PD-1 was investigated as a treatment for
melanoma-bearing mice.
Development of novel and effective clinical
protocols for cancer therapy is urgently required in modern
medicine (31). Previously,
immunotherapies against the growth and metastasis of tumor cells
have been revealed to be a potentially promising approach for the
treatment of human cancer (32). The
antitumor efficacy of the T cell immune response depends on the
exposure of tumor antigens and activities of antigen-presenting
cells, which may efficiently present tumor antigens leading to the
activation of cytotoxic T lymphocytes. Notably, oncolytic
adenovirus expressing interferon-γ (IFN-γ) resulted in significant
tumor growth suppression in a syngeneic Syrian hamster model for
the treatment of pancreatic cancer (33). This in turn, eradicates corresponding
tumor cells. In addition, immunotherapies are activated by
dendritic cells (DCs) loaded with tumor surface antigens including
tumor cell lysates, nucleic acids encoding tumor antigens,
tumor-specific proteins, apoptotic bodies and necrotic tumor cells.
A number of research papers have demonstrated a strategy for
producing mature DCs, influenced by tumor phenotype and immune
responses for eradicating tumor cells. Furthermore, targeting the
tumor specific antigens by treating the antibody specific to the
DC-restricted antigen may elevate exposure of the tumor specific
antigen. This may promote recognition of tumor specific antigens
resulting in a limitation in tumor capacity (34,35). The
present study demonstrated that the targeting of the melanoma
antigen to DC cells via anti-PD-1 expressed by recombinant
adenovirus expressing (rAd)-PD-1 markedly promoted the ability of
rAd to induce responses from melanoma antigen-specific cytotoxic T
lymphocytes.
Materials and methods
Cells and reagents
Melanoma cell lines, B16-F10 and SkMel-2, and a
normal human epidermal cell line (NHEC; NHEK) were purchased from
the American Type Culture Collection (ATCC; Manassas, VA, USA) and
cultured in minimum essential media (MEM) supplemented with 10%
fetal bovine serum (both from Gibco; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) and 2 mM penicillin/streptomycin
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) in a humidified
incubator at 37°C and 5% CO2.
Construction of recombinant
adenovirus
An adeno-X expression system (A4470; Sigma-Aldrich;
Merck KGaA) (36) was used to
construct the recombinant adenovirus constructs. DNA sequence
(GenBank, NM_008798.2) encoding full-length Anti-PD-1 linked with
the cell-penetrating peptide and Fc was cloned into the rAd-X
plasmids (A4470; Sigma-Aldrich; Merck KGaA) by polymerase chain
reaction (PCR) using KAPA Taq PCR kit with dNTPs (cat. no. TAQNTKB;
Sigma-Aldrich; Merck KGaA). The primers were as follows: PD-1
forward, 5′-TCGATCTGGAACTGTGGCCAT-3′ and reverse,
5′-TGGCCAGGGCGCCTGTGGATCTAA-3′. Then, the recombinant adenoviral
plasmid rAd-anti-PD-1 and rAd-enhanced green fluorescent protein
(rAd-EGFP, produced in our laboratory) were produced by PCR as
described previously for subsequent analysis. PCR and sequencing
(Invitrogen; Thermo Fisher Scientific, Inc.) were used to identify
recombinant adenovirus constructs. Thermocycling of PCR was
performed using PCR kit (11696505001; Sigma-Aldrich; Merck KGaA) as
follows: 95°C for 1 min; and 25 cycles of 94°C for 30 sec, 56°C for
1 min and 72°C for 1 min. The recombinant adenoviruses were
generated by transfecting into HEK293 cells using Lipofectamine
2000 (Sigma-Aldrich; Merck KGaA) according to manufacturers'
instructions and underwent culture in DMEM at 37°C in an atmosphere
containing 5% CO2. The recombinant adenovirus constructs
were purified as described in a previous study (18). Adenovirus titers were determined by
TCID50 as plaque-forming units/ml using the Reed-Muench
method (37).
MTT assay
B16-F10 and SkMel-2 cells (1×103) were
cultured and then inoculated with rAd-Anti-PD-1 [0.5 multiplicity
of infection (MOI)] or rAd-EGFP 0.5 (MOI) or PBS in 96-well plates
for 48 h at 37°C in triplicate for each condition. Following
culture, 20 µl MTT (5 mg/ml) in PBS solution was added to each
well, the plate was further incubated for 4 h at 37°C. The medium
was entirely removed and 100 µl dimethyl sulfoxide was added to the
wells to solubilize the crystals. The optical density (OD) was
measured using an ELISA microplate reader (Bio-Rad Laboratories,
Inc., Hercules, CA, USA) reader at a wavelength of 450 nm.
Cells morphology
B16-F10 and SkMel-2 cells (5×106) were
cultured in six-well plate and treated with rAd-Anti-PD-1 (0.5
MOI), rAd-EGFP, rAd or vehicle (Mock) in 6-well plates for 48 h at
37°C. The cells were cells morphology was observed by a microscope
(Nikon E400, Nikon Instrument Group, Japan). The EGFP fluorescence
was observed using a confocal microscope (magnification, ×40;
Olympus FV10; Olympus, Tokyo, Japan).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from B16-F10 and SkMel-2
cells using the RNAeasy Mini kit (Qiagen GmbH, Hilden, Germany).
The mRNA expression of PD-1 in B16-F10 and SkMel-2 cells was
measured using an RT-qPCR kit (Roche Diagnostics, Mannheim,
Germany) with β-actin expression as an endogenous control (Qiagen
GmbH) according to the manufacturer's instructions. All primers
were synthesized by Invitrogen (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The following primers were used: PD-1 forward,
5′-AAGTTTCAGGGAAGGTCAG-3′ and revers, 5′-CTGGGCATGTGTAAAGGT-3′; and
β-actin forward, 5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′ and reverse,
5′-CTAAGTCATAGTCCGCCTAGAAGCA-3′. PCR reactions contained 25 ng cDNA
template, 100 ng forward and reverse oligonucleotide primers, 2.5
µl Taq PCR buffer (Promega Corporation, Madison, WI, USA), 0.4 mM
dNTP mixture and 1 U Taq polymerase (Promega Corporation, Madison,
WI, USA) in a total reaction volume of 25 µl. After 120 sec
incubation at 95°C, PCR was performed using 25 cycles of
denaturation at 94°C for 30 sec, annealing at 54°C for 30 sec and
elongation at 72°C for 30 sec. Relative PD-1 expression level was
calculated using the 2−ΔΔCq method (38). The results were presented as the
n-fold change compared with β-actin using Quantiscan2.1 (Software
Demo of AB QuantStudio™ 12K Flex System; Thermo Fisher Scientific,
Inc.).
Cells invasion and migration
assays
B16-F10 and SkMel-2 cells were cultured in DMEM
medium at 37°C in an atmosphere containing 5% CO2 and
treated with rAd-Aiti-PD-1 (0.5 MOI), rAd-EGFP (0.5 MOI) or PBS as
a control. For the invasion assay, virus-treated cells were
suspended at a density of 1×106 cells/ml in serum-free
MEM and then transferred to the upper chamber of a BD BioCoat
Matrigel Invasion Chamber (BD Biosciences, San Jose, CA, USA)
according to the manufacturer's instructions. The cells were fixed
using methanol for 30 min at 37°C and stained for 30 min in a 0.1%
Crystal Violet solution in PBS at 37°C. Invasion and migration were
calculated in at least three random fields of view under a
microscope (Nikon E400; magnification, ×20; Nikon Instrument Group,
Tokyo, Japan).
Animal study
A total of 80 eight-week-old male
specific-pathogen-free C57BL/6 nude immunodeficiency mice (30–35 g)
were purchased from Shanghai Laboratory Animal Centre (Shanghai,
China). All animals were housed in a temperature-controlled
facility at 23±1°C (humidity, 50±5%) with a 12-h light/dark cycle.
All rats had free access to food and water. A total volume of 100
µl B16-F10 cells (1×107) were administered
subcutaneously into a site on the back of the C57BL/6 mice.
C57BL/6-bearing mice were randomly divided into four groups and
received treatment of rAd-Aiti-PD-1 (0.5 MOI), rAd-EGFP (0.5 MOI),
rAd (0.5 MOI) or PBS (n=20 in each group). The therapy (rAd,
rAd-EGFP or rAd-anti-PD-1, 5 MOI) was initiated on day 6 when tumor
diameters reached 5–6 mm and mice were treated 10 times for a
period of 20 days (i.e., every other day). Mice were sacrificed
when tumor diameters reached 12 mm. Tumor diameters were recorded
every 2 days and tumor volume was calculated as follows: 0.52 ×
smallest diameter2 × largest diameter (39). The present study was performed
according to the recommendations in the Guide for the Care and Use
of Laboratory Animals (40). All
experiments were completed in accordance with National Institutes
of Health and approved by Committee on the Ethics Committee of
Zhengzhou University (Zhengzhou, China). All surgeries and
euthanasia were performed under sodium pentobarbital anesthesia (40
mg/kg) when tumor diameter reached 16 mm.
Splenocyte collection and cytotoxic T
lymphocyte (CTL) responses
Splenocytes were extracted from spleens of the
experimental animals using cell separation method described
previously (41) (n=4 in each group)
on day 30 after tumor incubation. The monoplast suspension was
washed three times with PBS three times at 37°C. Then, inactivated
B16-F10 cells (1×106) were incubated with splenocytes
(1×104) for 12 h at 37°C. Release of interferon (IFN)
was evaluated by ELISA (DY485, Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) in the supernatants following culture for 72 h.
T cells (1×106) from the splenocytes were purified, as
previously described (32) and
co-cultured with fresh DMEM B16-F10 cells for 4 h at 37°C at the
effector, at target ratios of 5:1, 20:1 and 40:1, which were ratios
used in a previous study (42).
Specific CTL activity to the target cells was determined by MTT
cytotoxicity assays, as previously described (43).
Western blot analysis
B16-F10 and SkMel-2 cells (1×106) were
cultured, lysed and used to analyze PD-1 expression according to a
previous study (44). Cells were
homogenized in lysate buffer containing protease-inhibitor
(Sigma-Aldrich; Merck KGaA) and were centrifuged at 8,000 × g at
4°C for 10 min. Protein concentration was measured using the BCA
protein assay kit (Thermo Scientific Fisher Scientific, Inc.).
Protein samples (20 µg per lane) were separated using 15% SDS-PAGE
and transferred onto polyvinylidene difluoride membranes (EMD
Millipore, Billerica, MA, USA). Following this, blocking with 5%
bovine serum albumin was performed at 37°C for 1 h. The following
primary antibodies were used in immunoblotting assays: PD-1
(1:1,000, cat. no. ab63477) and β-actin (1:1,000, cat. no. ab8226;
all from Abcam, Cambridge, UK) for 12 h at 4°C. Horseradish
peroxidase-conjugated antibody (1:5,000; cat. no: HAF019, Bio-Rad
Laboratories, Inc.) was used as a secondary antibody for 2 h at
37°C. Subsequently, bands were detected using a western blotting
Luminol reagent (cat. no. 12015218001; Sigma-Aldrich; Merck KGaA).
The density of the bands was analyzed by Quantity one software
version 4.62 (Bio-Rad Laboratories, Inc.).
Histological analysis
Tumor tissues from experimental mice were fixed
using 10% formaldehyde for 2 h at 37°C followed by being embedded
in paraffin. Tumor samples were cut into sections (4 µm) and
antigen retrieval (at 95°C for 15 min) was also performed on tumor
sections. Tumor sections were incubated with primary antibodies:
PD-1 (1:1,000, ab214421, Abcam, Cambridge, MA, USA) and β-actin
(1:1,000, ab8226, Abcam). Subsequently, sections were incubated
with horseradish peroxidase (HRP)-conjugated polyclonal anti-rabbit
immunoglobulin G antibody (1:10,000; R&D Systems, Inc.,
Minneapolis, MN, USA) for 1 h at room temperature. A Ventana
Benchmark automated staining system (Ventana Medical Systems, Inc.;
Roche Holding AG, Basel, Switzerland) was used for observing the
protein expression (Olympus BX51, Olympus; Tokyo, Japan;
magnification, ×20).
Statistical analysis
All data were reported as means ± standard error of
the mean from triplicate experiments. Data was analyzed using
GraphPad Prism 6.0 software (GraphPad Software, Inc., La Jolla, CA,
USA). Unpaired data was analyzed by Student's t-test. Comparisons
of data between multiple groups were analyzed by one-way analysis
of variance followed by Tukey HSD test. P<0.05 was considered to
indicate statistically significant differences.
Results
Characterization of the recombinant
adenovirus constructs
The recombinant adenovirus constructs delivering the
Anti-PD-1 or EGFP were generated based on the adeno-X expression
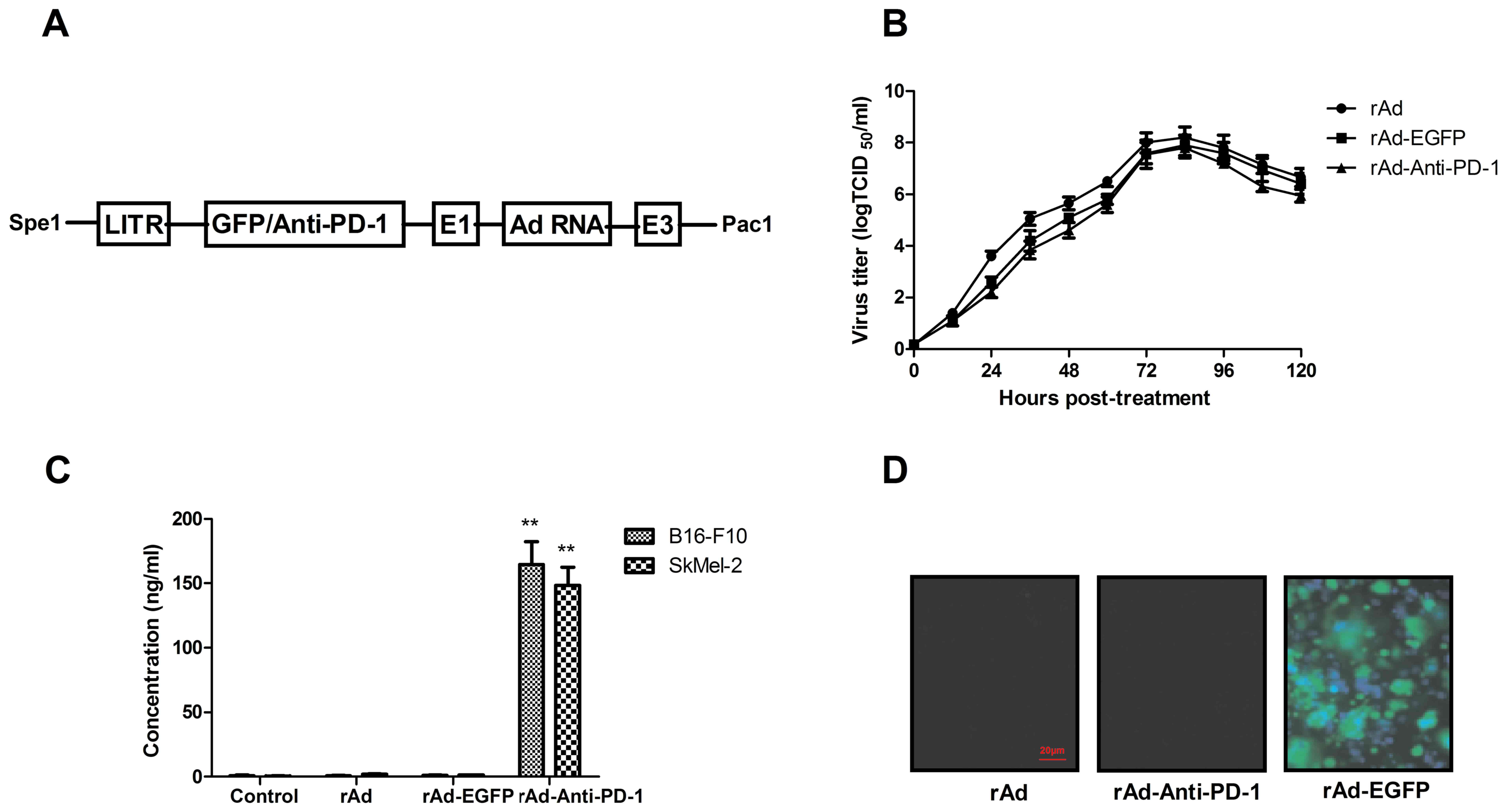
system. The description of adenovirus constructs is presented in
Fig. 1A. In order to investigate
whether the insertion of the foreign gene in the adeno-X expression
system affected the growth of the virus, the kinetic growth of
recombinant adenovirus constructs was analyzed. The result in
Fig. 1B demonstrated that similar
virus titers were observed; indicating that insertion of the
foreign gene in the adeno-X expression system did not significantly
affect replication and growth kinetics of recombinant adenovirus
constructs. In addition, the expression level of foreign gene
expression levels was analyzed in the virus-infected B16-F10 and
SkMel-2 cells. The results indicate that the Anti-PD-1 vector was
efficiently expressed and secreted into extracellular space 72 h
after infection (Fig. 1C).
Furthermore, EGFP syncytia formation by Anti-EGFP-infected cells
was observed by fluorescent microscopy (Fig. 1D). Collectively, the data suggests
that insertion of the foreign gene in the adeno-X expression system
do not affect viral growth and expression in tumor cells.
rAd expressing Anti-PD-1 targeted PD-1
and effectively enhanced the lysis of human and murine melanoma
cell lines
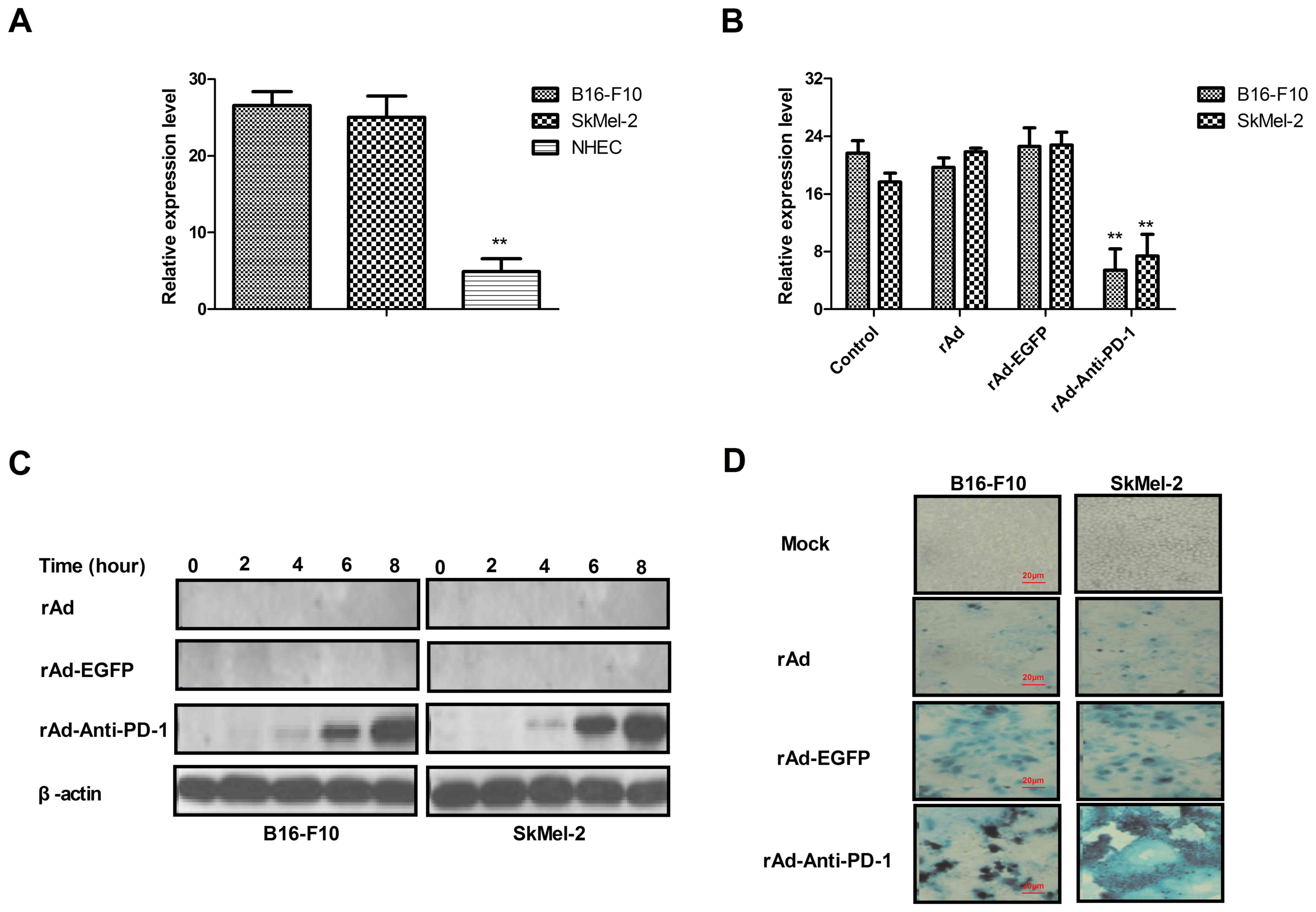
The PD-1 expression in B16-F10 and SkMel-2 cells was
further assessed using RT-qPCR. The results indicated that PD-1
expression was higher in B16-F10 and SkMel-2 cells compared with
NHECs (P<0.01; Fig. 2A). Notably,
rAd-anti-PD-1 significantly decreased the expression of PD-1 at 48
h following inoculation (P<0.05; Fig.
2B). To further confirm that the anti-PD-1 protein exhibited a
higher expression in tumor cells infected by rAd-anti-PD-1, the
present study performed a time course of rAd-anti-PD-1 infection in
B16-F10 cells. The results in Fig.
2C indicate that anti-PD-1 was detected as early as 6 h
following infection with rAd-anti-PD-1 at 0.1 MOI. Furthermore,
rAd-anti-PD-1 effectively lysed melanoma cell lines compared with
rAd-EGFP and rAd (Fig. 2D). Taken
together, these results suggest that rAd-anti-PD-1 is efficiently
expressed in tumor cells and was able to downregulate PD-1
expression.
rAd expressing Anti-PD-1 enhanced
antitumor efficacy in B16-F10-xenograft mice
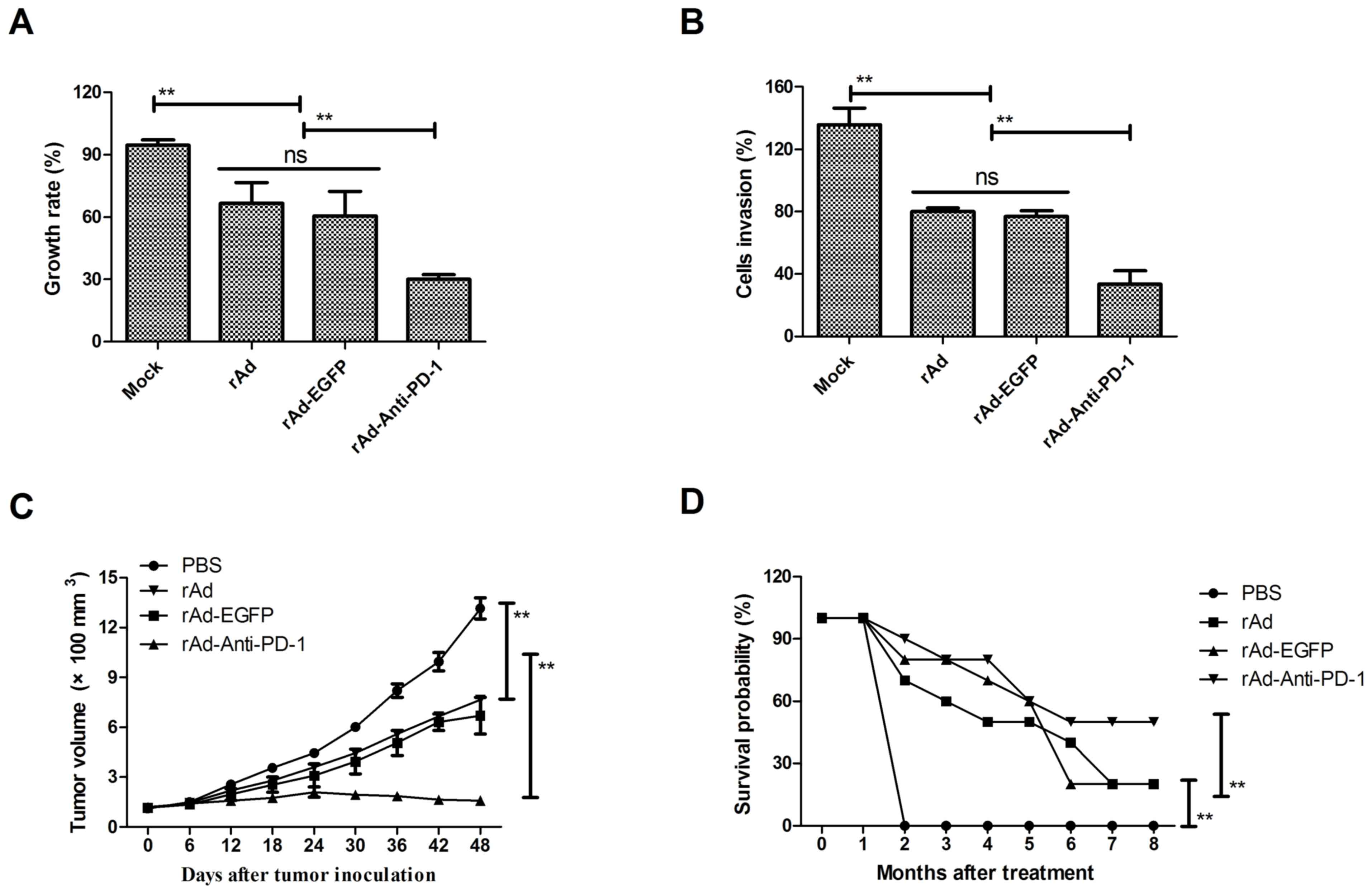
In order to detect the inhibitory effects of
rAd-Anti-PD-1, tumor cell growth was analyzed in vitro. The
growth of B16-F10 cells was significantly inhibited following
rAd-Anti-PD-1 treatment at 0.5 MOI for 48 h compared with rAd and
rAd-EGFP (P<0.01; Fig. 3A). In
addition, invasion of B16-F10 and SkMel-2 cells was significantly
suppressed when treated with rAd-Anti-PD-1 at 0.5 MOI for 48 h
compared with rAd and rAd-EGFP (P<0.01; Fig. 3B). Subsequently, the efficacy of
antitumor treatment with rAd-Anti-PD-1 was investigated in
B16-F10-bearing mice. Animals were administered treatment of rAd,
rAd-EGFP, rAd-PD-1 or PBS as control by intravenous injection.
These results demonstrate that rAd and rAd-EGFP treatment
significantly inhibited tumor growth, compared with PBS as a
control. Notably, a significant inhibition of tumor growth was
observed with rAd and compared with rAd-anti-PD-1 (P<0.01;
Fig. 3C). Furthermore, the 240-day
long-term survival following treatment with rAd-anti-PD-1 was
assessed and demonstrated that treatment with rAd-Anti-PD-1 (n=20)
significantly prolonged the survival of mice compared with other
groups (P<0.01; Fig. 3D).
Collectively, these results reveal that rAd-Anti-PD-1 significantly
inhibited melanoma cell growth in vitro and in vivo,
suggesting that the use of oncolytic therapy with rAd-anti-PD-1
against melanoma contributed to long-term survival of
melanoma-bearing mice.
Induction of rAd-Anti-PD-1-specific
cellular immune responses
A previous study indicated that presenting tumor
antigen by DC is essential in order to activate cytotoxic T
lymphocytes to inhibit the invasion of tumor cells (45). Therefore, the present study analyzed
the expression of B16-F10-sepcific tumor antigen and DC cells on
the surface of tumor cells 21 days following tumor inoculation. The
results indicated that the expression of tumor antigens and DC
cells was elevated in rAd-anti-PD-1-treated tumors (Fig. 4A). The present study also revealed
that PD-1 expression was markedly decreased, whereas apoptotic
bodies were increased in tumors following rAd-anti-PD-1 treatment
(Fig. 4B). In addition, the
cytotoxic T lymphocyte response in rAd-Anti-PD-1-treated mice was
also detected. As presented in Fig.
4C, the production of IFN-α and IFN-β increased, which
contributed to induce the maturation of DCs in
rAd-Anti-PD-1-treated mice. Furthermore, the results of the present
study demonstrated that rAd-Anti-PD-1 exhibited the ability to
generate significantly more CD4+ and CD8+ T
cells and induce a PD-1-specific CTL through DC-targeted surface
antigens in mice that resulted in further enhancing recognition
melanoma cells by the targeting of the rAd-anti-PD-1-encoded PD-1
to DCs (P<0.05; Fig. 4D and E).
The present study revealed that melanoma-bearing mice treated with
the rAd-Anti-PD-1-targeted PD-1 were significantly more protected
against apoptosis than tumor-bearing mice following challenge with
Anti-PD-1 (Fig. 4F). Collectively,
the results of the present study confirmed the hypothesis and
suggested that rAd-anti-PD-1 activated immunotherapy by inducing DC
maturation in melanoma, which contributed to tumor regression and
long-term survival.
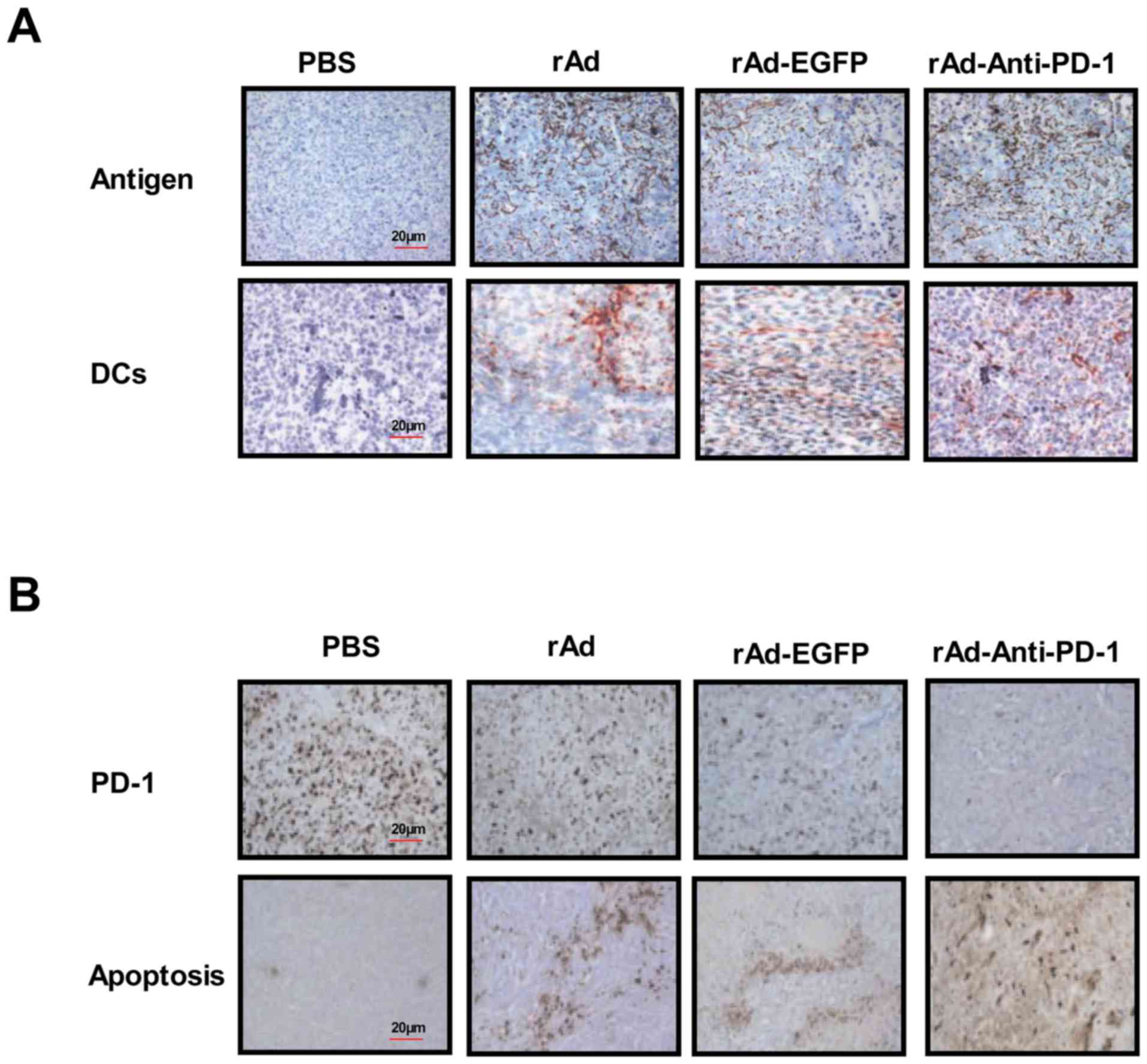 | Figure 4.rAd-anti-PD-1 enhanced DCs to present
melanoma tumor antigen. (A) Expression of tumor antigens and DC
cells was assessed in tumors from experimental mice on day 30
(magnification, ×40). (B) Expression of PD-1 and apoptotic bodies
in tumors were analyzed after treatment with recombinant adenovirus
(magnification, ×40). (C) IFN levels were analyzed after treatment
with recombinant adenovirus. (D) Level of
CD4+CD8+ T cells were assessed in tumors
during treatment on day 30. (E) Analysis of the CTL responses in
melanoma-bearing mice treated with the rAd-Anti-PD-1. (F) Tumor
challenge experiment analyzed the long-term efficacy of
rAd-anti-PD-1 treatment. Data are presented as mean ± standard
error of the mean. **P<0.01 vs. control. rAd, recombinant
adenovirus expressing; PD-1, programmed death-1; DC, dendritic
cells; IFN, interferon; CD, cluster of differentiation; CTL,
cytotoxic T lymphocyte; PBS, phosphate-buffered saline; EGFR,
enhanced green fluorescent protein. |
Discussion
Previous studies have indicated that gene therapy
may provide an improved clinical method for treating melanoma, and
these therapies were considered as potential adjuvants for other
cancer therapies (46,47). Adenovirus vectors are the most widely
used treatment and Adenovirus-mediated delivery of functional genes
or polypeptides into tumor cells is well understood (48). Additionally, gene transfer strategies
have led to more clinicians using immunotherapy to treat patients
with HCC, including inhibition of oncogenes and restoration of
tumor-suppressor genes, immunotherapy, anti-angiogenesis and
virotherapy (49). Therefore, the
present study may provide a more effective therapy for melanoma
tumors by delivering PD-1 via a recombinant adenovirus. The results
of the present study indicated that PD-1 expressed by recombinant
adenovirus induced the accumulation of DCs, which could expose
tumor antigens and promote T lymphocyte-induced tumor
cytotoxicity.
Gene therapy has previously identified potential
candidates for the treatment of human diseases including cancer,
cardiovascular disease, blood diseases, diabetes, genetic diseases
and other diseases that cannot be treated with conventional drug
therapies (50–52). Previously, the use of viral vectors
to deliver vectors to express functional genes has been applied in
different medical fields, including cancer therapy and
cardiovascular disease. These oncolytic viral vectors have been
demonstrated to be relatively safe due to a selectively to
replicate in cancer cells, but not in normal cells (53,54). The
benefits of these oncolytic viral vectors are that as they
replicate and lyse tumor cells, and functional genes delivered by
oncolytic viral vectors strengthen the effects of tumor eradication
and inhibit the chance of recurrence (55). Adenovirus, Newcastle disease virus
and herpes simplex virus are the most commonly used gene therapy
oncolytic viral vectors, as vectors that specifically mutate and
selectively replicate faster in cancer cells (56). The Adeno-X expression system is the
most commonly used and >250 patients have been treated with
ONYX-015 (a replicating adenovirus) (57). In the present study, the Adeno-X
expression system was used to deliver an antibody of PD-1 to assess
the oncolytic effects of a recombinant virus in melanoma in
vitro and in vivo.
CTL-associated PD-1 inhibitors are well understood
and represent one of the most important immunomodulating agents
(58). PD-1 receptor or antibody are
well tolerated and exhibit a low rate of adverse effect recurrence,
as demonstrated in a previous study (58). In addition, a study has demonstrated
that antibodies targeting PD-1 signaling promote a T-cell-mediated
antitumor therapy and prevent tumor invasion (59). The present study revealed that
anti-PD-1 demonstrated a significant inhibition of melanoma cells
growth and a marked increase of DC cell maturation to present
melanoma-specific antigens for cytotoxic T lymphocyte-associated
immunotherapy. A previous study indicated that DC cell maturation
is an essential step for the development of anti-cancer
immunotherapy and induction of the cytotoxic T-cell immune
responses in the majority of tumor cells (60). The data from the present study has
identified that rAd-Anti-PD-1 recognized PD-1 in tumor cells of
mice with melanoma and decreased the expression of PD-1 and
enhanced the infiltration of T cells. Although the induction of
tumor cell apoptosis due to PD-1 inhibition or antibodies in
patients with metastatic melanoma has been investigated, the PD-1
signaling pathway remains to be elucidated. Notably, the present
study revealed that anti-PD-1, delivered by rAd-anti-PD-1, induces
DC maturation and presents more melanoma-specific antigens for
antitumor responses, which contribute to improved long-term
survival. This may potentially lead to marked antitumor responses
through activation of the immune system.
In conclusion, the present study demonstrates that
full-length Anti-PD-1 may be expressed by rAd-Anti-PD-1-infected
tumor cells and inhibits the growth and invasion of melanoma cells.
In addition, the most notable finding in the present study is that
anti-PD-1 expressed by rAd-anti-PD-1-infected tumor cells induces
the maturation of DCs, which to the best of our knowledge has not
been reported in previous studies. The rAd-anti-PD-1 virus is
associated with a number of antitumor capacities and largely
inhibits tumor growth in vivo, indicating its potential
antitumor effects. Furthermore, rAd-anti-PD-1 induced the
DC-presenting tumor antigen and promoted a more potent CTL in the
immune response. Taken together, the use of DC-presenting tumor
antigens as a strategy of rAd-Anti-PD-1 may represent a potential
strategy of combining the oncolytic efficacy of adenovirus and
present tumor antigen in addition to CTL, which enhance the
anti-melanoma potency.
References
|
1
|
Ungureanu L, Botar Jid C, Candrea E,
Cosgarea R and Senila SC: The role of lymph node ultrasound
evaluation in melanoma-review of the literature. Med Ultrasono.
18:224–230. 2016. View Article : Google Scholar
|
|
2
|
Liu H, Yan Y and Jiang CM: Primary
malignant melanoma of the esophagus with unusual endoscopic
findings: A case report and literature review. Medicine
(Baltimore). 95:e34792016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Linertová R, Valcárcel-Nazco C and
Lacalle-Remigio JR: Management of benign melanocytic lesions as a
melanoma prevention. Systematic review. Med Clin (Barc).
147:162–170. 2016.(In Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cazes A and Ronai ZA: Metabolism in
melanoma metastasis. Pigment Cell Melanoma Res. 29:118–119. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martínez-Esteve A, Álvarez-Pérez RM,
Caballero-Gullón L, Sancho-Márquez MP and Borrego-Dorado I: Breast
metastasis from melanoma mimicking inflammatory breast cancer. Eur
J Nuc Med Mol Imaging. 43:389–390. 2016. View Article : Google Scholar
|
|
6
|
Margolin KA: Brain metastases in melanoma:
Moving toward curing the incurable. J Oncol Pract. 12:545–546.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chukwueke U, Batchelor T and Brastianos P:
Management of brain metastases in patients with Melanoma. J Oncol
Pract. 12:536–542. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schwartz H, Blacher E, Amer M, Livneh N,
Abramovitz L, Klein A, Ben-Shushan D, Soffer S, Blazquez R,
Barrantes-Freer A, et al: Incipient melanoma brain metastases
instigate astrogliosis and neuroinflammation. Cancer Res.
76:4359–4371. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sharma G, Lian CG, Lin WM, Amin-Mansour A,
Jané-Valbuena J, Garraway L, Bao W, Yoon CH and Ibrahim N: Distinct
genetic profiles of extracranial and intracranial acral melanoma
metastases. J Cutan Pathol. 43:884–891. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Luo M and Fu L: The effect of chemotherapy
on programmed cell death 1/programmed cell death 1 ligand axis:
Some chemotherapeutical drugs may finally work through immune
response. Oncotarget. 7:29794–29803. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fang XN and Fu LW: Predictive efficacy
biomarkers of programmed cell death 1/programmed cell death 1
ligand blockade therapy. Recent Pat Anticancer Drug Discov.
11:141–151. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dulos J, Carven GJ, van Boxtel SJ, Evers
S, Driessen-Engels LJ, Hobo W, Gorecka MA, de Haan AF, Mulders P,
Punt CJ, et al: PD-1 blockade augments Th1 and Th17 and suppresses
Th2 responses in peripheral blood from patients with prostate and
advanced melanoma cancer. J Immunother. 35:169–178. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sakthivel P, Gereke M and Bruder D:
Therapeutic intervention in cancer and chronic viral infections:
Antibody mediated manipulation of PD-1/PD-L1 interaction. Rev
Recent Clin Trials. 7:10–23. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee JJ, Chan A and Tang T: Tuberculosis
reactivation in a patient receiving anti-programmed death-1 (PD-1)
inhibitor for relapsed Hodgkin's lymphoma. Acta Oncol. 55:519–520.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Karakatsanis S, Bertsias G, Roussou P and
Boumpas D: Programmed death 1 and B and T lymphocyte attenuator
immunoreceptors and their association with malignant
T-lymphoproliferative disorders: Brief review. Hematol Oncol.
32:113–119. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Huang B, Chen L, Bao C, Sun C, Li J, Wang
L and Zhang X: The expression status and prognostic significance of
programmed cell death 1 ligand 1 in gastrointestinal tract cancer:
A systematic review and meta-analysis. Onco Targets Ther.
8:2617–2625. 2015.PubMed/NCBI
|
|
17
|
Ferris R: PD-1 targeting in cancer
immunotherapy. Cancer. 119:E1–E3. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yan F, Zheng Y and Huang L:
Adenovirus-mediated combined anti-angiogenic and pro-apoptotic gene
therapy enhances antitumor efficacy in hepatocellular carcinoma.
Oncol Lett. 5:348–354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sinkovics JG and Horvath JC: Natural and
genetically engineered viral agents for oncolysis and gene therapy
of human cancers. Arch Immunol Ther Exp (Warsz). 56 Suppl 1:S3–S59.
2008. View Article : Google Scholar
|
|
20
|
Zhang MM, Yan LN, Li DH, Gou XH, Liu JW,
Su Z, Han L and Zhao LY: Inhibition of adenovirus-mediated gene
transfer of antisense matrix metalloproteinase-2 on hepatocellular
carcinoma growth in vivo. Zhonghua Gan Zang Bing Za Zhi.
13:671–674. 2005.(In Chinese). PubMed/NCBI
|
|
21
|
Thomas AA, Ernstoff MS and Fadul CE:
Immunotherapy for the treatment of glioblastoma. Cancer J.
18:59–68. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Larsen CJ: Cellular immunotherapy and
glioblastoma: A hopeful treatment? Bull Cancer. 98:4572011.(In
French). PubMed/NCBI
|
|
23
|
Varghese S, Rabkin SD, Nielsen GP,
MacGarvey U, Liu R and Martuza RL: Systemic therapy of spontaneous
prostate cancer in transgenic mice with oncolytic herpes simplex
viruses. Cancer Res. 67:9371–9379. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Husain SR, Behari N, Kreitman RJ, Pastan I
and Puri RK: Complete regression of established human glioblastoma
tumor xenograft by interleukin-4 toxin therapy. Cancer Res.
58:3649–3653. 1998.PubMed/NCBI
|
|
25
|
Debinski W, Gibo DM, Obiri NI, Kealiher A
and Puri RK: Novel anti-brain tumor cytotoxins specific for cancer
cells. Nat Biotechnol. 16:449–453. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bera TK, Viner J, Brinkmann E and Pastan
I: Pharmacokinetics and antitumor activity of a bivalent
disulfide-stabilized Fv immunotoxin with improved antigen binding
to erbB2. Cancer Res. 59:4018–4022. 1999.PubMed/NCBI
|
|
27
|
Ghetie MA, Richardson J, Tucker T, Jones
D, Uhr JW and Vitetta ES: Antitumor activity of Fab' and
IgG-anti-CD22 immunotoxins in disseminated human B lymphoma grown
in mice with severe combined immunodeficiency disease: Effect on
tumor cells in extranodal sites. Cancer Res. 51:5876–5880.
1991.PubMed/NCBI
|
|
28
|
Neves Costa M, Giakoustidis A, Stamp G,
Gaya A and Mudan S: Extended survival after complete pathological
response in metastatic pancreatic ductal adenocarcinoma following
induction chemotherapy, chemoradiotherapy, and a novel
immunotherapy agent, IMM-101. Cureus. 7:e4352015.PubMed/NCBI
|
|
29
|
Li J, Chen QY, He J, Li ZL, Tang XF, Chen
SP, Xie CM, Li YQ, Huang LX, Ye SB, et al: Phase I trial of
adoptively transferred tumor-infiltrating lymphocyte immunotherapy
following concurrent chemoradiotherapy in patients with
locoregionally advanced nasopharyngeal carcinoma. Oncoimmunology.
4:e9765072015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang X, Bayer ME, Chen X, Fredrickson C,
Cornforth AN, Liang G, Cannon J, He J, Fu Q, Liu J, et al: Phase I
trial of active specific immunotherapy with autologous dendritic
cells pulsed with autologous irradiated tumor stem cells in
hepatitis B-positive patients with hepatocellular carcinoma. J Surg
Oncol. 111:862–867. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nazarkina ZhK and Laktionov PP:
Preparation of dendritic cells for cancer immunotherapy. Biomed
Khim. 61:30–40. 2015.(In Russian). View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dias JD, Hemminki O, Diaconu I, Hirvinen
M, Bonetti A, Guse K, Escutenaire S, Kanerva A, Pesonen S, Löskog
A, et al: Targeted cancer immunotherapy with oncolytic adenovirus
coding for a fully human monoclonal antibody specific for CTLA-4.
Gene Ther. 19:988–998. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
LaRocca CJ, Han J, Gavrikova T, Armstrong
L, Oliveira AR, Shanley R, Vickers SM, Yamamoto M and Davydova J:
Oncolytic adenovirus expressing interferon alpha in a syngeneic
Syrian hamster model for the treatment of pancreatic cancer.
Surgery. 157:888–898. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gerhardt A, Usener D, Keese M, Sturm J,
Schadendorf D and Eichmuller S: Tissue expression and
sero-reactivity of tumor-specific antigens in colorectal cancer.
Cancer Lett. 208:197–206. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Santin AD, Bellone S, Palmieri M, Bossini
B, Cane' S, Bignotti E, Roman JJ, Cannon MJ and Pecorelli S:
Restoration of tumor specific human leukocyte antigens class
I-restricted cytotoxicity by dendritic cell stimulation of tumor
infiltrating lymphocytes in patients with advanced ovarian cancer.
Int J Gynecol Cancer. 14:64–75. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Morotomi N, Fukuda K, Nakano M, Ichihara
S, Oono T, Yamazaki T, Kobayashi N, Suzuki T, Tanaka Y and
Taniguchi H: Evaluation of intestinal microbiotas of healthy
Japanese adults and effect of antibiotics using the 16S ribosomal
RNA gene based clone library method. Biol Pharm Bull. 34:1011–1020.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gustafsson RK, Engdahl EE and Fogdell-Hahn
A: Development and validation of a Q-PCR based TCID50 method for
human herpesvirus 6. Virol J. 9:3112012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fukui A, Muragaki Y, Saito T, Maruyama T,
Nitta M, Ikuta S and Kawamata T: Volumetric analysis using
low-field intraoperative magnetic resonance imaging for 168 newly
diagnosed supratentorial glioblastomas: Effects of extent of
resection and residual tumor volume on survival and recurrence.
World Neurosurg. 98:73–80. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
The Guide for the Care and Use of
Laboratory Animals. ILAR J 57: NP, 2016.40. Greaves MF and Brown G:
Purification of human T and B lymphocytes. J Immunol. 112:420–423.
1974.PubMed/NCBI
|
|
41
|
Shiono H and Ito Y: Novel method for
continuous cell separation by density gradient centrifugation:
Evaluation of a miniature separation column. Prep Biochem
Biotechnol. 33:87–100. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ting WH, Chien MN, Lo FS, Wang CH, Huang
CY, Lin CL, Lin WS, Chang TY, Yang HW, Chen WF, et al: Association
of cytotoxic t-lymphocyte-associated protein 4 (CTLA4) gene
polymorphisms with autoimmune thyroid disease in children and
adults: Case-control study. PloS One. 11:e01543942016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zamarin D, Vigil A, Kelly K, Garcia-Sastre
A and Fong Y: Genetically engineered Newcastle disease virus for
malignant melanoma therapy. Gene Ther. 16:796–804. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Allegra M, Zaragkoulias A, Vorgia E,
Ioannou M, Litos G, Beug H and Mavrothalassitis G: Semaphorin-7a
reverses the ERF-induced inhibition of EMT in Ras-dependent mouse
mammary epithelial cells. Mol Biol Cell. 23:3873–3881. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Youlin K, Jian K, Siming L, Li Z, Weiyang
H, Chaodong L and Xin G: Potent anti-prostate cancer immune
response induced by dendritic cells transduced with recombinant
adenoviruses encoding 4-1BBL combined with cytokine-induced killer
cells. Immunotherapy. 7:13–20. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Leisegang M, Kammertoens T, Uckert W and
Blankenstein T: Targeting human melanoma neoantigens by T cell
receptor gene therapy. J Clin Invest. 126:854–858. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Braybrooke JP, Slade A, Deplanque G,
Harrop R, Madhusudan S, Forster MD, Gibson R, Makris A, Talbot DC,
Steiner J, et al: Phase I study of MetXia-P450 gene therapy and
oral cyclophosphamide for patients with advanced breast cancer or
melanoma. Clin Cancer Res. 11:1512–1520. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zou W, Luo C, Zhang Z, Liu J, Gu J, Pei Z,
Qian C and Liu X: A novel oncolytic adenovirus targeting to
telomerase activity in tumor cells with potent. Oncogene.
23:457–464. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hernández-Alcoceba R, Sangro B and Prieto
J: Gene therapy of liver cancer. Ann Hepatol. 6:5–14.
2007.PubMed/NCBI
|
|
50
|
Moss JA: Gene therapy review. Radiol
Technol. 86:155–180; quiz 181–184. 2014.PubMed/NCBI
|
|
51
|
Eibel B, Markoski MM, Rodrigues CG, Dipp
T, de Salles FB, Giusti II, Nardi NB, Plentz RD and Kalil RA: VEGF
gene therapy cooperatively recruits molecules from the immune
system and stimulates cell homing and angiogenesis in refractory
angina. Cytokine. 91:44–50. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sharma A, Mathew Easow M, Sriganesh V and
Reiss UM: Gene therapy for haemophilia. Cochrane Database Syst Rev.
12:CD0108222016.PubMed/NCBI
|
|
53
|
Hammer A and Steiner S: Gene therapy for
therapeutic angiogenesis in peripheral arterial disease-a
systematic review and meta-analysis of randomized, controlled
trials. VASA. 42:331–339. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kumar MS, Masthan KM, Babu NA and Dash KC:
Gene therapy in oral cancer: A review. J Clin Diagn Res.
7:1261–1263. 2013.PubMed/NCBI
|
|
55
|
Koirala A, Conley SM and Naash MI: A
review of therapeutic prospects of non-viral gene therapy in the
retinal pigment epithelium. Biomaterials. 34:7158–7167. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kim YS, Hwang KA, Go RE, Kim CW and Choi
KC: Gene therapy strategies using engineered stem cells for
treating gynecologic and breast cancer patients (Review). Oncol
Rep. 33:2107–2112. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Watanabe M, Nasu Y and Kumon H:
Adenovirus-mediated REIC/Dkk-3 gene therapy: Development of an
autologous cancer vaccination therapy (Review). Oncol Lett.
7:595–601. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ludlow SP, Andrews S, Pasikhova Y and Hill
E: New-onset toxicity with programmed death-1 inhibitor
rechallenge. Melanoma Res. 26:316–318. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ansell SM: Where do programmed death-1
inhibitors fit in the management of malignant lymphoma? J Oncol
Pract. 12:101–106. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Villasboas JC and Ansell S: Checkpoint
inhibition: Programmed cell death 1 and programmed cell death 1
ligand inhibitors in Hodgkin lymphoma. Cancer J. 22:17–22. 2016.
View Article : Google Scholar : PubMed/NCBI
|