Introduction
Pancreatic cancer is frequently fatal, with a
generally poor prognosis (1).
Pancreatic adenocarcinoma (PAAD) is the most common subtype of
pancreatic cancer in humans and is responsible for >85% of
pancreatic cancer cases (2). It is
projected to become the second leading cause of cancer-associated
death in Western societies within a decade (3). Its dismal prognosis is due to rapid
disease progression and early metastasis, leading to late diagnosis
and a high level of resistance to treatment (4). Growing evidence has revealed a
relatively complex underlying mechanism of the disease in terms of
development and progression, including the interaction between
epigenomic and genomic alterations (5). Despite the understanding of aberrant
gene networks gained from previous studies, the epigenomic
interactions in PAAD remain to be fully elucidated (6).
At present, the genetic and phenotypic changes
associated with the pathogenesis of pancreatic cancer are
increasingly attracting attention (7). As an epigenetic modification, DNA
methylation is critical for maintaining genomic and chromosomal
structural stability, gene imprinting and gene silencing (8-10).
DNA methylation appears primarily on cytosine residues in CpG
sites, which exist throughout the genome. Areas with abundant CpG
sites are called CpG islands. Altered DNA methylation is linked to
the initiation and progression of various types of cancer, and high
levels of methylation in CpG islands and promoter regions may lead
to transcriptional silencing of tumor suppressor genes (8). By contrast, hypomethylation may lead to
overexpression of oncogenes and genomic instability.
High-throughput methods have been used to assess the
epigenetic changes associated with the development and progression
of PAAD (11). Studies have
indicated that a variety of genes in pancreatic cancer, including
Ras association domain family 1 isoform A (RASSF1A), p16,
suppressor of cytokine signalling 1 (SOCS-1) and neuronal pentraxin
2 (NPTX2), are abnormally methylated and are critical for the
pathogenesis of pancreatic cancer (12-14).
However, a large number of statistically significant methylation
events identified by high-throughput screening have no association
with gene expression changes. A high-throughput method is required
to integrate data across multiple platforms to determine the
epigenetic events that are most likely to be associated with
PAAD.
The Cancer Genome Atlas (TCGA) indicates a
significant diversity of genetic and epigenetic variations in PAAD
(15). Numerous platforms used in
the TCGA database enable the analysis of data integrated from
multiple sources to identify specific abnormalities that are most
likely to result in a carcinogenic process. However, no integrated
DNA methylation and transcriptome data are available and it is
currently not possible to identify DNA methylation-driven genes in
PAAD. MethylMix is a method of applying three criteria to identify
methylation-driven genes in diseases by integrating DNA methylation
and transcriptome data (16). First,
the determination of the degree of methylation should not depend on
any threshold that is normally used. Furthermore, the
hypermethylation or hypomethylation of a gene in cancer must be
evaluated in comparison with normal tissue. Finally, the
identification of highly methylated or hypomethylated genes should
be selective for those with predicted effects on transcription,
meaning that their methylation is functionally associated.
Despite the improvement in the ability to diagnose
PAAD over time, it remains difficult to differentiate malignant
tumors from benign disease. Endoscopic ultrasound-fine-needle
aspiration provides a tissue sample for cytological studies,
thereby helping resolve this issue; however, an improper biopsy
reduces the diagnostic value (17).
Adjuvant tests may be performed to screen for biomarkers in tissue
samples to reduce the requirement for biopsy. DNA methylation is
currently being assessed as a biomarker. Epigenetic changes do not
lead to DNA sequence mutations, but they serve as causal links
between genes and phenotypes. DNA methylation is one epigenetic
mechanism (18). Epigenetic changes
may activate or suppress numerous signaling pathways, thereby
causing cancer. Epigenetic changes may occur in the early
development stage of tumors; thus, they may serve as biomarkers
that may be able to help identify and prevent cancer in the early
stage (19). As PAAD-specific
biomarkers with high diagnostic value are not sufficient, the
identification of PAAD-specific DNA methylation markers with high
specificity and sensitivity will help further enhance the ability
of clinicians to diagnose PAAD.
PAAD is a painful and potentially fatal disease that
is an important health concern and poses numerous therapeutic
challenges. The significance of DNA methylation in the development
of PAAD is gaining attention. However, to date, no comprehensive
analysis of DNA methylation and transcriptome data has been
performed to identify DNA methylation-driven genes in PAAD.
Therefore, identifying DNA methylation-driven genes and
investigating the mechanisms underlying the tumorigenesis of PAAD
are of substantial importance to treatment planning and prognostic
prediction for PAAD. The present study reports on the analysis of
large-scale methylation data using Infinium 450 k methylation arrays
(Illumina) and expression profiles using RNA sequencing. A total of
7 DNA methylation-driven genes were identified, all of which may
serve as important markers for the diagnosis of PAAD. Furthermore,
6 of them were associated with cancer recurrence and patient
survival. These DNA methylation-driven genes are critical for
elucidating PAAD progression and may serve as future therapeutic
biomarkers.
Materials and methods
Screening for differentially expressed
genes (DEGs)
In total, 188 DNA methylation profiles (178 PAAD
samples together with 10 non-tumor samples), 182 RNA-sequencing
(RNA-seq) profiles (178 PAAD samples together with 4 non-tumor
samples), and the associated clinical PAAD data were downloaded
from the TCGA database (https://portal.gdc.cancer.gov/) (Table SI). Of the 178 patients with PAAD,
178 patients had overall survival (OS) data and 155 patients had
recurrence-free survival (RFS) data. To screen for the DEGs between
normal pancreatic tissue and PAAD, the ‘edgeR’ package in R was
used (20) England. A false
discovery rate (FDR)<0.05 and a |log2 fold change
(FC)|>2 were defined as the cut-off criteria to select genes for
further analysis.
Integrated analysis of DNA methylation
and gene expression
Level 3 DNA methylation data and clinical
information, including time of death, follow-up time and status of
PAAD, were downloaded from TCGA database on August 1, 2018. An
analysis combining gene expression (RNA-seq) data and methylation
data was performed using the ‘MethylMix’ package in R to identify
DNA methylation-driven genes (16).
MethylMix is a program designed to identify methylation events
associated with gene expression (16). MethylMix requires three datasets:
Disease DNA methylation data, matched disease gene expression data
and normal DNA methylation data. The MethylMix analysis has three
parts: First, disease DNA methylation data are combined with the
matched disease gene expression data to identify the methylation
events leading to changes in gene expression, and only genes that
pass the correlation filter are selected for further analysis;
second, a beta mixed model is used to define the methylation state
in a large number of patients, eliminating the need for any
threshold; and third, the Wilcoxon rank sum test is used to compare
the DNA methylation statuses between normal samples and cancer
samples. Multiple testing is calculated using a q cutoff of 0.05.
The final result is a differential methylation (DM) value, where a
positive DM value represents a high degree of methylation and a
negative DM value represents a low degree of methylation.
Receiver operating characteristic
(ROC) analyses
For the assessment of the diagnostic values of DNA
methylation-driven genes in PAAD, ROC analyses were performed using
the ‘pROC’ package in R. The ROC curve was generated and the area
under the curve (AUC) with the binomial exact confidence interval
was calculated. For AUC values >0.7, the hub gene was deemed
able to distinguish between normal pancreatic tissue and PAAD with
excellent specificity and sensitivity.
Survival analysis
For the survival analysis, 178 patients with DNA
methylation data and OS data were dichotomized into two groups
(high vs. low) based on the optimal cutoff value for each DNA
methylation-driven gene. Subsequently, the ‘survival’ package in R
was adopted to generate the Kaplan-Meier survival curve and perform
the log-rank test. Multivariate Cox regression analyses were then
performed to investigate whether the prognostic value of the gene
was independent of TNM stage.
Recurrence analysis
The correlation between the 7 DNA methylation-driven
genes and recurrence may provide a preliminary indication of the
role of these genes in the prognostic prediction and the aspects of
PAAD involved. In the recurrence analysis, 155 patients with DNA
methylation data and RFS data were dichotomized into two groups
(high vs. low) based on the optimal cutoff value for each DNA
methylation-driven gene. Recurrence analysis was performed as
described above.
Gene set enrichment analysis
(GSEA)
For the GSEA, 178 PAAD samples with DNA methylation
data were divided into two groups (high vs. low) according to the
DNA methylation level of each DNA methylation-driven gene and the
median DNA methylation value was used as the cut-off point. To gain
insight into the function of each DNA methylation-driven gene, GSEA
(version 3.0; http://software.broadinstitute.org/gsea/index.jsp) was
performed with these two groups. The annotated gene sets
c2.cp.kegg.v5.2.symbols.gmt were selected as the reference gene
sets. P<0.05 was considered to indicate a statistically
significant difference.
Results
DEGs in PAAD
The RNA expression levels in 178 PAAD tissues and 4
normal tissues were analyzed. Genes meeting the cut-off criteria of
a corrected P<0.05 and |log2FC|>2 were regarded as
differentially expressed. Thereby, 814 (65.92%) downregulated genes
and 30 (34.08%) upregulated genes were identified.
Identification of DNA
methylation-driven genes
DNA methylation data for 10 normal tissues and 178
PAADs and gene expression data for the DEGs identified in the 178
PAADs were included in the analysis. Thresholds of a corrected
P-value for differential expression between normal tissues and PAAD
of <0.05 and a correlation between gene expression and DNA
methylation of <-0.3 were used. A total of 7 DNA
methylation-driven genes, namely zinc finger protein 208 (ZNF208),
eomesodermin (EOMES), prostaglandin D2 receptor (PTGDR), chromosome
12 open reading frame 42 (C12orf42), integrin subunit α 4 (ITGA4),
dedicator of cytokinesis 8 (DOCK8) and protein phosphatase 1
regulatory inhibitor subunit 14D (PPP1R14D), were identified
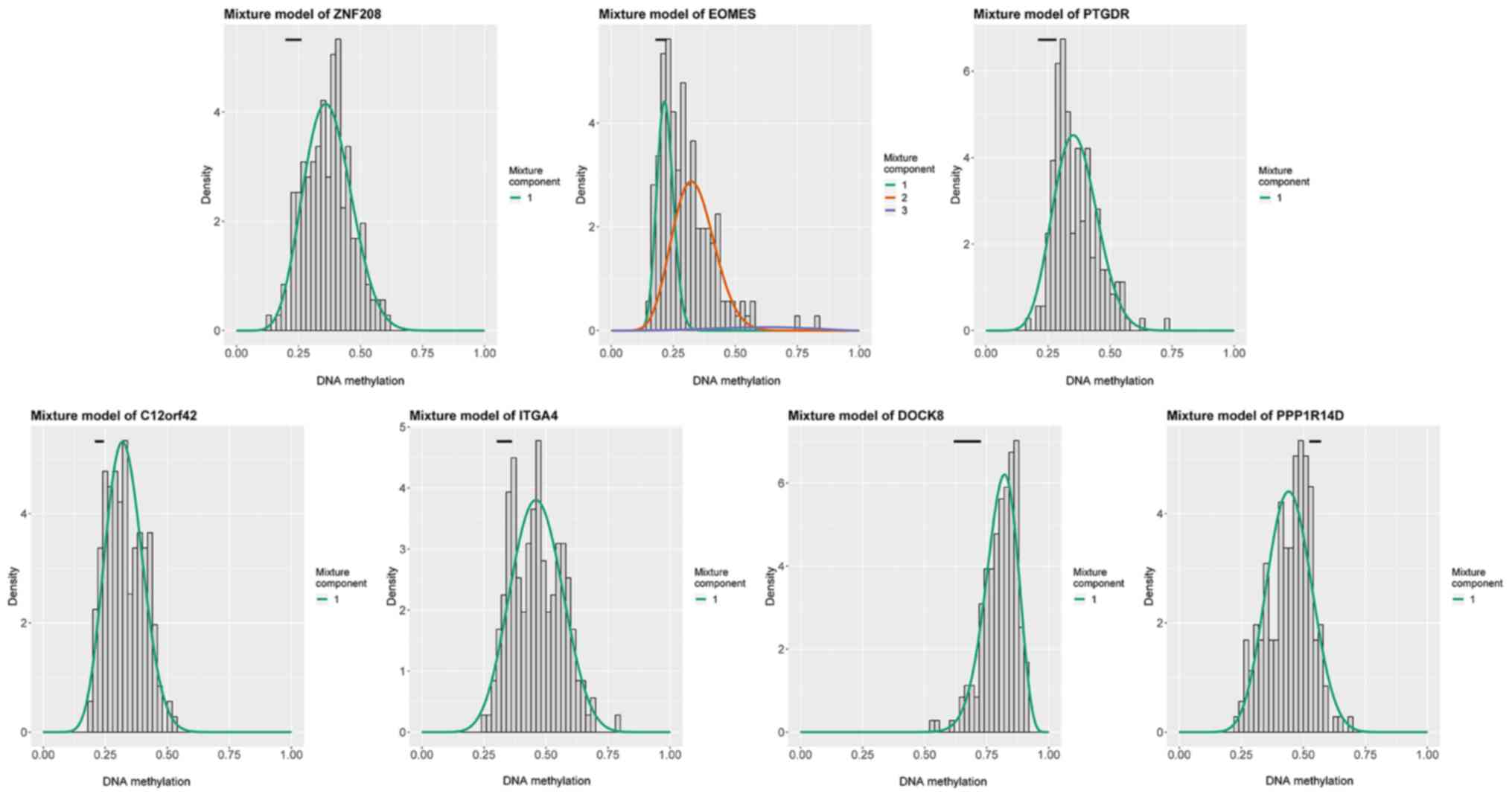
(Table SII). Of these genes,
ZNF208, EOMES, PTGDR, C12orf42, ITGA4 and DOCK8 were
hypermethylated, while PPP1R14D was hypomethylated (Fig. 1; Table
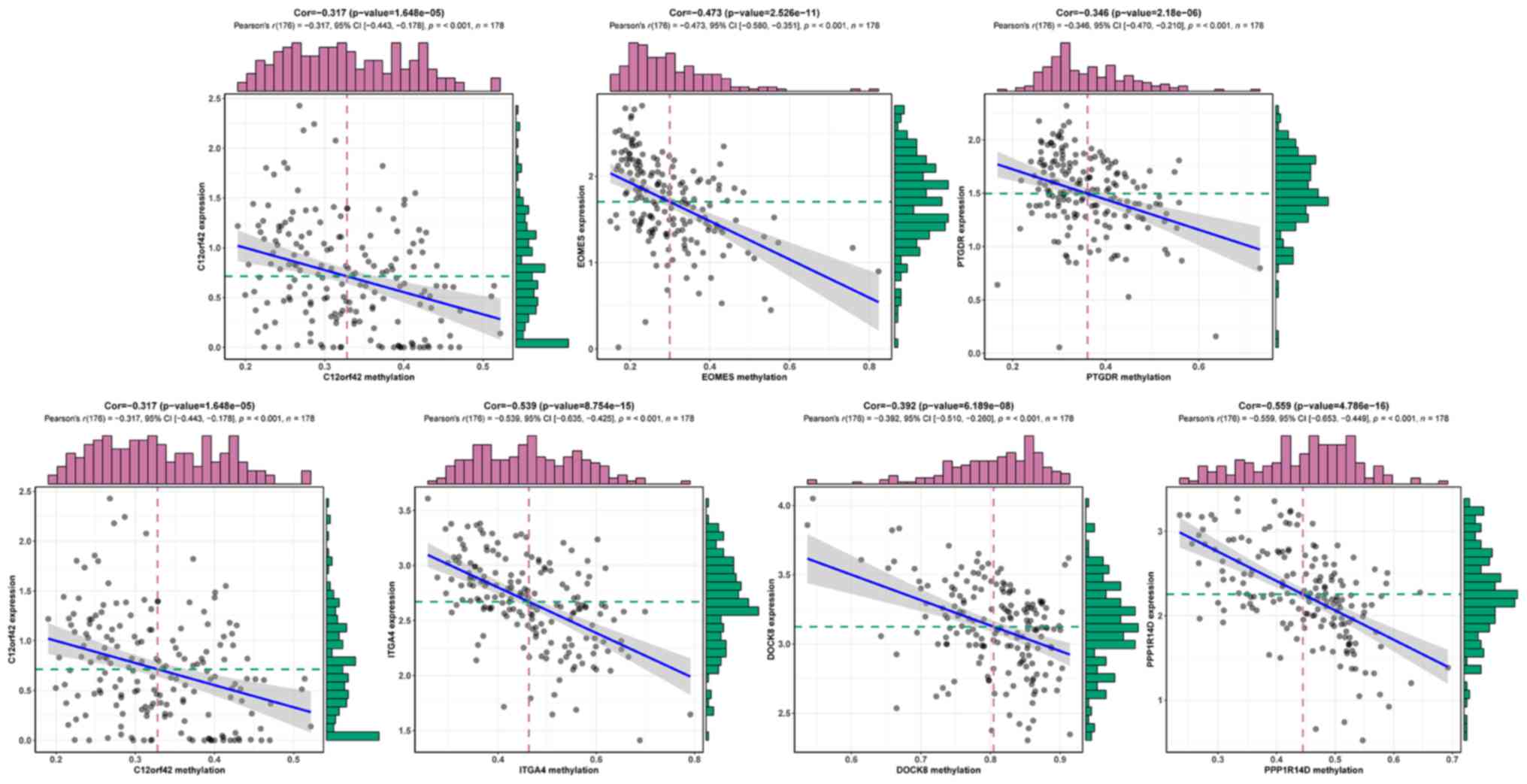
SII). Fig. 2 displays the
inverse correlation between DNA methylation and the matched gene
expression of the 7 methylation-driven genes.
Diagnostic efficiency of DNA
methylation-driven genes
To assess the diagnostic ability of the 7 DNA
methylation-driven genes in the TCGA dataset, ROC curve analyses
were performed and calculate the AUCs. The AUCs of ZNF208 (0.918),
EOMES (0.840), PTGDR (0.903), C12orf42 (0.918), ITGA4 (0.876),
DOCK8 (0.903) and PPP1R14D (0.888) were all >0.8, indicating
that the 7 DNA methylation-driven genes, particularly ZNF208,
PTGDR, C12orf42 and DOCK8, exhibited excellent diagnostic
efficiency for PAAD (Fig. 3). For
the diagnosis of PAAD, the sensitivity and specificity of ZNF208
were 88.6 and 90.0%, respectively; those of PTGDR were 83.8 and
90.0%, respectively; those of C12orf42 were 89.7 and 90.0%,
respectively; and the sensitivity and specificity of DOCK8 were
78.4 and 90.0%, respectively.
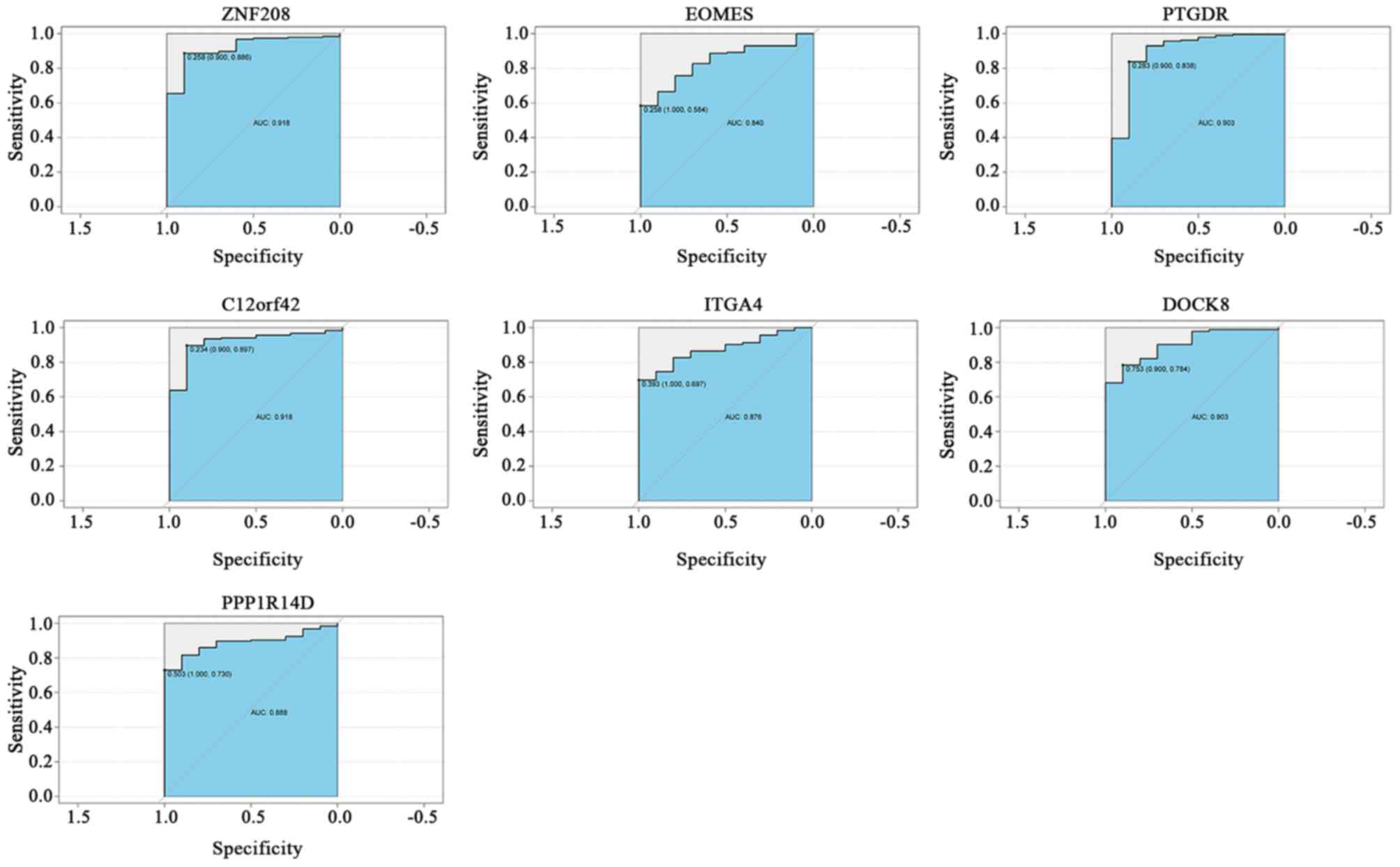 | Figure 3ROC curves of DNA methylation-driven
genes for distinguishing between pancreatic adenocarcinoma and
healthy controls. The cutoff value for determining the AUC was
obtained from the ‘roc’ function of the ‘pROC’ R package. The left
and right values in the brackets indicate the specificity and
sensitivity of the cutoff value, respectively. The ROC curves
indicate the diagnostic ability of the DNA methylation-driven
genes, with the x-axis and y-axis displaying the specificity and
sensitivity, respectively. ROC, receiver operating characteristic;
AUC, area under curve; ZNF208, zinc finger protein 208; EOMES,
eomesodermin; PTGDR, prostaglandin D2 receptor; C12orf42,
chromosome 12 open reading frame 42; ITGA4, integrin subunit α 4;
DOCK8, dedicator of cytokinesis 8; PPP1R14D, protein phosphatase 1
regulatory inhibitor subunit 14D. |
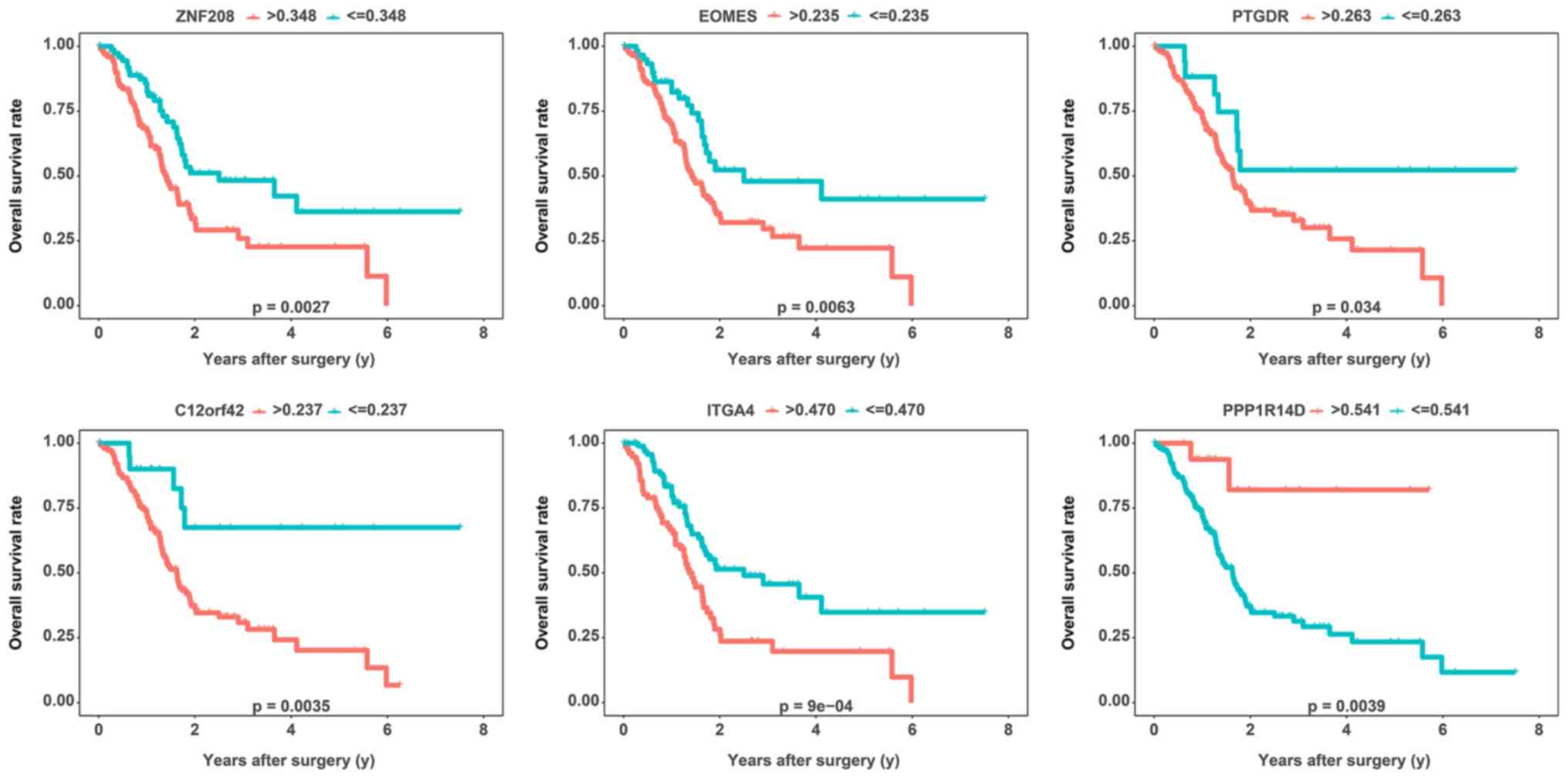
Survival prediction using the DNA
methylation-driven genes
For the analysis of the potential usefulness of DNA
methylation-driven genes for the prognostic prediction for PAAD,
Kaplan-Meier survival curves were generated and log-rank tests were
performed for each of the DNA methylation-driven genes. In total,
176 patients with clinical data, including OS and survival status,
were divided into high and low DNA methylation status groups with
respect to the 7 DNA methylation-driven genes according to their
respective optimal cutoffs based on the ‘surv_cutpoint’ function of
the ‘survminer’ R package. Finally, 5 DNA methylation-driven genes
(ZNF208, EOMES, PTGDR, C12orf42 and ITGA4) were considered to be
significantly negatively associated with the survival time of
patients with PAAD (P<0.05 by log-rank test), while PPP1R14D was
considered to be significantly positively associated with the
survival time of PAAD patients (P<0.01 by log-rank test)
(Fig. 4; Table SIII). However, DOCK8 was not
significantly associated with the OS of patients with PAAD
(Table SIII). To determine
whether these genes were independent of the TNM stage with regard
to the prediction of the prognosis of patients with PAAD, a
multivariate analysis was performed for each gene. Multivariate Cox
regression analysis revealed that after adjusting for TNM stage,
C12orf42 [P=0.012, hazard ratio (HR): 3.304, 95% CI: 1.302-8.383],
PPP1R14D (P=0.021, HR: 0.189, 95% CI: 0.046-0.774), EOMES (P=0.024,
HR: 1.744, 95% CI: 1.075-2.830), ZNF208 (P=0.006, HR: 1.885, 95%
CI: 1.202-2.954) and ITGA4 (P=0.012, HR: 3.304, 95% CI:
1.302-8.383) were still significantly associated with OS,
indicating that C12orf42, PPP1R14D, EOMES, ZNF208 and ZGA4 are
independent of the TNM stage. However, as the number of patients
was relatively small, PTGDR did not achieve statistical
significance (P=0.083, HR: 2.050, 95% CI: 0.910-4.617).
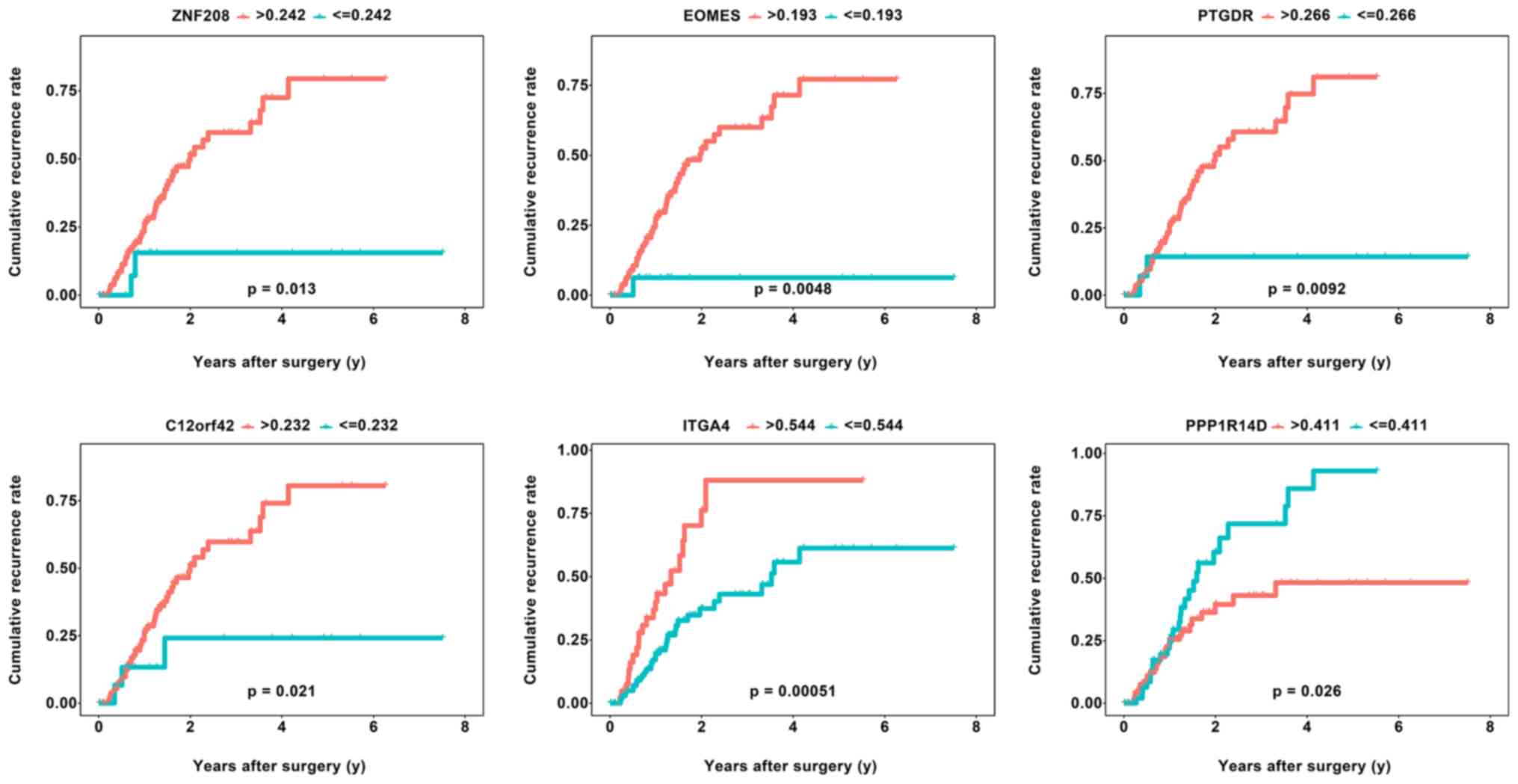
Recurrence prediction using DNA
methylation-driven genes
To explore the association between the 7 identified
DNA methylation-driven genes and the recurrence rate of PAAD, the
cumulative recurrence rates associated with the 7 DNA
methylation-driven genes in patients with PAAD were investigated
using Kaplan-Meier curve analysis. In total, 154 patients with
clinical data, including RFS and relapse status, were divided into
high and low DNA methylation status groups with respect to the
methylation status of each of the 7 DNA methylation-driven genes
according to their respective optimal cutoffs based on the
‘surv_cutpoint’ function of the ‘survminer’ R package. Of the 7 DNA
methylation-driven genes, 6 exhibited an obvious association with
the recurrence rate based on their respective optimal cut-offs. A
total of five DNA methylation-driven genes (ZNF208, EOMES, PTGDR,
C12orf42 and ITGA4) were positively associated with the recurrence
rate, while PPP1R14D was negatively associated with the recurrence
rate (log-rank P<0.05) (Fig. 5;
Table SIV). However, DOCK8 did not
have a significant correlation with the recurrence rate of PAAD
(Table SIV). This result is
consistent with the result of the OS analysis. To determine whether
these genes were predictors of RFS independent of the TNM stage, a
multivariate analysis was performed for each gene. The multivariate
Cox regression analyses revealed that after adjusting for TNM
stage, PTGDR (P=0.031, HR: 4.966, 95% CI: 1.159-21.271), EOMES
(P=0.002, HR: 2.298, 95% CI: 1.346-3.922), ZNF208 (P=0.045, HR:
4.474, 95% CI: 1.035-19.345) and ITGA4 (P=0.003, HR: 2.244, 95% CI:
1.317-3.822) were still significantly associated with RFS,
indicating that PTGDR, EOMES, ZNF208 and ITGA4 were predictors of
RFS independent of the TNM stage. However, as the number of
patients was small, no significant association was obtained for
C12orf42 (P=0.056, HR: 3.329, 95% CI: 0.970-11.424) and PPP1R14D
(P=0.076, HR: 0.612, 95% CI: 0.355-1.053), although the results
almost reached statistical significance.
Validation of the gene expression of
the 7 DNA methylation-driven genes
In the TCGA PAAD cohort (178 PAAD tissues and 4
normal tissues), EOMES (P<0.001), C12orf42 (P<0.001), ITGA4
(P<0.001), DOCK8 (P<0.001), PTGDR (P<0.001), ZNF208
(P<0.001) and PPP1R14D (P<0.001) were significantly
differentially expressed between PAAD tissues and normal tissues.
To verify these results, the expression levels of the 7 DNA
methylation-driven genes were assessed in the GSE15471 PAAD cohort
(35 PAAD tissues and 35 normal tissues). Consistent with the
results of the initial analysis, the mean expression levels of
EOMES (P=0.024), C12orf42 (P=0.002), ITGA4 (P<0.001) and DOCK8
(P<0.001) were significantly differentially expressed between
PAAD tissues and normal tissues according to the analysis with the
limma package in R. However, the differences in expression levels
of PTGDR (P=0.160), ZNF208 (P=0.178) and PPP1R14D (P=0.222) did not
reach statistical significance. In addition, the expression of
PTGDR, ZNF208 and PPP1R14D was also verified in a larger dataset
(GSE28735 PAAD cohort, including 45 PAAD tissues and 45 normal
tissues), and the results indicated that PPP1R14D (P=0.027) and
ZNF208 (P=0.033), but not PTGDR (P=0.307), were differentially
expressed between PAAD tissues and normal tissues.
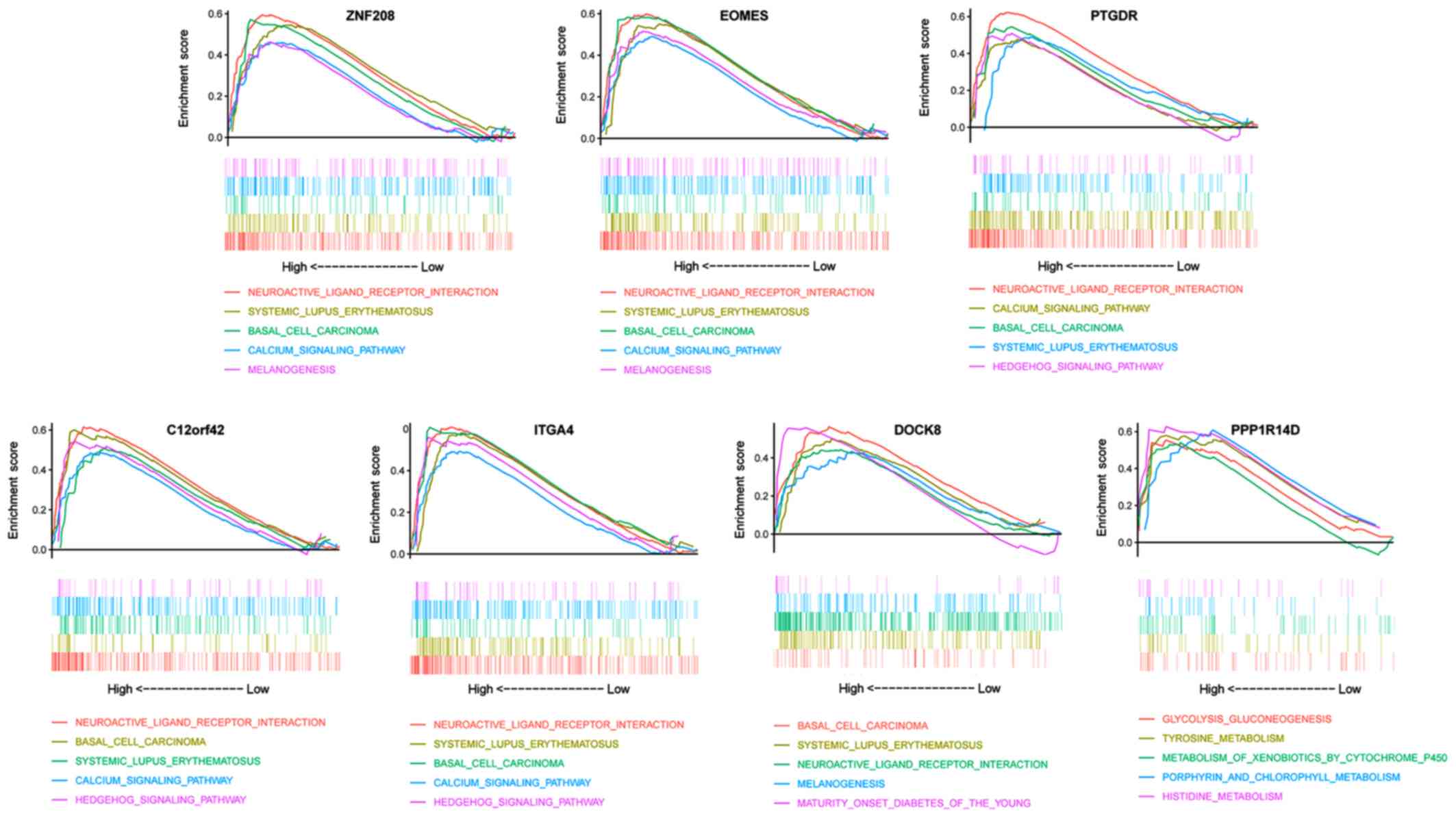
GSEA
To determine the potential function of the 7 DNA
methylation-driven genes, a GSEA was performed to map them using
the Encyclopedia of Genes and Genomes (KEGG) pathways database.
Under the cut-off criterion of P<0.05, PAAD samples in the high
DNA methylation groups for ZNF208, EOMES, PTGDR, C12orf42 and ITGA4
were all most significantly enriched in ‘neuroactive ligand
receptor interaction’ (Fig. 6;
Table SV). PAAD samples in the high
DNA methylation group for DOCK8 were most significantly enriched in
‘basal cell carcinoma’ (Fig. 6;
Table SV). PAAD samples in the high
DNA methylation group for PPP1R14D were most significantly enriched
in ‘glycolysis gluconeogenesis’ (Fig.
6; Table SV).
Discussion
DNA methylation is closely associated with the
occurrence and progression of cancer. In general, there are two
different states of abnormal DNA methylation, namely
hypomethylation and hypermethylation. It is now widely accepted
that abnormal DNA hypermethylation in and around the promoter
region results in gene silencing, while hypomethylation results in
gene activation. DNA analysis of different tumor cells suggested
that the probability of gene mutations in cancer cells was lower
than expected (21). However, at the
transcriptional level, up to 5% of known tumor suppressor genes
exhibit gene silencing caused by hypermethylation in the promoter
region in colorectal cancer, suggesting that abnormal DNA
methylation may be more responsible for malignant cell
transformation than gene mutations (22).
Compared with normal cells, the methylation levels
of tumor cell genomes change substantially; tumor cell genomes are
characterized by DNA hypomethylation of the whole genome and
abnormal hypermethylation of CpG islands in local promoter regions.
Numerous studies have already been performed regarding the abnormal
DNA methylation of the cancer cell genome, particularly the gene
silencing of tumor suppressor genes caused by DNA hypermethylation
in pancreatic cancer (23-27).
Studies have confirmed that DNA hypermethylation is closely linked
to PAAD cells evading apoptosis; obtaining sustained proliferation
signals; exhibiting insensitivity to growth inhibition signals,
tissue infiltration and metastasis; and obtaining infinite
replication potential (23-27).
The infinite replication potential of cancer cells is mainly linked
to increased telomerase activity. Telomerase solves the problem of
terminal end deletion during DNA replication at the transcriptional
level (28). Normally, telomerase is
active only in fertilized eggs and stem cells, while various types
of tumor cell have highly active telomerase (28). In the present study, 6 DNA
methylation-driven genes (ZNF208, EOMES, PTGDR, C12orf42, ITGA4 and
DOCK8) were hypermethylated. ZNF208 has been indicated to be
associated with leukocyte telomere length (LTL) (29). LTL-associated genes affect the cancer
risk in adults and increased telomere length is associated with an
elevated risk of glioma, melanoma and lung cancer in adults
(30-33).
In addition, downregulation of the mRNA levels of ZNF208, a key
tumor suppressor gene in gastric cancer, may contribute to the
extensive dissemination of cancer cells in the abdominal cavity
(34). The present study indicated
that ZNF208 is hypermethylated and has low mRNA expression levels
in PAAD compared to normal tissues, which may promote the
occurrence and progression of PAAD.
DNA hypomethylation includes DNA hypomethylation of
specific proto-oncogenes and DNA hypomethylation of the whole
genome. Recent studies have demonstrated that abnormal
hypomethylation of specific proto-oncogenes is also critical for
the progression and development of pancreatic cancer (35). In the present study, PPP1R14D was
hypomethylated and had a relatively low mRNA expression in PAAD. In
a previous study, PPP1R14D, a metabolic signaling protein, was
indicated to be differentially expressed between normal and
pathological conditions of the brain associated with diabetes
(36). Diabetes is considered to be
a crucial risk factor for PAAD (37). Therefore, PPP1R14D may be involved in
the development of PAAD.
Although numerous methods have been developed for
the diagnosis of PAAD, each method has its own strengths and
weaknesses, and no single method is absolutely superior to the
others. The method of choice depends on the resolution and genome
coverage requirements, and these two parameters dictate the
experimental cost (38). For
instance, sequencing approaches have the advantage of providing
quantitative information with regard to the methylation state of
each CpG and allowing for repeated analysis of methylation and rare
methylation variants, which cannot be performed via microarrays.
Furthermore, it is possible to apply sequencing approaches to
analyze the DNA methylation of regions without previous sequence
information. However, despite the quickly decreasing cost of
large-scale sequencing technologies, these technologies have
certain major weaknesses, including library bias, high cost and low
availability, as well as data management and analysis difficulties.
In addition, methods based on bisulfite conversion and sequencing
require a large number of bioinformatics resources for calling
bases, aligning sequence and performing the statistical analysis.
CpG-specific array technology, including DNA methylation arrays,
are alternative options that may be used to determine a genome-wide
DNA methylation profile. In comparison to sequencing technologies,
DNA methylation arrays are less expensive and allow for profiling
of a large number of samples, although the resolution is reduced.
DNA methylation assays take less time and are less labor-intensive
(39-41).
Advances in the technologies used to detect
genome-wide epigenetic alterations greatly assist us in developing
biomarkers for early-stage tumor diagnosis (42). DNA methylation maintains the genomic
structure while controlling gene expression. Aberrant DNA
methylation usually occurs in promoter regions of the transcription
factors that are involved in the development and proliferative
process of cancers (43). For
instance, hypomethylation of the mesothelin promoter causes
mesothelioma (44), and
hypomethylated Alu and long interspersed element-1 retrotransposons
may lead to lung cancer (45).
However, peritoneal mesothelioma has been indicated to feature DNA
hypermethylation (46). According to
all of these data, DNA methylation is able to perfectly represent
and reflect the molecular changes in human tumors in the early
stage. Therefore, it may be a useful biomarker facilitating the
diagnosis of malignant tumors in the early stage. The present study
focused on confirming the validity of highly sensitive biomarkers
that may be even more sensitive when used in combination with other
biomarkers for the detection of malignancies.
Epigenetic changes due to DNA methylation are one of
the important features of the progression of cells to malignancy.
Therefore, changes in DNA methylation may be an important
facilitator of the early diagnosis of cancer (47). Japanese scholars have collected the
fluid from normal pancreases and those with cancer and chronic
pancreatitis and used quantitative methylation-specific PCR to
detect the degree of methylation of 6 genes, including p16 and
cyclin D2(47). Experiments have
revealed that, compared with other methods, the measurement of
abnormal methylation of specific genes in the pancreatic fluid is
able to more accurately identify pancreatic lesions, leading to the
effective early detection of pancreatic cancer (47). In the present study, the AUCs for
distinguishing between normal pancreatic tissue and PAAD for the 7
DNA methylation-driven genes, ZNF208, EOMES, PTGDR, C12orf42,
ITGA4, DOCK8 and PPP1R14D, were >0.8, indicating that the 7 DNA
methylation-driven genes exhibited excellent diagnostic efficiency
for PAAD.
Unlike genetic mutations, epigenetic changes are
mostly reversible. Therefore, targeting epigenetic changes in cells
using methylation-associated drugs to alter the methylation status
may become a novel treatment for malignant tumors. At present,
there are mainly two types of reversal method: One is the use of an
antisense oligonucleotide, which is able to inhibit DNA
methyltransferase activity, leading to expression of the methylated
gene, and the second is the use of a cytidine analog that may
covalently bind to DNA methylase, reducing the biological activity
of the enzyme, thereby activating genes that are inactivated by
methylation (48). At present, the
more commonly used cytidine analogs include 5-azacytidine and
decitabine. Studies on different pancreatic cancer cell lines
cultured in vitro confirmed that 5-azacytidine changes the
methylation status of the tumor cell genome through demethylation,
thereby inhibiting tumor cell proliferation and promoting apoptosis
(48). In clinical trials, studies
on myelodysplastic syndromes and acute myeloid leukemia have
indicated that 5-azacytidine is able to increase complete response
rates by 10-17% and prolong patient survival (49,50). In
the present study, 6 of the 7 DNA methylation-driven genes
identified were significantly associated with OS and the RFS. Five
DNA methylation-driven genes (ZNF208, EOMES, PTGDR, C12orf42 and
ITGA4) were significantly negatively associated with the OS time
and positively associated with the recurrence rate. PPP1R14D was
significantly positively associated with the OS time and negatively
associated with the recurrence rate. These genes are likely to
become novel molecular targets in pancreatic cancer treatments
aimed at correcting abnormal DNA methylation to prevent or even
reverse cell cancerization.
The 7 DNA methylation-driven genes (ZNF208, EOMES,
PTGDR, C12orf42, ITGA4, DOCK8 and PPP1R14D) identified in the
present study were previously reported to be associated with other
cancer types. ZNF208 acts as a family member of ZNF proteins that
contain Kruppel-associated box domains, which may participate in
transcriptional regulation (51). In
a study by Hirbe et al (51),
immunohistochemistry helped detect ZNF208 protein expression in
metastatic tumors. In addition, very large genome-wide association
studies (GWAS) have identified an association between ZNF208 and
interindividual variation in LTL (52). A recently performed Mendelian
randomization study demonstrated the impact of single nucleotide
polymorphisms (SNPs) associated with LTL on adult cancer risk and
further indicated that the genetic predisposition to increased
telomere length may increase the risk of lung cancer, melanoma and
glioma in adults (52).
The expression of EOMES has been reported to be
negatively associated with tumor-infiltrating lymphocyte (TIL)
function in non-small cell lung cancers (NSCLCs) in the early
stage, suggesting that the functionality of TILs associated with
early-stage NSCLCs may be influenced by an exhaustion program
marked by EOMES expression (53).
Chang et al (54) indicated
that PTGDR was hypermethylated in endometrial cancer and ovarian
cancer tissues. Przybylski et al (55) identified a novel type of chromosomal
translocation, t(12;14)(q23; q11.2), in T-lymphoblastic lymphoma
between T-cell receptor delta-deleting elements (T-cell receptor
delta recombining element and T cell receptor alpha joining 61) and
the hypothetical gene C12orf42. In a recently performed melanoma
GWAS meta-analysis (involving 12,874 cases and 23,203 controls),
SNPs near DOCK8 reached global significance (56). Morandi et al (57) reported that ITGA4 was hypermethylated
in oral squamous cell carcinoma but not in samples from normal
healthy donors. With regard to PPP1R14D, the Pearson correlation
between the CpG sites with different methylation levels and the
gene expression values in NSCLCs in the early stage reached
statistical significance (58).
As reported by previous studies, RASSF1A, p16,
SOCS-1 and NPTX2 are abnormally methylated in pancreatic cancer and
are critical for the pathogenesis of this cancer type (12-14).
However, in the present study, neither RASSF1A nor p16 was present
in the TCGA database; thus, they were not included in the
prognostic analysis. As better sequencing approaches emerge in the
future, prognostic analyses will improve. In the present study, to
identify the most significant genes associated with PAAD,
thresholds for the screening of DEGs were set at an FDR<0.05 and
|log2FC|>2; hence, genes in the prognostic analysis
may exhibit more obvious statistical and biological significance.
However, SOCS-1 and NPTX2 did not reach the thresholds of
FDR<0.05 and |log2FC|>2.
Of note, several limitations of the present study
should be considered. First, the ethnicities of the population in
the TCGA database, which is from the US, are primarily Latino and
Caucasian, and it is necessary to substantiate the extrapolation of
the results of the present study to other ethnic groups. Second,
the prognostic analyses of these 7 DNA methylation-driven genes
were based on a retrospective study, so the use of these genes as
biomarkers requires prospective multicenter validation. Third,
further studies are required to investigate and validate the
functions and molecular mechanisms of the pathogenesis and
progression of PAAD with regard to these 7 DNA methylation-driven
genes in in vivo and in vitro experiments.
In summary, 7 DNA methylation-driven genes were
identified by a comprehensive analysis of DNA methylation and mRNA
expression data from 188 clinical samples. The 7 DNA
methylation-driven genes will contribute to the understanding of
how pancreatic cancer occurs and develops, as well as provide novel
ways to diagnose and treat pancreatic cancer.
Supplementary Material
Detailed clinical characteristics of
the TCGA pancreatic adenocarcinoma cohort.
MethylMix models for the seven DNA
methylation-driven genes.
Out of seven DNA methylation-driven
genes, six were associated with overall survival in pancreatic
adenocarcinoma.
Out of seven DNA methylation-driven
genes, six were associated with relapse-free survival in pancreatic
adenocarcinoma.
Gene enrichment in the DNA
methylation-driven genes hypermethylation group of patients with
pancreatic adenocarcinoma.
Acknowledgements
The authors would like to thank Dr Shijun Wei from
the Department of Medical Genetics, Shandong University School of
Medicine, Jinan, Shandong, China for providing technical
support.
Funding
The design of the current study and the collection,
analysis and interpretation of data in this work were supported by
the Basic Research Fund of the Ocean University of China (grant no.
201562018).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
XT and WZ were responsible for the conception and
design of this research. YY, PL, TW and XC were responsible for
data collection, collation and analysis. WZ and SS were responsible
for data analysis and interpretation, and manuscript content. XT
was responsible for approving the publication of the final version.
XT agreed to be accountable for all aspects of the work in ensuring
that questions related to the accuracy or integrity of any part of
the work are appropriately investigated and resolved.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rahib L, Smith BD, Aizenberg R, Rosenzweig
AB, Fleshman JM and Matrisian LM: Projecting cancer incidence and
deaths to 2030: The unexpected burden of thyroid, liver, and
pancreas cancers in the United States. Cancer Res. 74:2913–2921.
2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Higuera O, Ghanem I, Nasimi R, Prieto I,
Koren L and Feliu J: Management of pancreatic cancer in the
elderly. World J Gastroenterol. 22:764–775. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Crippa S, Capurso G, Camma C, Fave GD,
Castillo CF and Falconi M: Risk of pancreatic malignancy and
mortality in branch-duct IPMNs undergoing surveillance: A
systematic review and meta-analysis. Dig Liver Dis. 48:473–479.
2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Chiaravalli M, Reni M and O'Reilly EM:
Pancreatic ductal adenocarcinoma: State-of-the-art 2017 and new
therapeutic strategies. Cancer Treat Rev. 60:32–43. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kota J, Hancock J, Kwon J and Korc M:
Pancreatic cancer: Stroma and its current and emerging targeted
therapies. Cancer Lett. 391:38–49. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Giovannetti E, van der Borden CL, Frampton
AE, Ali A, Firuzi O and Peters GJ: Never let it go: Stopping key
mechanisms underlying metastasis to fight pancreatic cancer. Semin
Cancer Biol. 44:43–59. 2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Zhang ML, Lu S, Zhou L and Zheng SS:
Correlation between ECT2 gene expression and methylation change of
ECT2 promoter region in pancreatic cancer. Hepatobiliary Pancreat
Dis Int. 7:533–538. 2008.PubMed/NCBI
|
|
8
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Zhang M, Lv X, Jiang Y, Li G and Qiao Q:
Identification of aberrantly methylated differentially expressed
genes in glioblastoma multiforme and their association with patient
survival. Exp Ther Med. 18:2140–2152. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Pan R, Zhou C, Dai J, Ying X, Yu H, Zhong
J, Zhang Y, Wu B, Mao Y, Wu D, et al: Endothelial PAS domain
protein 1 gene hypomethylation is associated with colorectal cancer
in Han Chinese. Exp Ther Med. 16:4983–4990. 2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kisiel JB, Raimondo M, Taylor WR, Yab TC,
Mahoney DW, Sun Z, Middha S, Baheti S, Zou H, Smyrk TC, et al: New
DNA methylation markers for pancreatic cancer: Discovery, tissue
validation, and pilot testing in pancreatic juice. Clin Cancer Res.
21:4473–4481. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Pan FP, Zhou HK, Bu HQ, Chen ZQ, Zhang H,
Xu LP, Tang J, Yu QJ, Chu YQ, Pan J, et al: Emodin enhances the
demethylation by 5-Aza-CdR of pancreatic cancer cell
tumor-suppressor genes P16, RASSF1A and ppENK. Oncol Rep.
35:1941–1949. 2016.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Komazaki T, Nagai H, Emi M, Terada Y, Yabe
A, Jin E, Kawanami O, Konishi N, Moriyama Y, Naka T and Kishimoto
T: Hypermethylation-associated inactivation of the SOCS-1 gene, a
JAK/STAT inhibitor, in human pancreatic cancers. Jpn J Clin Oncol.
34:191–194. 2004.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhang L, Gao J, Li Z and Gong Y: Neuronal
pentraxin II (NPTX2) is frequently down-regulated by promoter
hypermethylation in pancreatic cancers. Dig Dis Sci. 57:2608–2614.
2012.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Integrated Genomic Characterization of
Pancreatic Ductal Adenocarcinoma. Cancer Cell 32: 185-203.e113,
2017.
|
|
16
|
Cedoz PL, Prunello M, Brennan K and
Gevaert O: MethylMix 2.0: An R package for identifying DNA
methylation genes. Bioinformatics. 34:3044–3046. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Cleveland P, Gill KR, Coe SG, Woodward TA,
Raimondo M, Jamil L, Gross SA, Heckman MG, Crook JE and Wallace MB:
An evaluation of risk factors for inadequate cytology in EUS-guided
FNA of pancreatic tumors and lymph nodes. Gastrointest Endosc.
71:1194–1199. 2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Li L, Li C, Mao H, Du Z, Chan WY, Murray
P, Luo B, Chan AT, Mok TS, Chan FK, et al: Epigenetic inactivation
of the CpG demethylase TET1 as a DNA methylation feedback loop in
human cancers. Sci Rep. 6(26591)2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Reis AH, Vargas FR and Lemos B: Biomarkers
of genome instability and cancer epigenetics. Tumour Biol.
37:13029–13038. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140.
2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Thomas RK, Baker AC, Debiasi RM, Winckler
W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L,
et al: High-throughput oncogene mutation profiling in human cancer.
Nat Genet. 39:347–351. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Schuebel KE, Chen W, Cope L, Glöckner SC,
Suzuki H, Yi JM, Chan TA, Van Neste L, Van Criekinge W, van den
Bosch S, et al: Comparing the DNA hypermethylome with gene
mutations in human colorectal cancer. PLoS Genet. 3:1709–1723.
2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Dammann R, Schagdarsurengin U, Liu L, Otto
N, Gimm O, Dralle H, Boehm BO, Pfeifer GP and Hoang-Vu C: Frequent
RASSF1A promoter hypermethylation and K-ras mutations in pancreatic
carcinoma. Oncogene. 22:3806–3812. 2003.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sato N, Fukushima N, Maitra A,
Matsubayashi H, Yeo CJ, Cameron JL, Hruban RH and Goggins M:
Discovery of novel targets for aberrant methylation in pancreatic
carcinoma using high-throughput microarrays. Cancer Res.
63:3735–3742. 2003.PubMed/NCBI
|
|
25
|
Ueki T, Walter KM, Skinner H, Jaffee E,
Hruban RH and Goggins M: Aberrant CpG island methylation in cancer
cell lines arises in the primary cancers from which they were
derived. Oncogene. 21:2114–2117. 2002.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Fukushima N, Sato N, Ueki T, Rosty C,
Walter KM, Wilentz RE, Yeo CJ, Hruban RH and Goggins M: Aberrant
methylation of preproenkephalin and p16 genes in pancreatic
intraepithelial neoplasia and pancreatic ductal adenocarcinoma. Am
J Pathol. 160:1573–1581. 2002.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Kumari A, Srinivasan R and Wig JD: Effect
of c-MYC and E2F1 gene silencing and of 5-azacytidine treatment on
telomerase activity in pancreatic cancer-derived cell lines.
Pancreatology. 9:360–368. 2009.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Nakamura TM, Morin GB, Chapman KB,
Weinrich SL, Andrews WH, Lingner J, Harley CB and Cech TR:
Telomerase catalytic subunit homologs from fission yeast and human.
Science. 277:955–959. 1997.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Ojha J, Codd V, Nelson CP, Samani NJ,
Smirnov IV, Madsen NR, Hansen HM, de Smith AJ, Bracci PM, Wiencke
JK, et al: Genetic variation associated with longer telomere length
increases risk of chronic lymphocytic leukemia. Cancer Epidemiol
Biomarkers Prev. 25:1043–1049. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Codd V, Nelson CP, Albrecht E, Mangino M,
Deelen J, Buxton JL, Hottenga JJ, Fischer K, Esko T, Surakka I, et
al: Identification of seven loci affecting mean telomere length and
their association with disease. Nat Genet. 45422–427.
(427e1-2)2013.PubMed/NCBI View
Article : Google Scholar
|
|
31
|
Walsh KM, Codd V, Rice T, Nelson CP,
Smirnov IV, McCoy LS, Hansen HM, Elhauge E, Ojha J, Francis SS, et
al: Longer genotypically-estimated leukocyte telomere length is
associated with increased adult glioma risk. Oncotarget.
6:42468–42477. 2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Iles MM, Bishop DT, Taylor JC, Hayward NK,
Brossard M, Cust AE, Dunning AM, Lee JE, Moses EK, Akslen LA, et
al: The effect on melanoma risk of genes previously associated with
telomere length. J Natl Cancer Inst. 106(pii:
dju267)2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhang C, Doherty JA, Burgess S, Hung RJ,
Lindström S, Kraft P, Gong J, Amos CI, Sellers TA, Monteiro AN, et
al: Genetic determinants of telomere length and risk of common
cancers: A Mendelian randomization study. Hum Mol Genet.
24:5356–5366. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhang J, Huang JY, Chen YN, Yuan F, Zhang
H, Yan FH, Wang MJ, Wang G, Su M, Lu G, et al: Whole genome and
transcriptome sequencing of matched primary and peritoneal
metastatic gastric carcinoma. Sci Rep. 5(13750)2015.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Fernandez-Zapico ME, Gonzalez-Paz NC,
Weiss E, Savoy DN, Molina JR, Fonseca R, Smyrk TC, Chari ST,
Urrutia R and Billadeau DD: Ectopic expression of VAV1 reveals an
unexpected role in pancreatic cancer tumorigenesis. Cancer Cell.
7:39–49. 2005.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Karthik D and Ravikumar S:
Characterization of the brain proteome of rats with diabetes
mellitus through two-dimensional electrophoresis and mass
spectrometry. Brain Res. 1371:171–179. 2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Rahn S, Zimmermann V, Viol F, Knaack H,
Stemmer K, Peters L, Lenk L, Ungefroren H, Saur D, Schäfer H and
Helm O: Diabetes as risk factor for pancreatic cancer:
Hyperglycemia promotes epithelial-mesenchymal-transition and stem
cell properties in pancreatic ductal epithelial cells. Cancer Lett.
415:129–150. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Jankowska AM, Millward CL and Caldwell CW:
The potential of DNA modifications as biomarkers and therapeutic
targets in oncology. Expert Rev Mol Diagn. 15:1325–1337.
2015.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Heyn H and Esteller M: DNA methylation
profiling in the clinic: Applications and challenges. Nat Rev
Genet. 13:679–692. 2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Olkhov-Mitsel E and Bapat B: Strategies
for discovery and validation of methylated and hydroxymethylated
DNA biomarkers. Cancer Med. 1:237–260. 2012.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Rivera CM and Ren B: Mapping human
epigenomes. Cell. 155:39–55. 2013.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Bird A: DNA methylation patterns and
epigenetic memory. Genes Dev. 16:6–21. 2002.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Razin A and Riggs AD: DNA methylation and
gene function. Science. 210:604–610. 1980.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Tan K, Kajino K, Momose S, Masaoka A,
Sasahara K, Shiomi K, Izumi H, Abe M, Ohtsuji N, Wang T, et al:
Mesothelin (MSLN) promoter is hypomethylated in malignant
mesothelioma, but its expression is not associated with methylation
status of the promoter. Hum Pathol. 41:1330–1338. 2010.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Daskalos A, Nikolaidis G, Xinarianos G,
Savvari P, Cassidy A, Zakopoulou R, Kotsinas A, Gorgoulis V, Field
JK and Liloglou T: Hypomethylation of retrotransposable elements
correlates with genomic instability in non-small cell lung cancer.
Int J Cancer. 124:81–87. 2009.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Hama R, Watanabe Y, Shinada K, Yamada Y,
Ogata Y, Yoshida Y, Tamura T, Hiraishi T, Oikawa R, Sakurai J, et
al: Characterization of DNA hypermethylation in two cases of
peritoneal mesothelioma. Tumour Biol. 33:2031–2040. 2012.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Matsubayashi H, Canto M, Sato N, Klein A,
Abe T, Yamashita K, Yeo CJ, Kalloo A, Hruban R and Goggins M: DNA
methylation alterations in the pancreatic juice of patients with
suspected pancreatic disease. Cancer Res. 66:1208–1217.
2006.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Missiaglia E, Donadelli M, Palmieri M,
Crnogorac-Jurcevic T, Scarpa A and Lemoine NR: Growth delay of
human pancreatic cancer cells by methylase inhibitor
5-aza-2'-deoxycytidine treatment is associated with activation of
the interferon signalling pathway. Oncogene. 24:199–211.
2005.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Fenaux P, Mufti GJ, Hellstrom-Lindberg E,
Santini V, Finelli C, Giagounidis A, Schoch R, Gattermann N, Sanz
G, List A, et al: Efficacy of azacitidine compared with that of
conventional care regimens in the treatment of higher-risk
myelodysplastic syndromes: A randomised, open-label, phase III
study. Lancet Oncol. 10:223–232. 2009.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Silverman LR, McKenzie DR, Peterson BL,
Holland JF, Backstrom JT, Beach CL and Larson RA: Cancer and
Leukemia Group B: Further analysis of trials with azacitidine in
patients with myelodysplastic syndrome: Studies 8421, 8921, and
9221 by the Cancer and Leukemia Group B. J Clin Oncol.
24:3895–3903. 2006.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Hirbe AC, Dahiya S, Miller CA, Li T,
Fulton RS, Zhang X, McDonald S, DeSchryver K, Duncavage EJ, Walrath
J, et al: Whole exome sequencing reveals the order of genetic
changes during malignant transformation and metastasis in a single
patient with NF1-plexiform neurofibroma. Clin Cancer Res.
21:4201–4211. 2015.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Walsh KM, Whitehead TP, de Smith AJ,
Smirnov IV, Park M, Endicott AA, Francis SS, Codd V, ENGAGE
Consortium Telomere Group , Samani NJ, et al: Common genetic
variants associated with telomere length confer risk for
neuroblastoma and other childhood cancers. Carcinogenesis.
37:576–582. 2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
O'Brien SM, Klampatsa A, Thompson JC,
Martinez MC, Hwang WT, Rao AS, Standalick JE, Kim S, Cantu E,
Litzky LA, et al: Function of human tumor-infiltrating lymphocytes
in early-stage non-small cell lung cancer. Cancer Immunol Res.
7:896–909. 2019.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Chang CC, Wang HC, Liao YP, Chen YC, Weng
YC, Yu MH and Lai HC: The feasibility of detecting endometrial and
ovarian cancer using DNA methylation biomarkers in cervical
scrapings. J Gynecol Oncol. 29(e17)2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Przybylski GK, Dittmann K, Grabarczyk P,
Dölken G, Gesk S, Harder L, Landmann E, Siebert R and Schmidt CA:
Molecular characterization of a novel chromosomal translocation
t(12;14)(q23;q11.2) in T-lymphoblastic lymphoma between the T-cell
receptor delta-deleting elements (TRDREC and TRAJ61) and the
hypothetical gene C12orf42. Eur J Haematol. 85:452–456.
2010.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Duffy DL, Zhu G, Li X, Sanna M, Iles MM,
Jacobs LC, Evans DM, Yazar S, Beesley J, Law MH, et al: Novel
pleiotropic risk loci for melanoma and nevus density implicate
multiple biological pathways. Nat Commun. 9(4774)2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Morandi L, Gissi D, Tarsitano A, Asioli S,
Gabusi A, Marchetti C, Montebugnoli L and Foschini MP: CpG location
and methylation level are crucial factors for the early detection
of oral squamous cell carcinoma in brushing samples using bisulfite
sequencing of a 13-gene panel. Clin Epigenetics.
9(85)2017.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Lokk K, Vooder T, Kolde R, Välk K, Võsa U,
Roosipuu R, Milani L, Fischer K, Koltsina M, Urgard E, et al:
Methylation markers of early-stage non-small cell lung cancer. PLoS
One. 7(e39813)2012.PubMed/NCBI View Article : Google Scholar
|