Introduction
Increasing age promotes the onset and development of
cardiovascular disease (CVD); the increase in the incidence of
age-associated CVDs causes a heavy burden on human health (1). The association between aging and CVDs
is thus receiving increasing attention (1-3).
Vascular endothelial cell (VEC) senescence is an important process
that contributes to the pathogenesis of age-associated CVDs
(4). Senescence impairs vascular
endothelial repair, compromises vascular regeneration, decreases
the bioavailability of nitric oxide and increases the expression of
pro-inflammatory factors and coagulation molecules, contributing to
the pro-inflammatory, pro-thrombotic and pro-atherogenic effects of
senescence on endothelial cells (2,5).
Homocysteine (HCY) is a cytotoxic methionine
metabolite and sulfur-containing amino acid and is a key
independent risk factor for age-associated CVDs (6,7). HCY
has been shown to induce senescence of endothelial cells by
reducing telomerase activity via induction of telomerase reverse
transcriptase DNA hypomethylation by decreasing telomerase
expression via increases in intracellular reactive oxygen species
(ROS) levels (8,9) and by inhibiting plasminogen activator
inhibitor-1 and integrin β-3(10).
It is thus of importance to explore the mechanism of HCY-induced
VEC senescence and therapeutic strategies for its treatment.
Autophagy, as a potential anti-aging treatment, has
been found to be regulated by HCY (11). Autophagy is an evolutionarily
conserved subcellular process that participates in lysosomal
degradation of proteins and damaged organelles (12). Under normal conditions, cells
maintain low levels of autophagy to maintain cellular homeostasis
(1). Some previous studies have
found that autophagy is involved in the regulation of endothelial
cell senescence (11,13,14).
The biomarkers of autophagy, such as beclin-1, p62, and LC-3, are
decreased in endothelial cells of the elderly (61-71 years) and
autophagy induction improves the endothelial-dependent diastolic
function and bioavailability of nitric oxide in elderly (27- to
28-month-old) mice (1). Compared
with a control group, the senescence of aortic endothelial cells in
mice lacking autophagy-related protein 5 or 7 is increased and the
senescence of human umbilical vein endothelial cells (HUVECs) is
induced by the drug-induced inhibition or gene knockout of
autophagy-related gene 5(13).
While induction of autophagy plays a protective role, it has been
suggested that it may contribute to endothelial cell injury
(15). For example, the inhibition
of autophagy with citrus flavones alleviates HUVEC injury induced
by high-glucose- and high-fat-mediated activation of the
PI3K/Akt/mTOR pathway (16).
Therefore, the beneficial effects of autophagy on endothelial cells
are dependent on conditions (17),
and these determinants need to be explored.
HCY induces autophagy in mouse liver cells and rat
brain cells (15,18) but also inhibits autophagy in mouse
brain cells (19). To the best of
our knowledge, the association between HCY and autophagy under
different conditions has not been fully explored. Moreover, the
role of autophagy in HCY-induced endothelial cell senescence has
not been reported to date. The present study used HCY to establish
a senescence model in HUVECs and the association between autophagy
and HCY-induced cell senescence was investigated to provide a novel
approach for the treatment of HCY-induced age-associated CVDs.
Materials and methods
Reagents
HCY and D-galactose (both Sigma-Aldrich; Merck KGaA)
were dissolved in cell culture media [complete medium containing
Dulbecco's modified Eagle's Medium/F-12 (HyClone; Cytiva), 10%
fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 1%
endothelial cell growth supplement (ScienCell Research
Laboratories, Inc.), 100 U/ml penicillin G and 100 µg/ml
streptomycin sulfate (both Hyclone; Cytiva)]. 3-Methyladenine
(3-MA) and rapamycin (both Gene Chem Co., Ltd., China) were
dissolved in dimethyl sulfoxide (DMSO; Beijing Solarbio Science
& Technology Co., Ltd.). All of the reagents were diluted with
cell culture media to experimental concentrations and were newly
prepared for each experiment.
Cell isolation and culture
Primary HUVECs were immediately isolated from fresh
umbilical cords of healthy pregnant individuals, as previously
described (20). The present study
was approved by the Ethics Committee of the First Affiliated
Hospital of Xinjiang Medical University (Urumchi, China; no.
20190225-13). Informed written consent was obtained from all
participants (age, 20-25 years) from whom HUVECs were obtained.
Human umbilical cords were collected into a sterile centrifuge tube
containing PBS (Shanghai Sangon Biotech Co, Ltd.), 100 U/ml
penicillin G and 100 ug/ml streptomycin sulfate and were isolated
immediately. All procedures were performed out on a cleaning bench.
Following washing with PBS, the umbilical vein was digested with
0.25% pancreatic enzyme at 37˚C for 10 min, centrifuged at 1,000 x
g at 28˚C for 5 min, resuspended and cultured in Dulbecco's
modified Eagle's medium/F-12 (Hyclone; Cytiva) containing 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 1%
endothelial cell growth supplement (ScienCell Research
Laboratories, Inc.), 100 U/ml penicillin G and 100 ug/ml
streptomycin sulfate (both Hyclone; Cytiva) in a humidified
atmosphere with 5% CO2 at 37˚C. The medium was changed
and the cells were passaged every 2 days. Cells obtained between
three and six passages were used to ensure cell line stability.
HUVECs were treated with various concentrations (0,
25, 100 and 500 µM) of HCY at 37˚C for 24 h, followed by incubation
at 37˚C for 24 h. Cells treated with 5 g/l D-galactose at 37˚C for
24 h were used as a classic senescence model control. Then, cells
were treated with 100 µM 3-MA or 5.5 nM rapamycin for 1 h at 37˚C
before culture with 500 µM HCY for 24 h at 37˚C to test changes in
autophagy and its role in HCY-induced senescence. The final
concentration of DMSO used for dilution did not exceed 0.2%.
Cell Counting Kit-8 (CCK-8) assay
CCK-8 (Dojindo Laboratories, Inc.) assay was used to
assess cell proliferation. Untreated HUVECs were plated at a
density of 1x103/well in 96-well plates and incubated as
aforementioned. Following culture period, cells were washed with
PBS three times. The cells were then cultured with 100 µl fresh
cell culture media and 10 µl CCK-8 solution. After 3 h incubation
at 37˚C with 5% CO2, the absorbance at 450 nm was
measured using a microplate reader (Thermo Fisher Scientific,
Inc.). The cell proliferation was calculated using the following
formula: Proliferation rate=[optical density (OD)experimental
group-ODblank group]/(ODcontrol
group-ODblank group).
Cell cycle assay
Propidium iodide (PI)/ribonuclease (RNase) staining
buffer (BD Pharmingen; BD Biosciences) was used to measure cell
cycle arrest. Cells in 100-mm petri dishes were cultured to 50-60%
confluence and treated with HCY, D-galactose, 3-MA and rapamycin as
aforementioned. Following treatment, cells were collected in 5 ml
Eppendorf (EP) tubes, fixed at -4˚C for 24 h with precooled 70%
ethanol and incubated at -4˚C overnight. Cells were centrifuged at
1,000 x g for 5 min at 28˚C, the supernatant was discarded and the
cells were resuspended in PBS. The procedure was repeated three
times. The cells were stained with PI/RNase staining buffer for 15
min at 28˚C in the dark. Each sample was analyzed using the BD
LSRII Analyzer (BD Pharmingen; BD Biosciences) and Modfit version 5
(Becton, Dickinson and Company).
Senescence-associated β-galactosidase
(SA-β-Gal) staining
Cells in 6-well plates were cultured to 50-60%
confluence. The cells were washed with PBS once after treatment
with HCY, D-galactose, 3-MA and rapamycin as aforementioned, and
stained with SA-β-Gal staining reagent (Beyotime Institute of
Biotechnology) according to the manufacturer's instruction. The
cells were still mounted. Blue-stained cells were manually counted
in three randomly selected fields of view using light microscopy
(x100 magnification).
ROS test
Cells in 6-well plates were cultured to 50-60%
confluence. The cells were washed with PBS. Cells were cultured for
20 min with DCFH-DA (Beyotime Institute of Biotechnology), which
was diluted 1:1,000 in cell culture media without FBS, at 37˚C and
5% CO2 in the dark. After washing the cells three times
with Dulbecco's modified Eagle's Medium/F-12, images were obtained
using a Leica DMI6000B fluorescence microscope (Leica Microsystems
GmbH; x100 magnification) and the mean fluorescence intensity/cell
was measured using ImageJ 1.53e (National Institutes of
Health).
Cell autophagic flux measurement
Cells in 96-well plates were cultured to 10-20%
confluence. The cells were transfected with 100 µl mixed liquid
containing 10 µl autophagy-related lentivirus, 4 µl HitransG A
(both Shanghai GeneChem Co., Ltd) and 86 µl cell culture media,
followed by incubation for 12-16 h at 37˚C. The
Stub-RFP-Sens-GFP-LC3 autophagy-related double fluorescence
lentivirus is a lentivirus expressing red fluorescent and green
fluorescent protein and autophagy marker protein LC3 that was used
according to the manufacturer's instructions. The Sens-GFP is an
acid-sensitive protein and the Stub-RFP is a stable fluorescent
protein. When the autophagic flux increases, the expression of GFP
and RFP increases simultaneously. Then autophagosomes fuse with
lysosomes, changing the environment to acid, which quenches green
fluorescence. The fluorescence color change from yellow to red
indicates autophagy flux change (21). The transfected cells were cultured
in a 100-mm Petri dish for 36-72 h at 37˚C. Successfully
transfected cells were cultured for 24 h at 37˚C in confocal dishes
and treated with HCY, D-galactose, 3-MA and rapamycin as
aforementioned. Images were captured using a confocal laser
microscope (Leica Microsystems GmbH; x400 magnification). The
number of autophagy-related fluorescent dots/cell was manually
counted in three randomly selected fields of view.
Statistical analysis
All data are presented as the mean ± standard
deviation of three independent experimental repeats. SPSS version
20.0 (IBM Corp.) was used to perform statistical analysis.
Comparisons between two independent samples were performed using an
unpaired t-test. One-way analysis of variance followed by Dunnett's
post hoc test was used for comparisons between >2 groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
HCY induces senescent phenotype of
HUVECs
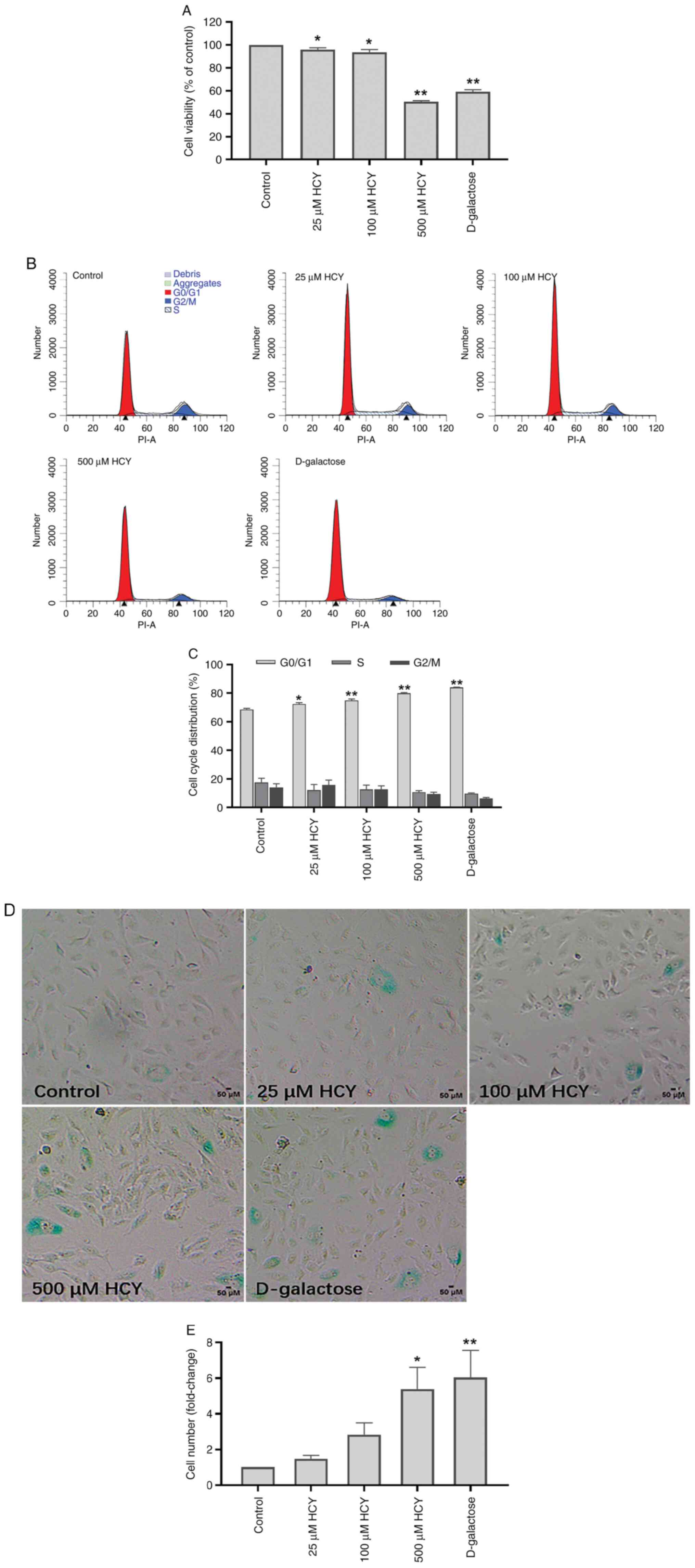
D-galactose is a classical senescence inducer
(4). D-galactose was used to
establish the classical senescence model. The cell proliferation
rate in each group was tested by CCK-8 and examined via assessing
cell viability. Compared with that in the control group, the cell
proliferation rate of D-galactose group was reduced by 40.87%, and
that of 500 µM HCY group was decreased by 49.46%. HCY decreased
cell proliferation activity significantly in a dose-dependent
manner (Fig. 1A). The cell cycle
arrest was tested by flow cytometry. Compared with that in the
control group, the number of G1/G0 stage
cells increased by 15.46% in the D-galactose intervention group and
11.40% in the 500µM HCY group. Cell cycle in the HCY-treated group
was arrested in a dose-dependent manner and HCY significantly
arrested HUVECs in the G1/G0 stage (Fig. 1B and C).
The activity of SA-β-Gal is upregulated during cell
senescence and the test of SA-β-Gal activation is used as a
senescence marker (9).
Blue-stained cells represent senescent cells. The number of
senescent cells increased significantly in HUVECs subjected to HCY
treatment (Fig. 1D and E). Higher HCY concentration resulted in
more senescent cells. Compared with that in the control group, the
number of senescent cells increased 6.04 times in the D-galactose
intervention group and 5.39 times in the 500 µM HCY group.
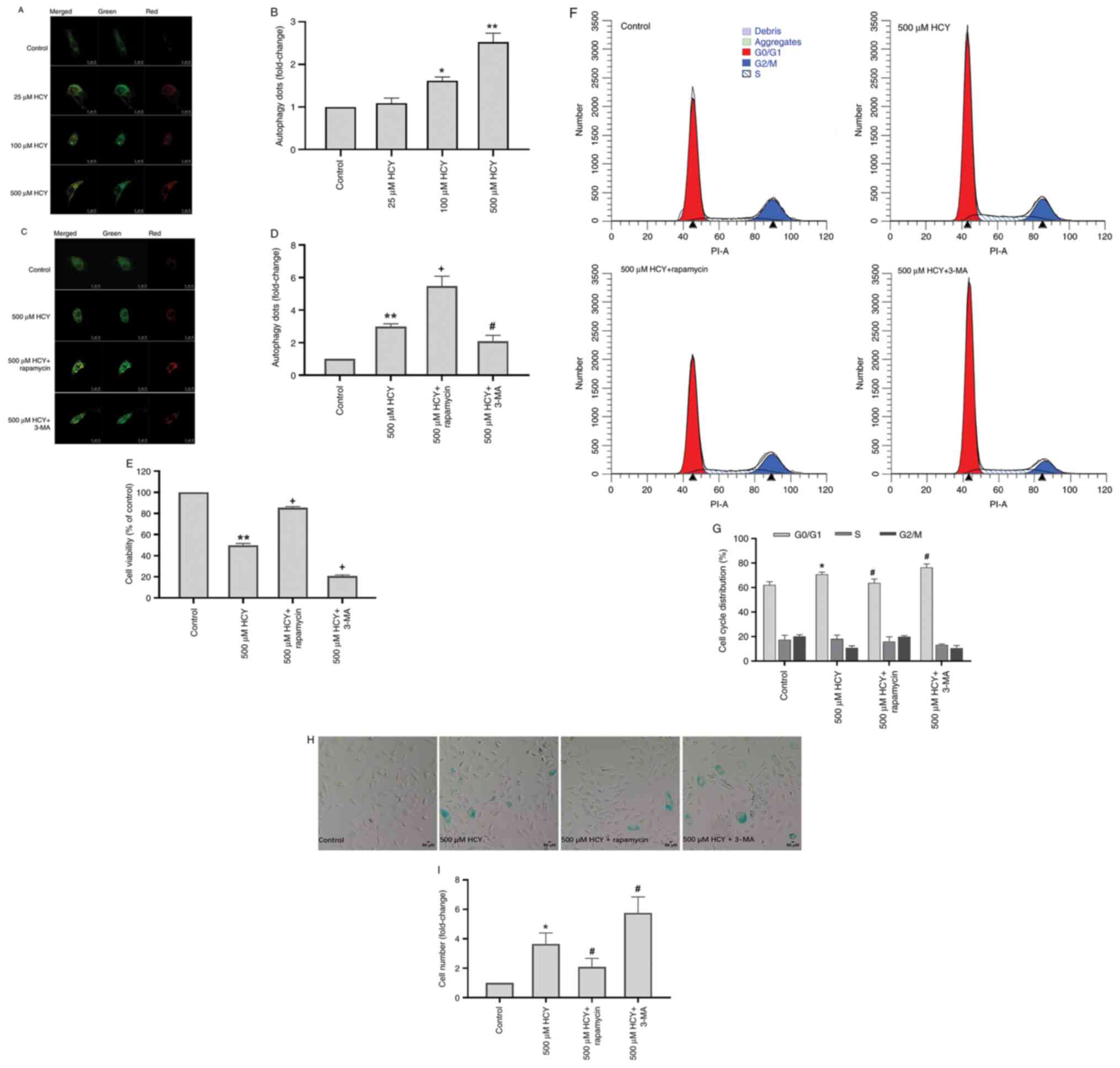
Autophagy alleviates the senescent
phenotype of HCY-induced HUVECs
LC3 is an autophagy marker protein (22). To observe intracellular autophagic
flux, the Stub-RFP-Sens-GFP-LC3 autophagy-related double
fluorescence lentivirus was used. Autophagy-associated yellow dots
increased significantly in HUVECs treated with HCY (Fig. 2A and B). The autophagy dots in the 500 µM HCY
group increased by 2.53 times compared with those of the control
group.
To investigate the effect of autophagy on
HCY-induced HUVEC senescence, 3-MA, a commonly used autophagy
inhibitor, and rapamycin, an autophagy inducer, were used to
inhibit and induce autophagy, respectively (22). Compared with the 500 µM HCY group,
3-MA decreased the number of autophagic dots in HUVECs treated with
HCY by 30.10%, while rapamycin increased these by 82.94% (Fig. 2C and D). The HVUECs treated with 3-MA exhibited
decreased cell viability and cell cycle progression (Fig. 2E-G). Treatment with rapamycin
increased cell viability and alleviated cell cycle arrest. Compared
with the 500 µM HCY group, 3-MA reduced the cell viability of
HUVECs treated with HCY by 29.13%, and increased the number of
G1/G0 stage cells by 5.66%, while rapamycin
increased the cell viability of HUVECs treated with HCY by 35.56%,
and reduced the number of G1/G0 stage cells
by 6.94%. Furthermore, compared with the 500 µM HCY group, 3-MA
treatment increased the number of senescent cells in the
HCY-treated group by 57.81%, while rapamycin decreased the number
of senescent cells by 42.74% (Fig.
2H and I).
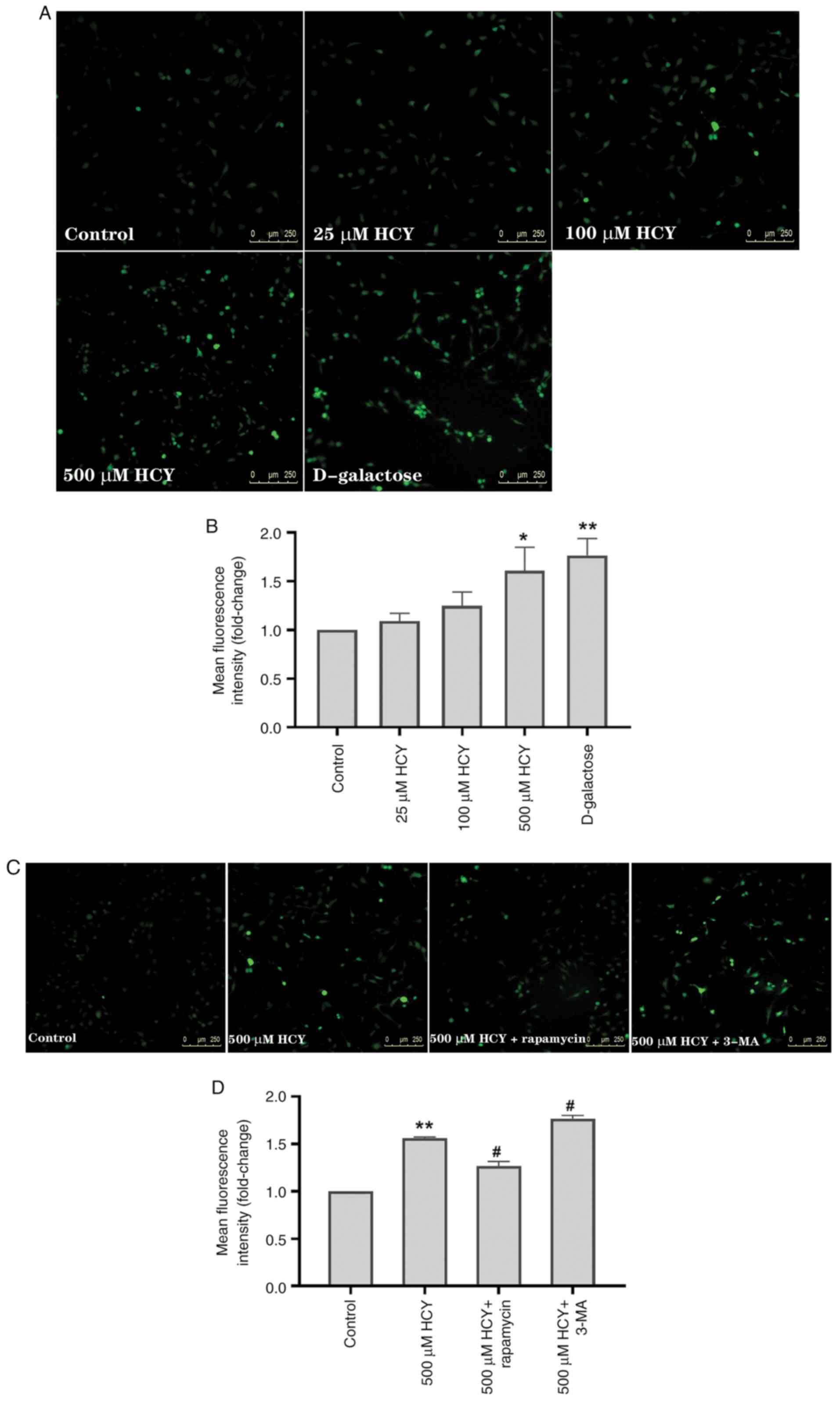
Autophagy alleviates HCY-induced
senescence by reducing intracellular ROS
ROS are increased during cell senescence and
participate in the mechanisms of senescence (1). To test ROS in HCY-treated HUVECs, an
inverted fluorescence microscope was used. The mean fluorescence
intensity, indicative of the ROS concentration, was significantly
higher in HUVECs treated with HCY and D-galactose than in the
control group (Fig. 3A and
B). Compared with that in the
control group, the mean fluorescence intensity in the 500 µM HCY
intervention group increased by 1.61 times, and in the D-galactose
intervention group, by 1.76 times.
Previous studies have shown that ROS induces
autophagy, while autophagy can delay cell senescence by decreasing
intracellular ROS (4,23). To investigate the role of ROS in
autophagy and HCY-induced endothelial cell senescence, 3-MA and
rapamycin were used. Compared with the 500 µM HCY group, exposure
to 3-MA increased ROS levels in HUVECs treated with HCY by 13.46%,
while exposure to rapamycin decreased ROS levels in HUVECs treated
with HCY by 18.59% (Fig. 3C and
D).
Discussion
VECs are the primary cell type in blood vessels.
Aging endothelial cells exhibit the decreased ability to synthesize
NO, increased secretion of pro-inflammatory and pro-thrombotic
factors and impaired vascular regenerative functions, which
contribute to development of hypertension, atherosclerosis and the
other age-associated CVDs (4).
Age-associated vascular changes are promising targets for
preventive therapy and lifestyle interventions, including drug
therapies, must be implemented to prevent the development of
subclinical manifestation of senescence before the pathology
becomes apparent (3).
HCY is a non-classical independent risk factor for
age-related CVDs and risk of chronic heart disease increases by
~20% for each 5 µM increase in the serum HCY concentration
(7,24). The mechanism by which HCY
accelerates vascular disease has not been fully clarified, but it
may be related to induced endothelial dysfunction, increased
platelet adhesion, induced production of low-density lipoprotein
and enhanced coagulation cascade reactions (25). HCY can cause endothelial
dysfunction by inducing endothelial cell senescence. For example,
HCY can induce endothelial cell senescence by inducing DNA
hypomethylation (8), increasing
intracellular ROS levels (9) and
inhibiting plasminogen activator inhibitor-1 and integrin
β-3(10). Folic acid and adenosine
methionine reverse the HCY-induced upregulation of the senescence
markers P21, P16 and P53 in endothelial cells, preventing
HCY-induced endothelial cell senescence (8). In the present study, HCY was used to
establish an endothelial cell senescence model, which demonstrated
decreased cell proliferation, inhibition of the cell cycle at
G1/G0 phase and an increase in the activation
of SA-β-Gal. This was similar to the classical cell senescence
model established by D-galactose.
As a widely recognized anti-aging mechanism,
autophagy has received increasing attention in previous years
(4,11,26).
To study the association between autophagy and HCY-induced
endothelial cell senescence, double-fluorescent-labeled LC3
lentivirus was used to detect autophagic flux. HCY increased
autophagy in endothelial cells. Then, 3-MA and rapamycin were used
to inhibit and induce autophagy, respectively. The inhibition of
autophagy by 3-MA increased HCY-induced endothelial cell
senescence, while the upregulation of autophagy by rapamycin
alleviated HCY-induced endothelial cell senescence. In previous
studies, HCY has been reported to both induce and inhibit autophagy
(15,19,27,28).
In mouse hepatocytes, HCY inhibits expression of cystic fibrosis
transmembrane conductance regulator protein via interaction between
histone H3 lysine methylation and DNA methylation, thus activating
mouse liver autophagy and aggravating liver injury (18). However, HCY activates mTOR in mouse
brain neurons and in cultured forebrain neurons derived from
pluripotent stem cells, inhibiting autophagy and inducing
dysfunction of neuronal clearance, thus contributing to the
pathogenesis of Alzheimer's disease-like spongiform
neurodegeneration via accumulation of phosphorylated τ and amyloid
aggregates (19). However, the
reason for the discrepancy between HCY and autophagy effects is not
known. From a macroscopic perspective, variations in experimental
conditions stemming from cell type, animal model, reagents,
intervention and sample collection may affect initial autophagic
state of the cells, intensity of autophagic responses to stress and
the degree to which the intervention influences autophagy. From the
perspective of microscopic genes, proteins and other molecules,
researches of autophagy in tumors may provide further mechanistic
insight into the differences between autophagy effects. Different
types of intracellular autophagy, substrate and autophagy receptor,
time of autophagy activation and the cell sites of autophagy
activation determine the final autophagic effect (26). In summary, the cytoprotective
effect of autophagy is dependent on the environmental conditions
(17). Considering that excessive
autophagy leads to cell damage and death (29) and the complexity of the autophagy
regulatory network, the specific mechanism and conditions of
autophagic protection require further study. Further, a method to
regulate autophagy at an appropriate level without cell damage is
key to alleviate cell aging.
ROS, including superoxide and hydroxyl radicals and
H2O2, are highly active molecules involved in
signal transduction, gene expression, cell proliferation and other
cellular pathophysiological processes (22). ROS are increased in senescent
cells. Although the increase in ROS is not a landmark event for
cell senescence, its increase is one indicator of this process and
a potential key factor in inducing and maintaining cell senescence
(22). HCY has been reported to
induce cell senescence via induction of intracellular ROS (9). Consistent with results of previous
studies (7,9,25),
the present found that HCY induced HUVEC senescence by inducing the
production of intracellular ROS. It was reported previously that
oxidative stress promotes autophagy and autophagy might decrease
oxidative damage through the engulfment and degradation of
oxidative substances (23).
Therefore, the present study used rapamycin to induce autophagy;
autophagy decreased intracellular ROS levels, while the use of 3-MA
increased intracellular ROS levels. Therefore, it was hypothesized
that the decrease in ROS production is one of the underlying
mechanisms by which autophagy alleviates HCY-induced HUVEC
senescence.
The present study explored the relationship between
autophagy and HCY-induced HUVEC senescence and confirmed that HCY
increased HUVEC senescence and autophagy by inducing production of
intracellular ROS. The induction of autophagy decreased production
of ROS and alleviated HCY-induced HUVEC senescence. However, the
present study only performed phenotypical observations based on
in vitro cell experiments. The lack of mRNA and protein
analysis of senescence and autophagy markers is a limitation of
this study; these experiments should be performed in future.
Manipulating autophagy in vivo is complicated; therefore,
further experiments should be conducted to elucidate the regulatory
mechanism of autophagy in tissue homeostasis. The present findings
may provide insight into the underlying mechanism of HCY-induced
VEC senescence and potential treatments for age-associated
CVDs.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the First Affiliated
Hospital of Xinjiang Medical University (grant no.
2022B03009-2).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YZ, JO, HW and XZ contributed to the design of the
present study. YZ, JO, LZ, YL, SL and YH performed the experiments
and analyzed the data. HW and XZ drafted the manuscript. YZ, JO, HW
and XZ confirm the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of the First Affiliated Hospital of Xinjiang Medical
University (approval no. 20190225-13). Written informed consent was
obtained from all participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Donato AJ, Machin DR and Lesniewski LA:
Mechanisms of dysfunction in the aging vasculature and role in
age-related disease. Circ Res. 123:825–848. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ashapkin VV, Kutueva LI and Vanyushin BF:
Aging as an epigenetic phenomenon. Curr Genomics. 18:385–407.
2017.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lakatta EG and Levy D: Arterial and
cardiac aging: Major shareholders in cardiovascular disease
enterprises: Part I: Aging arteries: A ‘set up’ for vascular
disease. Circulation. 107:139–146. 2003.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sun C, Diao Q, Lu J, Zhang Z, Wu D, Wang
X, Xie J, Zheng G, Shan Q, Fan S, et al: Purple sweet potato color
attenuated NLRP3 inflammasome by inducing autophagy to delay
endothelial senescence. J Cell Physiol. 234:5926–5939.
2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wang Y, Boerma M and Zhou D: Ionizing
radiation-induced endothelial cell senescence and cardiovascular
diseases. Radiat Res. 186:153–161. 2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Moselhy SS and Demerdash SH: Plasma
homocysteine and oxidative stress in cardiovascular disease. Dis
Markers. 19:27–31. 2003.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Scalera F, Martens-Lobenhoffer J, Täger M,
Bukowska A, Lendeckel U and Bode-Böger SM: Effect of L-arginine on
asymmetric dimethylarginine (ADMA) or homocysteine-accelerated
endothelial cell aging. Biochem Biophys Res Commun. 345:1075–1082.
2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhang D, Sun X, Liu J, Xie X, Cui W and
Zhu Y: Homocysteine accelerates senescence of endothelial cells via
DNA hypomethylation of human telomerase reverse transcriptase.
Arterioscler Thromb Vasc Biol. 35:71–78. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wang CJ, Hu CP, Xu KP, Tan GS and Li YJ:
Effects of selaginellin on homocysteine-induced senescence in human
umbilical vein endothelial cells. J Cardiovasc Pharmacol.
55:560–566. 2010.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sun T, Ghosh AK, Eren M, Miyata T and
Vaughan DE: PAI-1 contributes to homocysteine-induced cellular
senescence. Cell Signal. 64(109394)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Abdellatif M, Sedej S, Carmona-Gutierrez
D, Madeo F and Kroemer G: Autophagy in cardiovascular aging. Circ
Res. 123:803–824. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Boya P, Reggiori F and Codogno P: Emerging
regulation and functions of autophagy. Nat Cell Biol. 15:713–720.
2013.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Vion AC, Kheloufi M, Hammoutene A, Poisson
J, Lasselin J, Devue C, Pic I, Dupont N, Busse J, Stark K, et al:
Autophagy is required for endothelial cell alignment and
atheroprotection under physiological blood flow. Proc Natl Acad Sci
USA. 114:E8675–E8684. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Song S, Wu S, Wang Y, Wang Z, Ye C, Song
R, Song D and Ruan Y: 17β-estradiol inhibits human umbilical
vascular endothelial cell senescence by regulating autophagy via
p53. Exp Gerontol. 114:57–66. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhang JW, Yan R, Tang YS, Guo YZ, Chang Y,
Jing L, Wang YL and Zhang JZ: Hyperhomocysteinemia-induced
autophagy and apoptosis with downregulation of hairy enhancer of
split 1/5 in cortical neurons in mice. Int J Immunopathol
Pharmacol. 30:371–382. 2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wang K, Peng S, Xiong S, Niu A, Xia M,
Xiong X, Zeng G and Huang Q: Naringin inhibits autophagy mediated
by PI3K-Akt-mTOR pathway to ameliorate endothelial cell dysfunction
induced by high glucose/high fat stress. Eur J Pharmacol.
874(173003)2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Jiang F: Autophagy in vascular endothelial
cells. Clin Exp Pharmacol Physiol. 43:1021–1028. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Guo ZX, Zhou FZ, Song W, Yu LL, Yan WJ,
Yin LH, Sang H and Zhang HY: Suppression of microRNA-101 attenuates
hypoxia-induced myocardial H9c2 cell injury by targeting
DIMT1-Sp1/survivin pathway. Eur Rev Med Pharmacol Sci.
22:6965–6976. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Khayati K, Antikainen H, Bonder EM, Weber
GF, Kruger WD, Jakubowski H and Dobrowolski R: The amino acid
metabolite homocysteine activates mTORC1 to inhibit autophagy and
form abnormal proteins in human neurons and mice. FASEB J.
31:598–609. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Nakano E, Taiwo FA, Nugent D, Griffiths
HR, Aldred S, Paisi M, Kwok M, Bhatt P, Hill MH, Moat S and Powers
HJ: Downstream effects on human low density lipoprotein of
homocysteine exported from endothelial cells in an in vitro system.
J Lipid Res. 46:484–493. 2005.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhou C, Zhong W, Zhou J, Sheng F, Fang Z,
Wei Y, Chen Y, Deng X, Xia B and Lin J: Monitoring autophagic flux
by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3)
reveals that high-dose rapamycin impairs autophagic flux in cancer
cells. Autophagy. 8:1215–1226. 2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Saxena S, Vekaria H, Sullivan PG and
Seifert AW: Connective tissue fibroblasts from highly regenerative
mammals are refractory to ROS-induced cellular senescence. Nat
Commun. 10(4400)2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Li L, Tan J, Miao Y, Lei P and Zhang Q:
ROS and autophagy: Interactions and molecular regulatory
mechanisms. Cell Mol Neurobiol. 35:615–621. 2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Clarke R, Daly L, Robinson K, Naughten E,
Cahalane S, Fowler B and Graham I: Hyperhomocysteinemia: An
independent risk factor for vascular disease. N Engl J Med.
324:1149–1155. 1991.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Esse R, Barroso M, Tavares de Almeida I
and Castro R: The contribution of homocysteine metabolism
disruption to endothelial dysfunction: State-of-the-art. Int J Mol
Sci. 20(867)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kwon Y, Kim JW, Jeoung JA, Kim MS and Kang
C: Autophagy is pro-senescence when seen in close-up, but
anti-senescence in long-shot. Mol Cells. 40:607–612.
2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Tripathi M, Singh BK, Zhou J, Tikno K,
Widjaja A, Sandireddy R, Arul K, Abdul Ghani SAB, Bee GGB, Wong KA,
et al: Vitamin B(12) and folate decrease inflammation and fibrosis
in NASH by preventing syntaxin 17 homocysteinylation. J Hepatol.
77:1246–1255. 2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wang M, Liang X, Cheng M, Yang L, Liu H,
Wang X, Sai N and Zhang X: Homocysteine enhances neural stem cell
autophagy in in vivo and in vitro model of ischemic stroke. Cell
Death Dis. 10(561)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Zhang Y, Zhang Y, Tang J, Zhao S, Li C,
Huang YP and Yi M: Oxymatrine inhibits homocysteine-mediated
autophagy via MIF/mTOR signaling in human umbilical vein
endothelial cells. Cell Physiol Biochem. 45:1893–1903.
2018.PubMed/NCBI View Article : Google Scholar
|