Introduction
Multiple molecular dysfunctions have been associated
with glioblastoma multiforme (GBM) formation and growth (1–3).
Among these, EGFR plays a vital role in various aspects of
malignancy, particularly in tumor cell proliferation, survival and
metastasis (4,5). Glioma-associated EGFR mutant forms
show constitutive kinase activity that chronically stimulates Ras
signaling to drive cell cycle progression and also activates the
PI3K/AKT pathway to promote cellular proliferation and migration
(6–8).
MicroRNAs modulate protein expression by binding to
the 3′UTR of mRNA and promoting RNA degradation, inhibiting mRNA
translation, and affecting transcription (9–12).
The miR-7 gene is found in most sequenced urbilateria species, and
the sequence of its mature miRNA product is perfectly conserved
from annelids to humans, indicating strong functional conservation
(13–15). Increasing evidence has indicated
that miR-7 is a potential tumor suppressor in several human breast
and non-small cell lung cancers (16,17).
However, thus far, a limited number of target genes have been
identified, and the biological function of miR-7 in GBM remains to
be further elucidated.
miR-7, which is expressed only in normal brain and
pancreatic tissue, demonstrates a high degree of tissue specificity
(18) and may be an ideal target
for cancer therapy. To further characterize the potential of miR-7
as a target for treating GBM and to clarify its role in the
response, we used both in vitro and in vivo systems
to investigate the effect of miR-7 suppression in GBM cell lines.
We found that overexpression of miR-7 not only suppressed GBM cell
proliferation, induced cell apoptosis, and inhibited cell migration
in vitro but also reduced tumorigenicity in vivo.
Bioinformatics predictions revealed at least four potential binding
sites of miR-7 in the 3′-UTR of EGFR, PI3K, and Raf-1. Further
research, however, by luciferase assay, confirmed that the PI3K and
Raf-1 mRNAs are both direct targets of miR-7, but no clear
targeting relationship between EGFR and miR-7 was identified in
subsequent experiments. Therefore, we postulate that miR-7 inhibits
simultaneously the PI3K/ATK and Raf/MEK/ ERK pathways through its
two direct targets, the transcription factors PI3K and Raf-1, which
are both located downstream of EGFR. These findings provide a basic
rationale for the use of miR-7 in the treatment of malignant brain
tumors, such as GBM.
Materials and methods
Reagents, animals, and patient
tissues
The 2′-O-methyl (2′-OMe) oligonucleotides were
chemically synthesized by SBS Genetech (Beijing, China). The
2′-O-Me oligos were composed entirely of 2′-O-Me bases with the
following sequences: scrambled sequence:
5′-AAGGCAAGCUGACCCUGAAGU-3′; 2′-O-Me-miR-7:
5′-UGGAAGACUAGUGAUUUUGUUGU-3′. The pGL3-WT-EGFR (or PI3K or
Raf-1)-3′-UTR-Luc reporter was created by ligation of the
polymerase chain reaction (PCR) products of the 3′-UTR of EGFR (or
PI3K or Raf-1) into the XbaI site of the pGL3 control vector
(Promega, Madison, WI, USA). The pGL3-MUT-EGFR (or PI3K or
Raf-1)-3′-UTR-Luc reporter was generated from pGL3-WT-EGFR (or PI3K
or Raf-1)-3′-UTR-Luc reporter by deleting the binding site for
miR-7. BALB/c-A nude mice at 6 weeks of age were purchased from the
animal center of the Cancer Institute of the Chinese Academy of
Medical Sciences. All experimental procedures were carried out
according to the regulations and internal biosafety and bioethics
guidelines of Tianjin Medical University and the Tianjin Municipal
Science and Technology Commission. Tissue specimens of GBM patients
were collected after informed consent was received and immediately
frozen in liquid nitrogen. The patients or their family member
agreed and signed a consent to enroll in the study.
Cell culture and transfection
The human CHG5 glioblastoma cell lines were
purchased from Chinese Academy of Sciences. Human glioblastoma cell
lines TJ899 and TJ905 were established and characterized in Tianjin
Neurological Institute. All cell lines were maintained in
Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen, Carlsbad,
CA, USA) supplemented with 10% fetal bovine serum (Invitrogen), 100
U/ml penicillin (Sigma, St. Louis, MO, USA), and 100 μg/ml
streptomycin (Sigma), at 37°C with 5% CO2.
Oligonucleotides (50 nm/l) were transfected into CHG5, TJ899 and
TJ905 cells at 70% confluence using Lipofectamine 2000 (Invitrogen)
following the manufacturer’s instructions.
MicroRNA array analysis
Total RNA was extracted from GBM cells (CHG5, TJ866
and TJ905) and human normal glial cells using TRIzol reagent
(Invitrogen) for miRNA profile examination. The miRNA microarray
was obtained from CapitalBio Corp. (Beijing, China). Raw data were
normalized to the normal brain cell RNA level and analyzed using
the significance analysis of microarrays (SAM) software (version
2.1; http://www-stat.stanford.edu/Btibs/SAM, Stanford
University, Stanford, CA, USA).
Real-time reverse transcription
(RT)-PCR
Real-time RT-PCR was carried out using the miRNA
detection kit (Ambion). Amplification was performed for 40 cycles,
each consisting of 95°C for 3 min, 95°C for 15 sec, and 60°C for 30
sec. Both the RT and PCR primers were purchased from Ambion, and 5S
RNA was used for normalization.
Cell viability assay
Cell viability was determined using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT,
Sigma) assay according to the manufacturer’s instructions.
Apoptosis assays
Cells were plated in 12-well plates and transfected
with oligonucleotides. The apoptosis ratio was analyzed 24 h after
transfection using an Annexin V FITC apoptosis detection kit (BD
Biosciences, Franklin Lakes, NJ, USA) according to the
manufacturer’s instructions.
Cell cycle analysis
Cells were collected by trypsinization, washed in
PBS, and fixed in 70% ethanol for 30 min at 4°C. Next, cells were
incubated with the DNA-binding dye PI (50 μg/ml) and RNase
(1.0 mg/ml) for 30 min at 37°C in the dark. Finally, cells were
washed, and red fluorescence was analyzed by a FACSCalibur flow
cytometer (BD Biosciences, San Jose, CA, USA) using a peak
fluorescence gate to discriminate aggregates.
Cell invasion assessment
Cell invasion abilities were examined using 6-well
Transwell chambers and a reconstituted extracellular matrix
membrane (BD Biosciences, San Jose, CA, USA). After allowing
Matrigel polymerization, the cells treated with miR-7 mimic or
scrambled sequences were added to the upper chambers. The migrated
cells were counted microscopically (x400) in five fields per
filter.
Colony formation assay
Colony formation was evaluated as described
previously (19). Briefly, 250–300
cells were placed in a 6-well plate and maintained in DMEM
containing 10% FBS for 7–14 days. Colonies were fixed with methanol
and stained with 0.1% crystal violet in 20% methanol for 15 min,
and representative colonies were photographed.
Luciferase assays
For reporter assays, TJ899 cells were transfected
with miR-7 mimic or scrambled sequences for 48 h and subsequently
cultured in 96-well plates and transfected with pGL3-WT-EGFR (or
PI3K or Raf-1)-3′-UTR-Luc, or pGL3-MUT-EGFR (or PI3K or
Raf-1)-3′-UTR-Luc. After 48 h, luciferase assays were performed
using the Luciferase System kit, and luminescence was measured on a
Promega GloMax 20/20 luminometer and normalized as previously
described (20).
Western blotting
Western blotting was performed as previously
described (21). Proteins were
incubated with primary antibodies against PI3K, Raf-1, and cyclin
D1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), as well as
EGFR, p-AKT, and p-MEK1/2 (Zhongshan Bio Corp., Beijing, China)
followed by incubation with an HRP-conjugated secondary antibody
(Zymed, San Francisco, CA, USA). Proteins were detected using a
SuperSignal protein detection kit (Pierce, Rockford, IL, USA).
Xenograft tumor assay
Thirty-two mice were randomly divided into 4 groups.
Glioma subcutaneous model was established as previously described
(22). A mixture of 10 μl
of Lipofectamine 2000 and 10 μg of oligonucleotides was
injected into the xenograft tumor model in a multi-site injection
manner. Mice in the control group received only 10 μl of
Lipofectamine 2000. In the removal group, the agents were removed
after 10 days of the treatment. Tumor growth was monitored by
caliper measurement every 3 days for 25 days. Tumor volume (V) was
calculated as follows: V = L × W2 × 0.5; L, length; W,
width. Tumor weight was detected at the end of the study.
In situ hybridization assay
Using a locked nucleic acid (LNA)-modified antisense
oligonucleotide probe, in situ hybridization was performed
using the In Situ hybridization kit (Wuhan Boster Biological
Technology, Ltd., Wuhan, China). After sections were prehybridized
for 2 h in hybridization liquid, they were incubated with 20
μl of LNA-miR-7 hybridization solution at 42°C for 16 h, and
Cy3-avidin was used to label miR-7 at a concentration of 0.5
mg/ml.
In situ terminal deoxynucleotidyl
transferase dUTP nick-end labeling (TUNEL) analysis
Apoptotic cell death was examined by TUNEL analysis
using an In Situ Cell Death kit (Roche Carolina Inc.,
Florence, SC, USA) according to the manufacturer’s
instructions.
Immunohistochemistry
The Ki67, EGFR, PI3K, p-AKT-2, Raf-1, p-MEK1/2 and
cyclin D1 protein levels were determined by immunohistochemistry as
previously described (22).
Statistical analysis
Results were analyzed using the SPSS software
version 11.0. Data are presented as the mean ± standard deviation
(SD). A P-value of <0.05 was taken to indicate statistical
significance.
Results
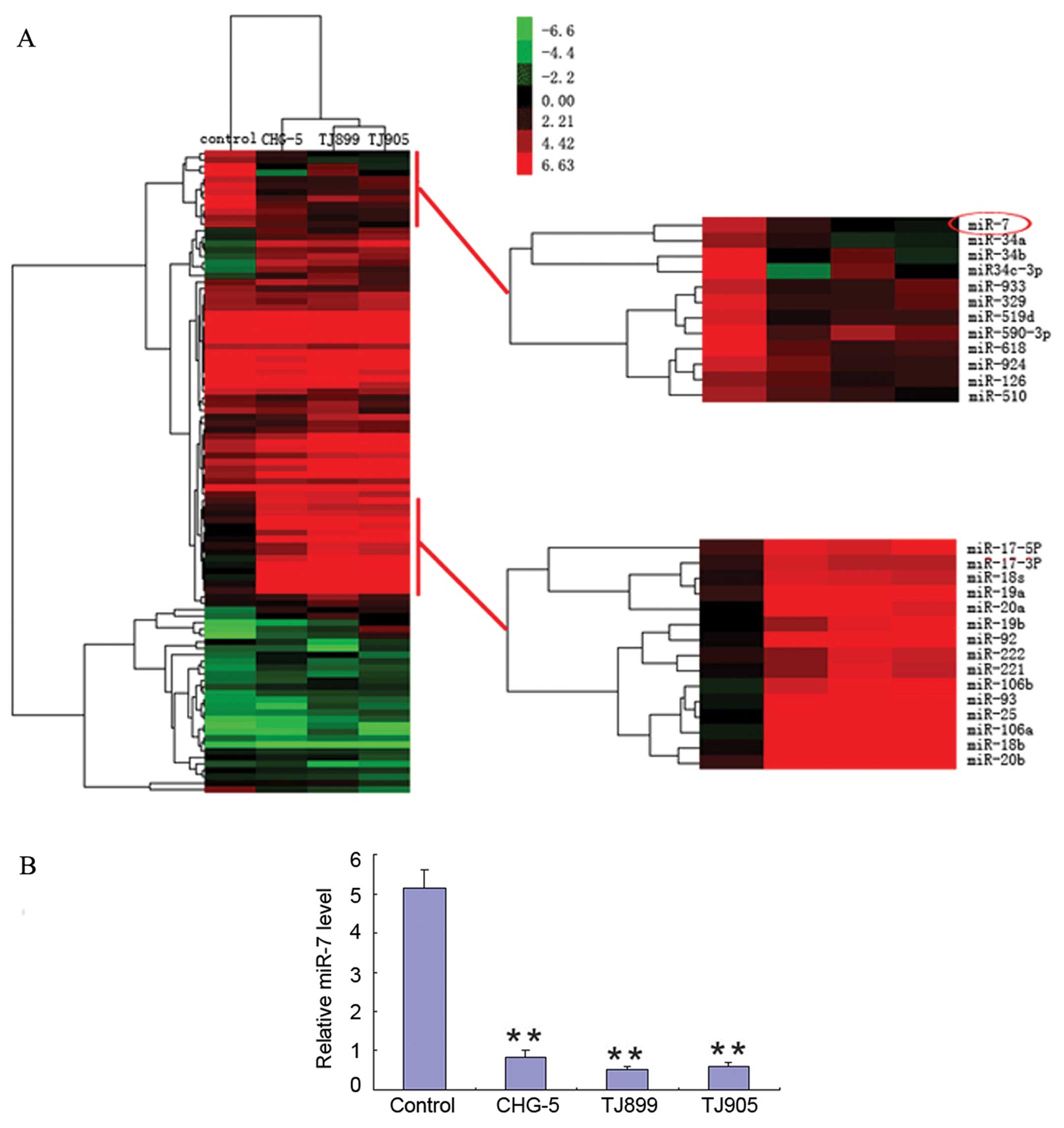
miR-7 expression was detected in normal
brain tissue and at low levels in glioblastoma
Previous studies have reported that miR-7 expression
showed high tissue specificity and was expressed in only normal
brain tissue, normal pancreatic tissue, and pancreatic tumor
tissue. In glioma cell lines, our analysis showed that expression
of 7 of the 118 human miRNAs (5.93%) was more than 2-fold lower,
and that of 11 of 118 (9.32%) 2-fold higher relative to levels in
normal tissue (Fig. 1A). miR-7
showed the most significant decrease relative to normal tissue
(6.4-fold), particularly in the TJ899 GBM cell line (7.9-fold). The
miR-7 expression data from the microarray analysis were confirmed
using quantitative RT-PCR (Fig.
1B).
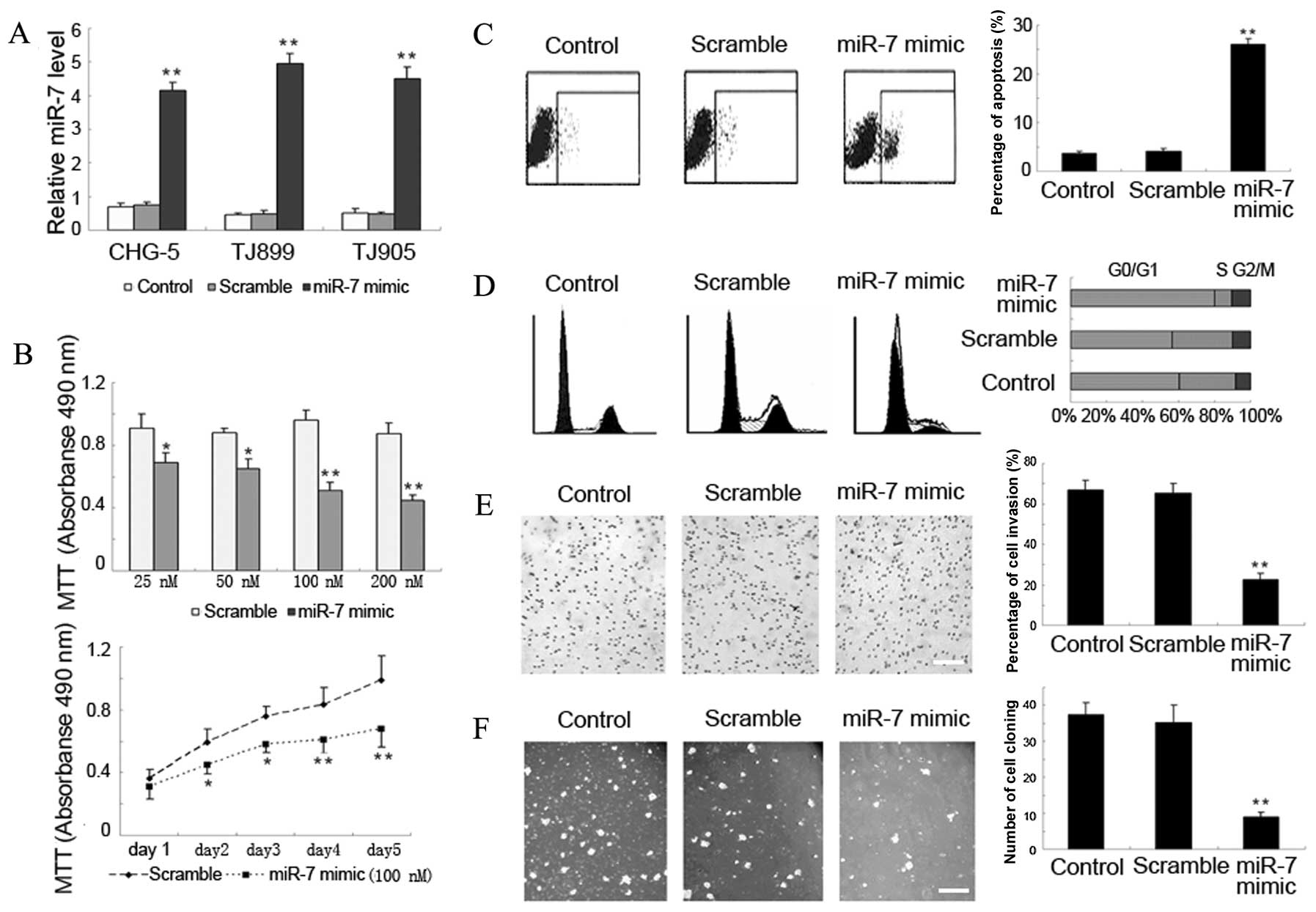
miR-7 inhibits GBM malignancy
We assessed the effects of miR-7 on GBM malignancy
variables, including cell proliferation, cell cycle, cell death,
and cell invasion in three GBM cell lines from Chinese patients. We
first performed in vitro gain-of-function analyses by
introducing miR-7 mimics into CHG-5, TJ899 and TJ905 cells. The
high expression levels of miR-7 in all the three cell lines were
found to be at least 5 times that of the control or scramble group
72 h after the miR-7 mimics were transiently transfected into the
cells (Fig. 2A). Ectogenic miR-7
may significantly inhibit cell proliferation in a dose- and
time-dependent manner (Fig. 2B),
induce apoptosis (Fig. 2C) and
cell cycle arrest (Fig. 2D), and
suppress cell invasiveness (Fig.
2E) as well as colony formation (Fig. 2F). On the third day of the miR-7
mimic transfection at doses of 25, 50, 100 or 200 nM, the cell
proliferation decreased gradually with the increase in dose. After
2 days of miR-7 mimic transfection at 100 nM dose, miR-7
restoration inhibited cell proliferation significantly. The miR-7
expression induced cell death from 3.6±0.6% to 21.4±2.1% in CHG-5
cells (n=3; P<0.05), from 3.7±0.4% to 26.1±1.1% in TJ899 cells
(n=3; P<0.05), and from 1.8±0.2% to 16.4±0.5% in TJ905 cells
(n=3; P<0.05). MiR-7 also significantly induced cell cycle
arrest and increased the G0–G1 fraction in the three Chinese cell
lines. Moreover, miR-7 restoration also resulted in a remarkable
increase of cell invasiveness from 73.9±4.0% to 91.7±2.1% in CHG-5
cells (n=3; P<0.05), from 67.1±4.4% to 22.5±3.4% in TJ899 cells
(n=3; P<0.05), and from 68.8±1.1% to 86.4±1.4% in TJ905 cells
(n=3; P<0.05). Ectogenic miR-7 significantly promoted colony
formation from 36.9±2.8% to 11.7±2.2% in CHG-5 cells (n=3;
P<0.05), from 37.4±3.4% to 8.9±1.4% in TJ899 cells (n=3;
P<0.05), and from 28.8±1.7% to 11.4±1.2% in TJ905 cells (n=3;
P<0.05).
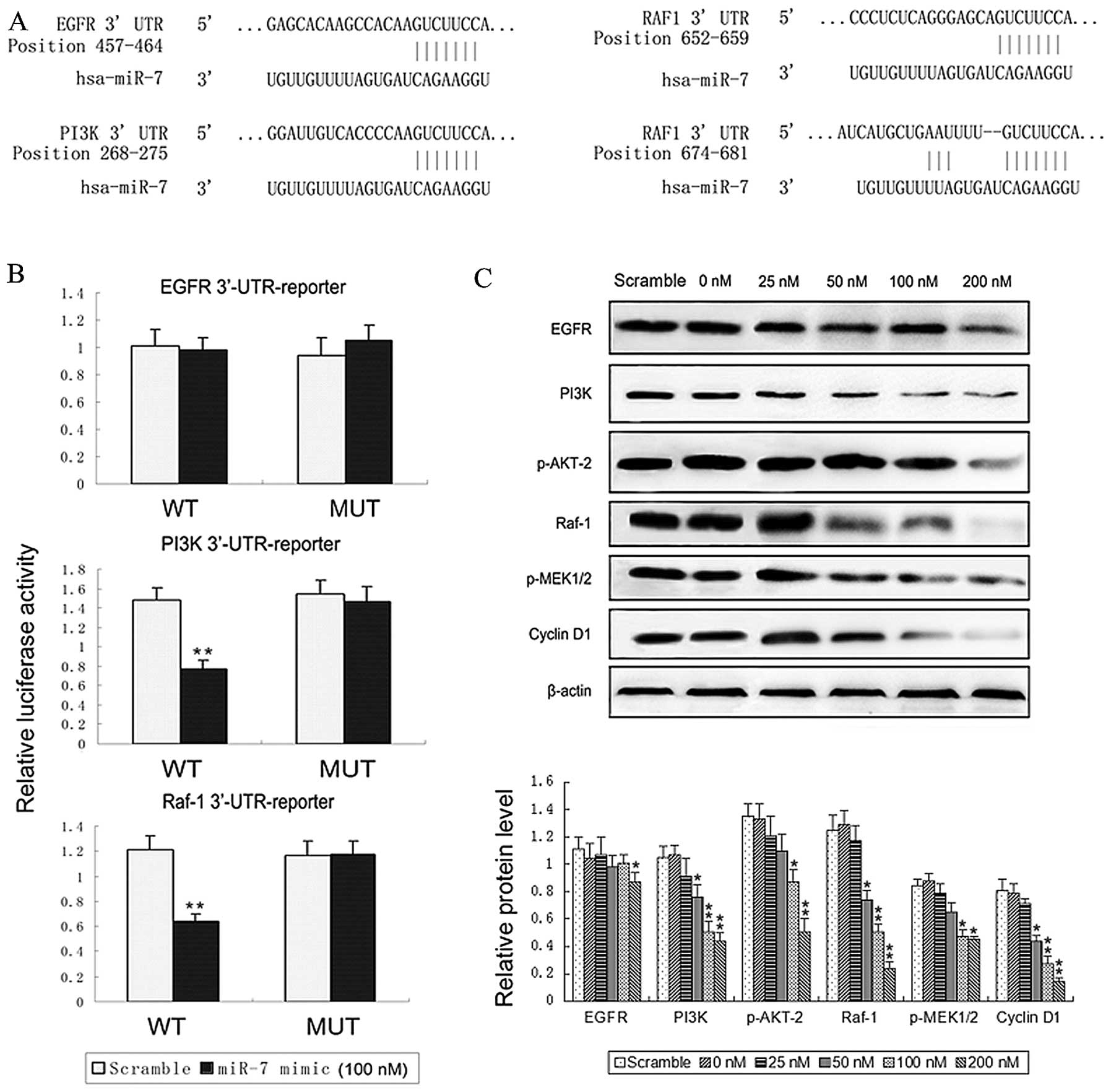
miR-7 inhibits the expression of PI3K and
Raf-1 and binds to their 3′-UTR
The TargetScan and Pictar target prediction
databases found the 3′-UTR of EGFR (PCT=0.61), PI3K
(PCT=0.74), and Raf-1 (PCT=0.97) to contain
the highly conserved putative miR-7 binding sites (Fig. 3A). To determine whether miR-7 could
directly inhibit EGFR, PI3K, and Raf-1 protein expression by
binding to their 3′-UTR, we created pGL3-WT-EGFR (or PI3K or
Raf-1)-3′-UTR and pGL3-MUT- EGFR (or PI3K or Raf-1)-3′-UTR
plasmids. Reporter assays revealed that miR-7 restoration led to a
marked decrease in the luciferase activity of the pGL3-WT-PI3K (or
Raf-1)-3′-UTR plasmid, but did not affect the luciferase activity
of the pGL3-MUT-PI3K (or Raf-1)-3′-UTR plasmid. By contrast, there
was no statistically significant difference between the
pGL3-WT-EGFR-3′-UTR and pGL3-MUT-EGFR-3′-UTR plasmids in terms of
luciferase activity (Fig. 3B).
Furthermore, we assessed the effects of miR-7 mimic
transfection on the protein levels of all the above oncogenes and
several accompanying downstream genes (23–30).
Western blotting showed that expression of PI3K and Raf-1 was
down-regulated in TJ899 cells in a dose-dependent manner after 3
days of miR-7 mimic transfection. EGFR, however, showed only
slightly and non-dose-dependently reduced expression. Moreover,
both phosphorylated AKT2 (p-AKT2) and p-MEK1/2 levels were
downregulated, and the expression of cyclin D1 was decreased
(Fig. 3C and D), compared with
that in cells treated with scrambled oligonucleotide or vector
alone (control group) (data not shown).
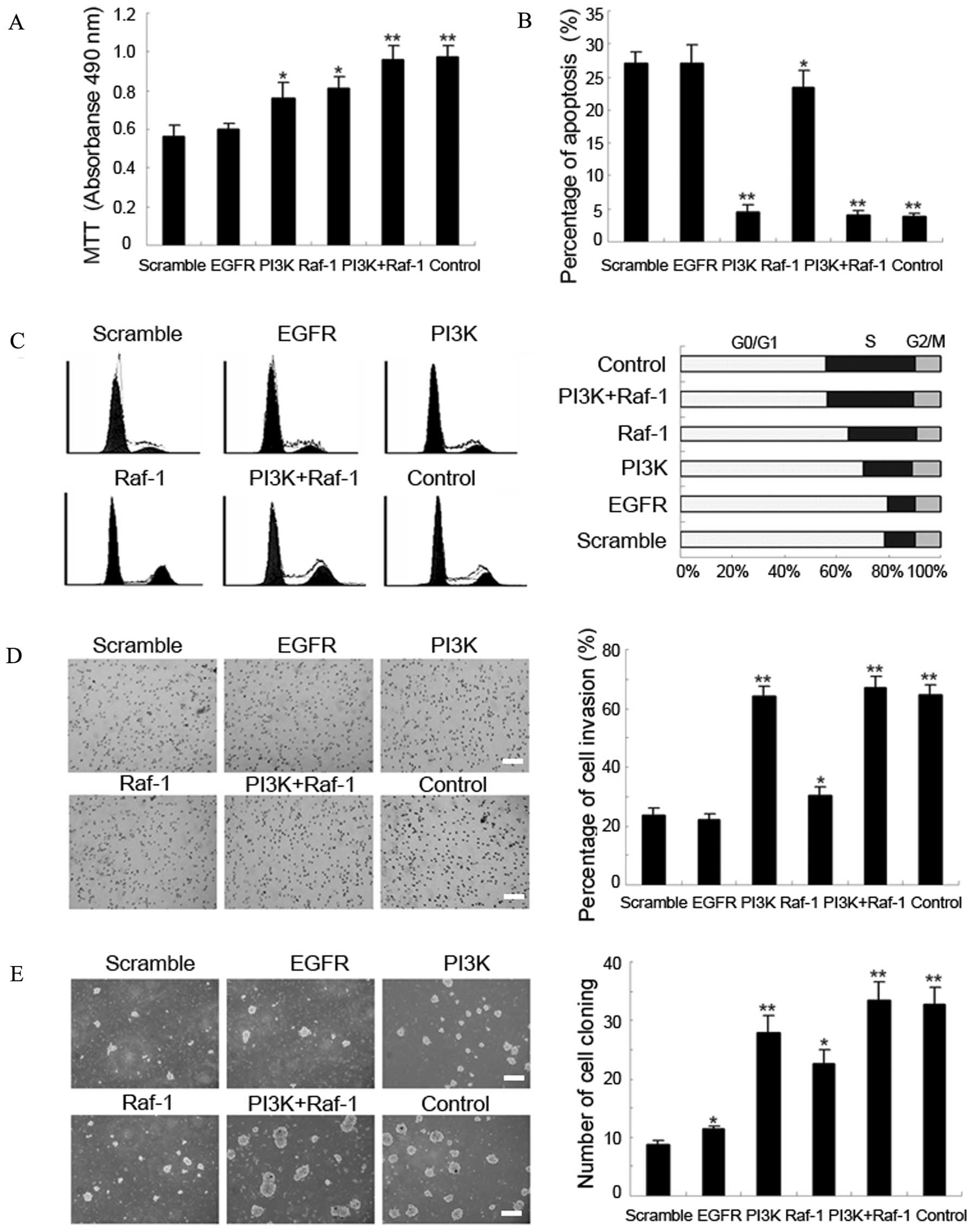
Recovery of PI3K and Raf-1 expression
overrides the effect of miR-7 in GBM
To further assess the regulatory role of miR-7 in
the PI3K/ATK and Raf/MEK/ERK pathways, we transfected PI3K lacking
the 3′-UTR and Raf-1 lacking the 3′-UTR into TJ899 cells. Two days
later, the expression of PI3K recovered most of the functions
abolished by miR-7 mimic transfection in TJ899 cells, including
tumor cell proliferation, inhibition of apoptosis, and cell
invasion (Fig. 4A–E). However,
only slight recovery of the cell cycle was observed, as indicated
by a small shortening of the G0/G1 phase and extension of the S
phase, as well as slightly increased cyclin D1 expression (Figs. 4C, 5A
and B). The expression of Raf-1 largely abrogated the effects
of miR-7 on the cell cycle of TJ899 cells, with an obvious
shortening of the G0/G1 phase and prominent extension of the S
phase, as well as dramatically increased cyclin D1 expression
(Figs. 4C, 5A and B). Surprisingly, when PI3K lacking
the 3′-UTR and Raf-1 lacking the 3′-UTR were transfected
simultaneously into TJ899 cells, the tumor cells were almost
completely restored to their original state, with high
proliferative activity, obvious apoptosis inhibition, high
invasiveness and rapid cell cycle progression (Fig. 4A–E). Equally surprisingly, trans
fection of EGFR lacking the 3′-UTR into TJ899 cells had little
effect (Fig. 4A–E). These findings
further indicated that miR-7 directly modulates PI3K and Raf-1, but
not EGFR, expression by binding to the mRNA 3′-UTR.
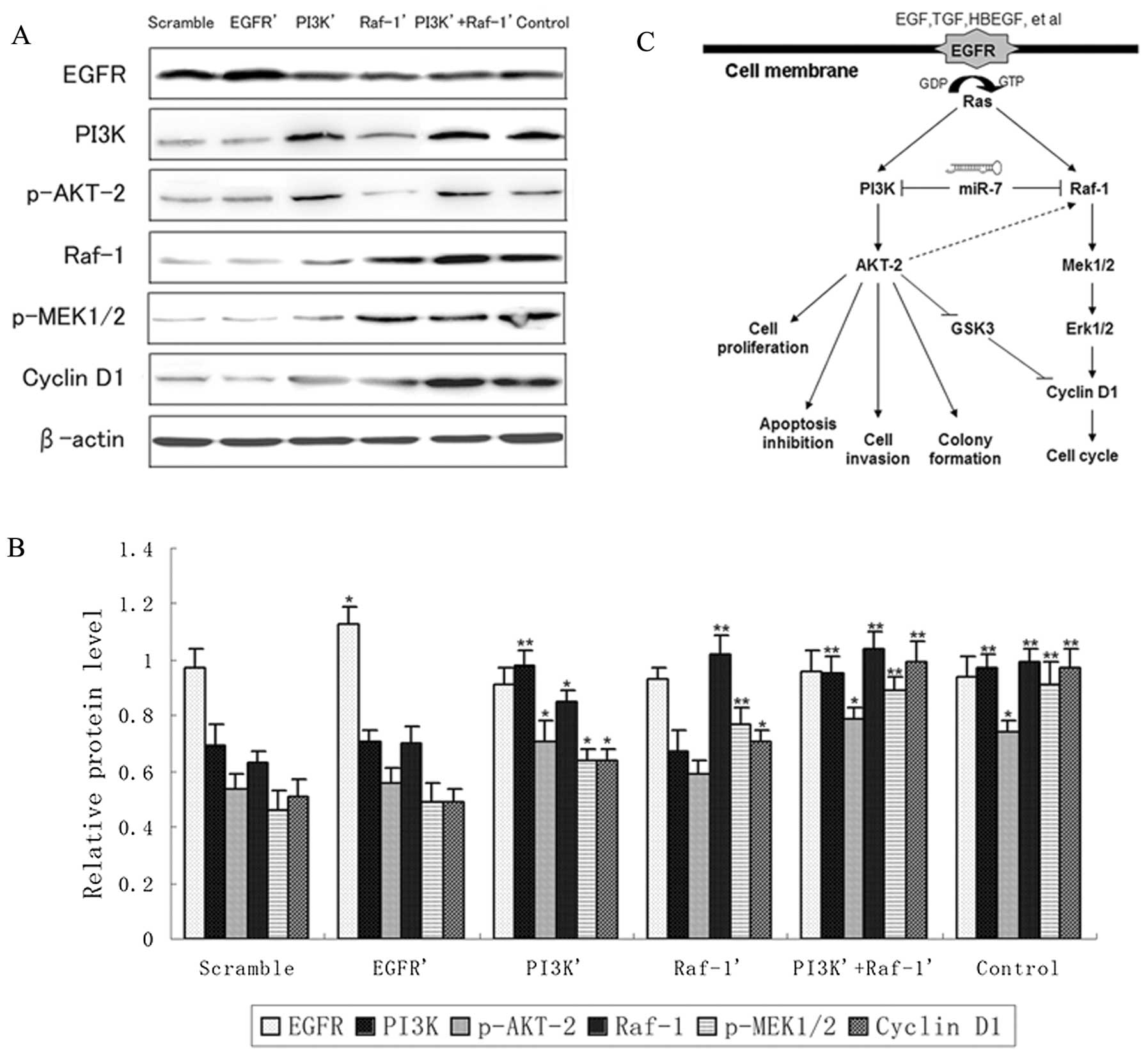
To clarify the regulatory mechanism, we determined
the effect of EGFR, PI3K, and Raf-1 (not including the 3′-UTR)
expression on several proteins involved in the PI3K/ATK and
Raf/MEK/ERK pathways in human GBM TJ899 cells. Forced PI3K
expression significantly increased the level of p-AKT-2 and
slightly increased the level of cyclin D1; additionally, Raf-1
expression significantly increased the level of p-MEK1/2 and
slightly increased the level of cyclin D1. Of note, forced PI3K
expression also slightly increased Raf-1 and p-MEK1/2 protein
expression, but forced Raf-1 expression had no effect on PI3K and
p-AKT-2 expression levels. We also found that EGFR expression had
little impact on the expression levels of downstream proteins,
including PI3K, p-AKT-2, Raf-1, p-MEK1/2 and cyclin D1, indicating
that miR-7 plays a key role in silencing the downstream pathways of
EGFR (Fig. 5A–C).
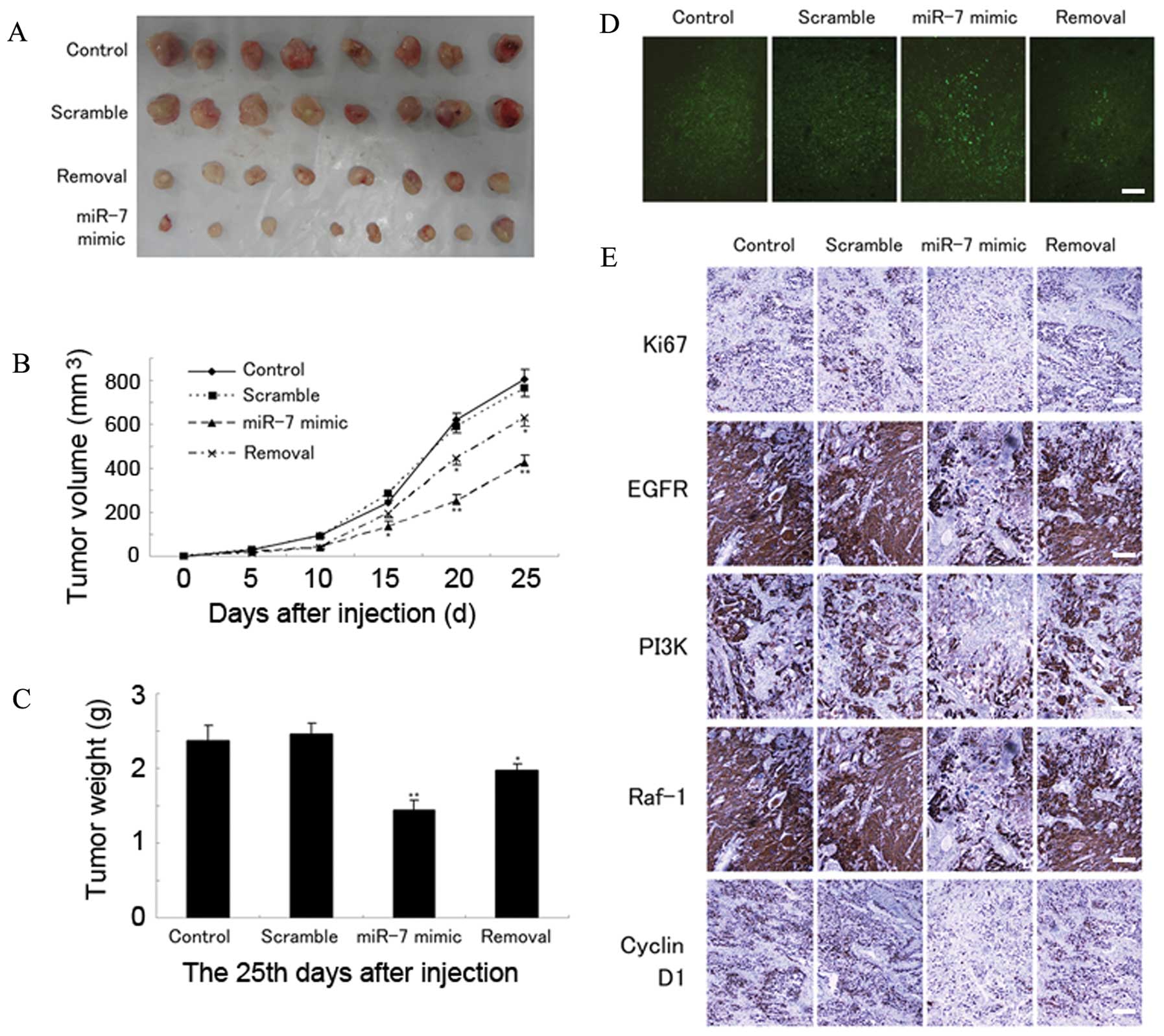
miR-7 inhibits glioblastoma xenograft
growth
Since miR-7 plays an important role in GBM tumor
progression, we further examined the effects of miR-7 on tumor
growth in vivo. Compared with tumors that continued growing
in both the scramble and control groups, the miR-7 group
demonstrated a significant reduction in both tumor volume and
weight (Fig. 6A–C) (P<0.05). In
another group (the removal group), the tumors started to regrow 10
days after cessation of treatment (Fig. 6A–C). TUNEL assay of xenograft
tumors performed 25 days after treatment revealed markedly greater
apoptosis in the miR-7 group compared with the scramble and control
groups (χ2=21.74; P<0.01; Fig. 6D). Ki-67 staining showed that
miR-7-treated tumors had a lower proliferation index than control
tumors (χ2=17.35; P<0.05; Fig. 6E). Additionally, immuno
histochemical analysis revealed a marked decrease in the PI3K,
Raf-1, and cyclin D1 protein levels in the miR-7-treated group
(P<0.05; Fig. 6E). These
findings confirm that miR-7 targets PI3K and Raf-1 and so could be
used for treatment of GBM.
Discussion
GBM is a leading cause of cancer death involving the
central nervous system (31).
Unfortunately, the underlying molecular mechanisms of unlimited
proliferation and progressive local invasion remain obscure.
Previous work has demonstrated that GBM frequently harbors
mutations that activate EGFR and launch downstream signaling
pathways, including the PI3K/ATK and Ras/Raf/MEK/ERK pathways
(6,8,32).
However, EGFR-Ras or PI3K mutations alone are not sufficient to
transform glial cells; instead, multiple mutations that coactivate
the EGFR-Ras and PI3K/Akt pathways are necessary to induce glioma
(33). Recently, much attention
has focused on miRNAs, a newly discovered family of genes encoding
small RNA molecules that bind through partial sequence homology to
the 3′-UTRs of target genes, which play key roles in the regulation
of gene expression and the protein network (34–38).
In the present study, we reviewed the relevant
literature and found that miR-7 demonstrated high tissue
specificity, being expressed in only normal brain tissue and
pancreatic tissue (18). We
hypothesized that miR-7 may have important research value in brain
diseases. To identify the mechanism by which miR-7 regulates GBM
malignancy, we identified the 3′-UTR of EGFR, PI3K, and Raf-1 to
contain highly conserved putative miR-7 binding sites by
bioinformatics analysis. However, the expression of PI3K and Raf-1,
but not EGFR, were regulated by miR-7 as determined by luciferase
activity reporter and western blot assays. We also investigated
p-Akt-2, p-MEK1/2 and cyclin D1 levels. Glioma-associated EGFR
mutant forms showed constitutive kinase activity that chronically
stimulates Ras/Raf/MEK/ERK signaling to drive cell proliferation
and cell cycle progression; this is mediated mainly by cyclin
D.
Other common genetic lesions include activating
mutations in PIK3CA, which encodes the p110a catalytic subunit of
PI3K. AKT, a major PI3K effector, regulates the cell cycle and cell
proliferation through its direct action on the cyclin-dependent
kinase inhibitors p21 and p27, and its indirect effect on the
levels of cyclin D1 and p53, as well as cell survival, through
direct inhibition of pro-apoptotic signals such as Bad and the
Forkhead family of transcription factors (39,40).
Our data show that miR-7 expression reduces AKT2 and MEK1/2
phosphorylation levels, as well as cyclin D1 expression.
Furthermore, recovery of the PI3K or Raf-1 expression level by
transfection of pcDNA-PI3K or pcDNA-Raf-1 (not including the
3′-UTR), respectively, partially abrogated the inhibition by miR-7
of the malignant progression of TJ899 cells. When PI3K and Raf-1,
both lacking the 3′-UTR, were transfected simultaneously, the TJ899
tumor cells were almost completely restored to their original
state. However, transfection of EGFR lacking the 3′-UTR had little
effect on TJ899 cells. We also found that forced PI3K expression
slightly increased Raf-1 and p-MEK1/2 protein levels, but forced
Raf-1 expression had no effect on PI3K and p-AKT-2 levels.
Therefore, we hypothesized a potential regulatory mechanism between
PI3K and Raf-1 or MEK1/2. Taken together, these results revealed
that miR-7 is a common regulator of the PI3K/ATK and Ras/Raf/
MEK/ERK pathways, an effect mediated by its targeting of the
transcription factors PI3K and Raf-1, but not EGFR. The end result
is inhibition of the malignant transformation of GBM cells.
Kefas and others have shown quite convincingly that
EGFR is targeted by miR-7 (41).
However, our data are not contradictory with their findings, for
Kefas has also reported that transfection with the pri-miR-7 vector
did not result in decreased EGFR protein in U87MG, and furthermore
that there might have a processing defect in generating pre-miR-7
from pri-miR-7 (41). Therefore,
there may be a case that the single stranded miR-7 mimic cannot
been processed into the RISC complex to target EGFR. However, there
are also positive facts that the single stranded miR-7 mimic can
effectively silence the two other target genes PI3K and Raf-1,
which are both located downstream of EGFR. Moreover, IRS2 has been
previously shown to be a target of miR-7 and lies upstream of Akt
signaling (41). Therefore, there
may be a complex regulatory network involving miR-7, and PI3K/ATK
and Ras/Raf/ MEK/ERK pathways that are both launched by EGFR. Taken
together, our data provide evidence that miR-7 may represent a key
therapeutic target for glioblastoma.
Acknowledgements
This study was supported by the China
National Natural Scientific Fund (81000901), Tianjin Health Bureau
Science and Technology Projects (10KG210, 2011KZ24 and 2011KZ26),
Tianjin Binhai New Area Health Bureau Science and Technology
Projects (2011BHKZ004, 2012BHKZ001 and 2012BHKZ003), and Tianjin
Binhai New Area Science and Technology Projects (2012MS05-04).
References
|
1.
|
Raizer JJ, Grimm S, Chamberlain MC, et al:
A phase 2 trial of single-agent bevacizumab given in an
every-3-week schedule for patients with recurrent high-grade
gliomas. Cancer. 116:5297–5305. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Bonavia R, Inda MM, Cavenee WK and Furnari
FB: Heterogeneity maintenance in glioblastoma: a social network.
Cancer Res. 71:4055–4060. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Masica DL and Karchin R: Correlation of
somatic mutation and expression identifies genes important in human
glioblastoma progression and survival. Cancer Res. 71:4550–4561.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Yang RY, Yang KS, Pike LJ and Marshall GR:
Targeting the dimerization of epidermal growth factor receptors
with small-molecule inhibitors. Chem Biol Drug Des. 76:1–9. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Tabunoki H, Saito N, Suwanborirux K, et
al: Molecular network profiling of U373MG human glioblastoma cells
following induction of apoptosis by novel marine-derived
anti-cancer 1,2,3,4-tetrahydroisoquinoline alkaloids. Cancer Cell
Int. 12:142012. View Article : Google Scholar
|
|
6.
|
Ivliev AE, ‘t Hoen PA and Sergeeva MG:
Coexpression network analysis identifies transcriptional modules
related to proastro cytic differentiation and sprouty signaling in
glioma. Cancer Res. 70:10060–10070. 2010. View Article : Google Scholar
|
|
7.
|
Rong Y, Belozerov VE, Tucker-Burden C, et
al: Epidermal growth factor receptor and PTEN modulate tissue
factor expression in glioblastoma through JunD/activator protein-1
transcriptional activity. Cancer Res. 69:2540–2549. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Mazzoleni S, Politi LS, Pala M, et al:
Epidermal growth factor receptor expression identifies functionally
and molecularly distinct tumor-initiating cells in human
glioblastoma multiforme and is required for gliomagenesis. Cancer
Res. 70:7500–7513. 2010. View Article : Google Scholar
|
|
9.
|
Martinez I and Dimaio D: B-Myb, cancer,
senescence, and microRNAs. Cancer Res. 71:5370–5373. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Ibrahim AF, Weirauch U, Thomas M, et al:
MicroRNA replacement therapy for miR-145 and miR-33a is efficacious
in a model of colon carcinoma. Cancer Res. 71:5214–5224. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Farazi TA, Horlings HM, Ten Hoeve JJ, et
al: MicroRNA sequence and expression analysis in breast tumors by
deep sequencing. Cancer Res. 71:4443–4453. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Liu N, Landreh M, Cao K, et al: The
microRNA miR-34 modulates ageing and neurodegeneration in
Drosophila. Nature. 482:519–523. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Wu DG, Wang YY, Fan LG, et al: MicroRNA-7
regulates glioblastoma cell invasion via targeting focal adhesion
kinase expression. Chin Med J. 124:2616–2621. 2011.PubMed/NCBI
|
|
14.
|
Nguyen HT, Dalmasso G, Yan Y, et al:
MicroRNA-7 modulates CD98 expression during intestinal epithelial
cell differentiation. J Biol Chem. 285:1479–1489. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Reddy SD, Ohshiro K, Rayala SK and Kumar
R: MicroRNA-7, a homeobox D10 target, inhibits p21-activated kinase
1 and regulates its functions. Cancer Res. 68:8195–8200. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Webster RJ, Giles KM, Price KJ, et al:
Regulation of epidermal growth factor receptor signaling in human
cancer cells by microRNA-7. J Biol Chem. 284:5731–5741. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Xiong S, Zheng Y, Jiang P, et al:
MicroRNA-7 inhibits the growth of human non-small cell lung cancer
A549 cells through targeting BCL-2. Int J Biol Sci. 7:805–814.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Lu J, Getz G, Miska EA, et al: MicroRNA
expression profiles classify human cancers. Nature. 435:834–838.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Hu BS, Tan JW, Zhu GH, et al: WWOX induces
apoptosis and inhibits proliferation of human hepatoma cell line
SMMC-7721. World J Gastroenterol. 18:3020–3026. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Yoo AS, Sun AX, Li L, et al:
MicroRNA-mediated conversion of human fibroblasts to neurons.
Nature. 476:228–231. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Xiong F, Wu C, Chang J, et al: Genetic
variation in an miRNA-1827 binding site in MYCL1 alters
susceptibility to small-cell lung cancer. Cancer Res. 71:5175–5181.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Zhou X, Ren Y, Moore L, et al:
Downregulation of miR-21 inhibits EGFR pathway and suppresses the
growth of human glioblastoma cells independent of PTEN status. Lab
Invest. 90:144–155. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Dong H, Luo L, Hong S, et al: Integrated
analysis of mutations, miRNA and mRNA expression in glioblastoma.
BMC Syst Biol. 4:1632010. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Huang PH, Miraldi ER, Xu AM, et al:
Phosphotyrosine signaling analysis of site-specific mutations on
EGFRvIII identifies determinants governing glioblastoma cell
growth. Mol Biosyst. 6:1227–1237. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Lal B, Goodwin CR, Sang Y, et al: EGFRvIII
and c-Met pathway inhibitors synergize against PTEN-null/EGFRvIII+
glioblastoma xenografts. Mol Cancer Ther. 8:1751–1760.
2009.PubMed/NCBI
|
|
26.
|
Reznik TE, Sang Y, Ma Y, et al:
Transcription-dependent epidermal growth factor receptor activation
by hepatocyte growth factor. Mol Cancer Res. 6:139–150. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Huang PH, Cavenee WK, Furnari FB and White
FM: Uncovering therapeutic targets for glioblastoma: a systems
biology approach. Cell Cycle. 6:2750–2754. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Chou YT, Lin HH, Lien YC, et al: EGFR
promotes lung tumorigenesis by activating miR-7 through a
Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor
ERF. Cancer Res. 70:8822–8831. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Kirubakaran P, Kothapalli R, Singh KhD, et
al: In silico studies on marine actinomycetes as potential
inhibitors for Glioblastoma multiforme. Bioinformation. 6:100–106.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Kakkar A, Suri V, Jha P, et al: Loss of
heterozygosity on chromosome 10q in glioblastomas, and its
association with other genetic alterations and survival in Indian
patients. Neurol India. 59:254–261. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Halatsch ME, Löw S, Hielscher T, et al:
Epidermal growth factor receptor pathway gene expressions and
biological response of glioblastoma multiforme cell lines to
erlotinib. Anticancer Res. 28:3725–3728. 2008.PubMed/NCBI
|
|
32.
|
Motomura K, Natsume A, Kishida Y, et al:
Benefits of interferon-β and temozolomide combination therapy for
newly diagnosed primary glioblastoma with the unmethylated MGMT
promoter: a multicenter study. Cancer. 117:1721–1730. 2011.
|
|
33.
|
Griffero F, Daga A, Marubbi D, et al:
Different response of human glioma tumor-initiating cells to
epidermal growth factor receptor kinase inhibitors. J Biol Chem.
284:7138–7148. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Cesana M, Cacchiarelli D, Legnini I, et
al: A long noncoding RNA controls muscle differentiation by
functioning as a competing endogenous RNA. Cell. 147:358–369. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Patterson EE, Holloway AK, Weng J, et al:
MicroRNA profiling of adrenocortical tumors reveals miR-483 as a
marker of malignancy. Cancer. 117:1630–1639. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Paroo Z, Ye X, Chen S and Liu Q:
Phosphorylation of the human microRNA-generating complex mediates
MAPK/Erk signaling. Cell. 139:112–122. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Hide T, Takezaki T, Nakatani Y, et al:
Combination of a ptgs2 inhibitor and an epidermal growth factor
receptor-signaling inhibitor prevents tumorigenesis of
oligodendrocyte lineage-derived glioma-initiating cells. Stem
Cells. 29:590–599. 2011. View Article : Google Scholar
|
|
38.
|
Fu S and Kurzrock R: Development of
curcumin as an epigenetic agent. Cancer. 116:4670–4676. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Zhu H, Acquaviva J, Ramachandran P, et al:
Oncogenic EGFR signaling cooperates with loss of tumor suppressor
gene functions in gliomagenesis. Proc Natl Acad Sci USA.
106:2712–2716. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Löw S, Vougioukas VI, Hielscher T, et al:
Pathogenetic pathways leading to glioblastoma multiforme:
association between gene expressions and resistance to erlotinib.
Anticancer Res. 28:3729–3732. 2008.PubMed/NCBI
|
|
41.
|
Kefas B, Godlewski J, Comeau L, et al:
microRNA-7 inhibits the epidermal growth factor receptor and the
Akt pathway and is down-regulated in glioblastoma. Cancer Res.
68:3566–3572. 2008. View Article : Google Scholar : PubMed/NCBI
|