Introduction
Breast cancer is one of the most common malignancies
in women worldwide, including Japan, and its incidence is still
increasing (1). Over the past few
decades, a great deal of effort has been made to improve the
diagnosis and treatment of breast cancer, however, some patients
still experience recurrence and clinical progression despite
appropriate adjuvant therapy. To identify such patients, many
researchers and clinicians have been trying to explore the
molecular signaling pathway and gene expression pattern involved in
the development of breast cancer, leading to the generation of
several prediction tools (2–4).
Representative gene sets such as Oncotype DX® (4), MammaPrint® (3), and PAM50 (2) enable us to predict survival in breast
cancer patients more accurately than using only established
biomarkers, namely, estrogen receptor (ER), progesterone receptor
(PR), and human epidermal growth factor receptor 2 (HER2).
Additionally, the identification of several novel biomarkers or
pathways related to breast cancer has led to the development of
molecular therapies targeting these pathways. Among these target
therapies are tyrosine kinase inhibitors, agents directed against
insulin-like growth factor-1 receptor (IGF-1R),
mesenchymal-epithelial transition factor (c-Met) (5) and fibroblast growth factor receptor
(FGFR) (6), angiogenesis
inhibitors directed against vascular endothelial growth factor
(VEGF), and agents that interfere with DNA repair (7). Understanding breast cancer diversity,
together with accurate categorization of a patient’s subgroup,
should allow the effective use of molecular-based targeted
therapy.
The metastasis-associated in colon cancer 1
(MACC1), was first identified in primary and metastatic
tumors of colon cancer by differential display reverse
transcription-polymerase chain reaction (PCR) by Stein et al
(8). MACC1 was revealed to be a
novel key regulator of tumor growth and metastasis mediated via
hepatocyte growth factor (HGF)/c-Met (mesenchymal-epithelial
transition factor gene) signaling in colorectal cancers. Increased
MACC1 expression has been shown to be correlated with worse
survival in several types of cancers, such as colorectal (8,9),
hepatocellular (10), gastric
(11), and ovarian (12). The HGF/c-Met pathway regulates
diverse biological activities, including proliferation, motility,
and invasion, many of which are hallmarks of cancer development
(13,14). Aberrant signaling of the c-Met
pathway has been associated with a poor prognosis in various tumors
(15–17), including breast cancer (18–20).
Stein et al, provided evidence that MACC1 binds to the
endogenous c-Met promoter, resulting in increased c-Met
transcription and activation of its downstream signaling (8,21).
The significance of MACC1 expression in some cancers
has been emphasized, but in human breast cancer it has barely been
investigated. We therefore performed comprehensive gene expression
analysis of MACC1 in a series of 300 breast cancers to investigate
whether MACC1 is a crucial prognostic factor across the cancers. In
addition, we evaluated the biological function of MACC1 in
regulating c-Met by means of an in vitro study.
Materials and methods
Subjects and tissues
A total of 300 female patients with primary breast
cancer who received both surgery and adjuvant treatment at Kumamoto
University Hospital (Kumamoto, Japan) between 2001 and 2009 were
selected. Breast cancer tissues were snap-frozen in liquid nitrogen
at pretherapeutic biopsy or surgical treatment and stored at −80°C
until RNA extraction. Adjuvant and neoadjuvant treatment was
assigned to each patient according to their risk on the basis of
tumor biology and clinical parameters, also in accordance with the
recommendations of the St. Gallen International Expert Consensus on
primary therapy of early breast cancer (22); that included 74% treated with
hormonal therapy, 26% with chemotherapy and 6.8% with targeted
therapy using trastuzumab. Forty-one (13%) patients did not receive
any treatment. Patients received either breast-conserving surgery
or total mastectomy and sentinel lymph node biopsy or axillary
lymph node dissection. The median follow-up period was 61
months.
Approval for the analyses conducted in the study was
received from the Ethics Committee of Kumamoto University Graduate
School of Medical Sciences (Kumamoto, Japan). Written informed
consent was obtained from all patients.
Immunohistochemical analysis
Histological sections (4 μm) were deparaffinized and
incubated for 10 min in methanol containing 0.3% hydrogen peroxide.
They were then immunostained with monoclonal antibodies against ERα
(SP1), PR (1E2) (both from Ventana Japan, Tokyo, Japan), HER2 (4B5;
Roche Diagnostics K.K., Tokyo, Japan), Ki67 (MIB1; Dako Japan,
Kyoto, Japan), and c-Met (D1C2; Cell Signaling Technology Japan
K.K., Tokyo, Japan), and a polyclonal antibody against MACC1
(ProSci, Inc., Poway, CA, USA). The staining was carried out in a
NexES IHC immunostainer (Ventana Medical Systems, Inc., Tucson, AZ,
USA), in accordance with the manufacturer’s instructions. ER and PR
were regarded as positive if >1% of nuclei were stained. HER2
expression was also determined by IHC staining based on the Hercep
test. We considered a tumor to be HER2-positive if the specimen
either scored 3+ by IHC, or showed a >2.2-fold increase in
fluorescence in situ hybridization (FISH). Tumor subtypes
were defined according to the expression of ER, PR and HER2. Ki67
was scored as the percentage of nuclear-stained cells out of all
cancer cells in the invasive front of the tumor regardless of the
intensity in a ×400 high-power field [Ki67 labeling index (23)]. We counted 500–1,000 tumor cells as
recommended by the International Ki67 in Breast Cancer Working
Group (24). For MACC1 and c-Met
expression, the H-score was calculated by multiplying the
percentage of positive cells (0–100) by the staining intensity
score (0–3).
RNA isolation and quantitative reverse
transcription-polymerase chain reaction (RT-qPCR)
Total RNA from tissue samples was isolated using the
AllPrep® DNA/RNA Mini kit (Qiagen, Valencia, CA, USA)
and that from cells was isolated using ReliaPrep™ RNA Cell Miniprep
System (Promega K.K., Tokyo, Japan). RNA was quantified by
measuring the A260/A280 absorbance ratios (Nano Drop Technologies,
Inc., Wilmington, DE, USA). Total RNA (0.5 μg) was reverse
transcribed to cDNA using PrimeScript® RT Master Mix
(Takara Bio, Inc., Shiga, Japan), according to the manufacturer’s
instructions. Each quantitative PCR was performed with 2 μl of the
cDNA and 0.2 μmol/l of each probe in the ABI Prism 7500 (Applied
Biosystems, Carlsbad, CA, USA) with SYBR Premix Dimer Eraser
(Takara Bio, Inc.). Each reaction (20 μl samples) was performed
under the following conditions: initialization for 10 sec at 95°C,
and then 45 cycles of amplification, with 5 sec at 95°C for
denaturation and 20 sec at 60°C for annealing and elongation. The
PCR primer sequences are shown in Table I. The expression of target gene was
normalized against GAPDH mRNA.
 | Table IOligonucleotide sequences for PCR,
siRNA, and ChIP. |
Table I
Oligonucleotide sequences for PCR,
siRNA, and ChIP.
| Sequence
(5′→3′) |
|---|
| RT-qPCR |
| MACC1 | F:
TTCTTTTGATTCCTCCGGTGA
R: ACTCTGATGGGCATGTGCTG |
| c-Met | F:
GAGAAGCCCAAGCCCATCC
R: GCCCAGGGCTCAGAGCTT |
| HGF | F:
GAATGACACTGATGTTCCTTTGG
R: GGATACTGAGAATCCCAACGC |
| GAPDH | F:
GCACCGTCAAGGCTGAGAAC
R: ATGGTGGTGAAGACGCCAGT |
| siRNA |
| MACC1-1 | F:
AGGUAAGAUUGGACUUGUAtt
R: UACAAGUCCAAUCUUACCUct |
| MACC1-2 | F:
AGUUAGUACGACUCACAAAtt
R: UUUGUGAGUCGUACUAACUtt |
| Control | F:
UUCUCCGAACGUGUCACGUdTdT
R: ACGUGACACGUUCGGAGAAdTdT |
| ChIP |
| c-Met
promoter | F:
CTAACTTCAGACTGCCTGAGC
R: CACCACCCAGAGGGAAATC |
Cell culture
The human breast carcinoma cell line MCF7 was
maintained in Eagle’s Minimum Essential Medium (EMEM) (Wako Pure
Chemical Industries, Ltd., Osaka, Japan), and the cell lines
MDA-MB-231, MDA-MB-468, and SW480 were maintained in Dulbecco’s
modified Eagle’s medium (DMEM) (Gibco, Grand Island, NY, USA), and
T47D, MDA-MB-453, and DLD-1 cells were maintained in RPMI-1640
medium (Gibco). All media were supplemented with 10% fetal bovine
serum (SAFC Biosciences, Inc., Lenexa, KS, USA) and all cell lines
were maintained at 37°C in an incubator with 5% CO2.
Cell motility and proliferation
assay
Cell migration was estimated by scratch motility
assay. Aliquots of 20×104 cells were seeded into wells
of 6-well plate and grown overnight to confluence. After 24 h,
transfection was performed, and after another 24 h, the monolayer
was scratched with a pipette tip, then washed with PBS to remove
floating cells. The number of cells that migrated into the
scratched area was photographed on days 0–3. Cell proliferation was
determined using Cell Counting kit-8 (Dojindo Laboratories,
Kumamoto, Japan). WST-8 reagent solution (10 μl) was added to each
well, then the microplate was incubated for 2 h in an incubator at
37°C. Absorbance at 450 nm was then measured using an EMax
Precision Microplate Reader (Molecular Devices Japan K.K., Tokyo,
Japan). Each experimental group contained three replicate wells,
and the experiment was repeated three times.
Generating of plasmid DNA containing the
MACC1 ORF
Plasmid DNA containing the MACC1 ORF was generated
using the Flexi® Vector System (http://www.promega.com/tbs/tm254/tm254.html).
Full-length human MACC1 was transferred from pFN21AE2447 to pFN28K
HaloTag® CMV-neo Flexi® Vector (both from
Promega K.K.), using Sgf I and Pme. A control vector harboring only
HaloTag was constructed using pFN28K HaloTag® CMV-neo
Flexi® Vector (Promega K.K.). We generated some
mutations in the Barnase coding region of the vector using the
PrimeSTAR® Mutagenesis Basal kit (Takara Bio, Inc.) to
abolish the function of the Barnase gene. E. coli DH5α
competent cells (Takara Bio, Inc.) were used for transformation
according to the manufacturer’s instructions. Plasmid purification
was performed using a HiSpeed™ Plasmid Maxi kit (Qiagen). We
confirmed expression of each protein by western blotting and
performed immunofluorescence assays to evaluate the transfection
efficacy.
Transfections and treatments
Transfections into MDA-MB- 231 and SW480 cells were
carried out using FuGENE® HD (Promega K.K.) according to
the manufacturer’s instructions. MCF7 cells were transfected using
a NEPA21 Electroporator (Nepa Gene Co., Ltd., Chiba, Japan)
according to the manufacturer’s instructions. The transfected cells
were cultured for 48 h before harvesting total RNA or protein.
MACC1 siRNA and control siRNA (both from Qiagen)
were transfected into the cells with a cocktail of 20 nM each using
Lipofectamine™ RNAiMAX (Life Technologies Japan, Tokyo, Japan)
according to the manufacturer’s instructions. These sequences are
described in Table I. For
expression analysis, cells were harvested 48 h after siRNA
transfection.
Cell cultures were treated with HGF (Sigma, St.
Louis, MO, USA) at 20 ng/ml for 24 h.
Protein extraction and western
blotting
Cultured cells were washed in ice-cold PBS and lysed
in Mammalian Protein Extraction Buffer (GE Healthcare Ltd.,
Buckinghamshire, UK). The protein concentration was determined
using a Pierce Protein 660 nm Protein Assay (Thermo Fisher
Scientific K.K., Kanagawa, Japan). Equal amounts of protein were
separated on Mini-PROTEAN® TGX™ gel and
electrophoretically transferred onto Trans-Blot® Turbo™
Mini Nitrocellulose membranes (both from Bio-Rad Laboratories,
Inc., Hercules, CA, USA). The membranes were blocked and incubated
overnight at 4°C with primary antibody; anti-MACC1 (1:200; Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA), anti-c-Met
(1:1,000), anti-β-actin (1:500) (both from Cell Signaling
Technology Japan K.K.) or anti-HaloTag® (1:1,000;
Promega K.K.). Proteins were visualized with horseradish
peroxidase-conjugated secondary antibodies (anti-rabbit IgG,
HRP-linked antibody, Cell Signaling Technology Japan K.K.;
anti-goat IgG-HRP, Santa Cruz Biotechnology, Inc.) followed by
chemiluminescence detection (Pierce Western Blotting Substrate
Plus; Thermo Fisher Scientific K.K.).
Chromatin immunoprecipitation (ChIP)
Immunoprecipitation was performed using the
HaloCHIP™ System, which is an antibody-free alternative to the ChIP
method that utilizes the HaloTag® fusion protein
(25). The detailed protocol can
be found at: http://www.promega.com/tbs/tm075/tm075.html. Briefly,
24 h before transfection, the cells were seeded and allowed to
attach, then transfections with HaloTag®-MACC1 fusion
constructs and control vector were performed as described above.
Forty-eight hours after transfection, the cells were crosslinked
with formaldehyde (Sigma-Aldrich Japan K.K., Tokyo, Japan) at a
final concentration of 1% and quenched with 0.125 M glycine.
Chromatin was sheared by sonication using a Bioruptor®
(Cosmo Bio Co., Ltd., Tokyo, Japan), and the
HaloTag®-MACC1 fusion protein and DNA complex was
captured by incubation with HaloLink™ Resin for 2 h at room
temperature. This allows complete covalent linkage between the
resin, protein and DNA. Subsequently, DNA was released by reversal
of crosslinking for 6–8 h at 65°C. Isolated DNA was further
purified using a QIAquick PCR Purification kit (Qiagen) and
amplified using AmpliTaq Gold® 360 Master Mix (Life
Technologies Japan). It has been shown that c-Met is a
transcriptional target of MACC1 in colorectal cancer, we therefore
generated a c-Met primer whose amplicon included an Sp1-1 site in
the c-Met promoter, where MACC1 might bind. The primer sequences of
c-Met promoter are shown in Table
I.
Statistical analysis
Comparisons between two groups were performed by
two-sided Student’s t-test. The significance of differences in
categorized demographic variables as a result of MACC1 expression
was evaluated using the Chi-square or Fisher’s exact test, and the
non-parametric Mann-Whitney U test. Relapse-free survival (RFS) and
breast cancer-specific survival (BCSS) curves were generated using
the Kaplan-Meier method and verified by the Wilcoxon test. Cox’s
proportional hazards model was used for univariate and multivariate
analyses of survival. All statistical analyses were carried out
using STATA ver.12 (StataCorp LP, College Station, TX, USA). All
tests were two-sided and P<0.05 was considered statistically
significant.
Results
Reduced MACC1 expression in breast cancer
is associated with worse prognosis
We first compared the expression levels of MACC1,
c-Met, and HGF mRNA in breast cancers and normal breast tissues
(n=172, Fig. 1). Expression of
both c-Met and HGF was higher in malignant tissues than in normal
tissues (each Wilcoxon P<0.001), whereas there was no difference
in MACC1 expression between them.
In a series of 300 breast cancer patients, MACC1
expression was analyzed by IHC and RT-qPCR. We calculated the
cut-off points for the corresponding expressions using a ROC curve
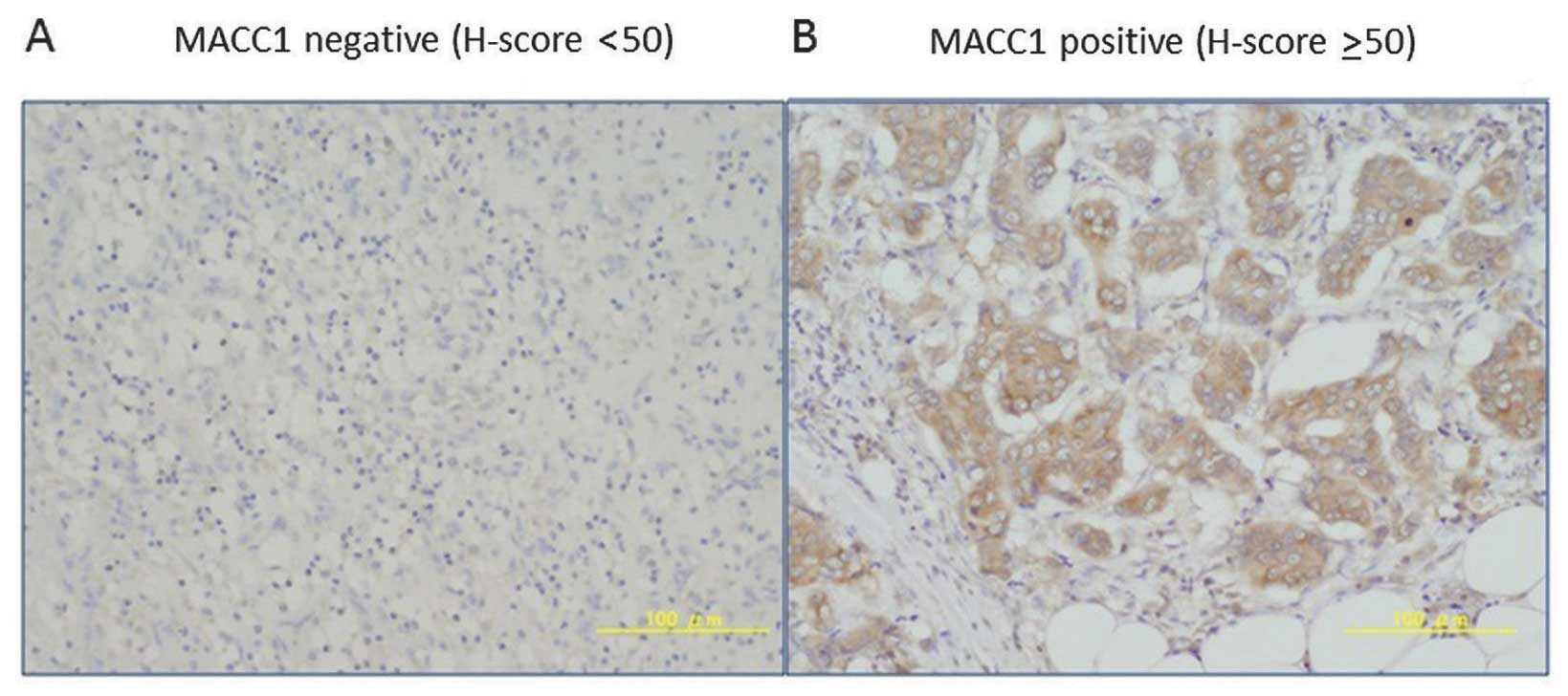
analysis. The IHC analysis showed that MACC1 staining was
detectable mainly in the cytoplasm and that 71% of the patients
exhibited high levels of MACC1 expression in their tumor samples
when the H-score cut-off point was set at 50 (Fig. 2). In this analysis, correlation
between MACC1 mRNA and protein expressions was weak (Spearman’s
γ=0.25, P<0.001). For the survival analysis, 43 patients (14%)
experienced recurrence and 27 patients (8.9 %) died as a result of
breast cancer during a median follow-up period of 61 months. As
shown in Fig. 3, the patients with
low levels of MACC1 (both mRNA and protein) were more likely to
have reduced RFS and BCSS compared to those with higher levels. In
multivariate analysis using a Cox proportional hazard model, MACC1
mRNA (HR=0.25, P=0.001), MACC1 protein (HR=0.37, P=0.016), axillary
nodal status (HR=5.09, P=0.003), and ER status (HR=0.09,
P<0.001) were independent predictors of mortality (Table II).
| Table IIUnivariate and multivariate analysis
for BCSS (Cox proportional hazard model). |
Table II
Univariate and multivariate analysis
for BCSS (Cox proportional hazard model).
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|---|
| Variable | HR (95% CI) | P | HR (95% CI) | P |
|---|
| Tumor size |
| >2 cm vs. ≤2
cm | 2.92
(1.23–6.92) | 0.015 | 1.44
(0.50–4.14) | 0.495 |
| Axillary lymph
node |
| Positive vs.
negative | 3.39
(1.48–7.76) | 0.004 | 5.09
(1.74–14.8) | 0.003 |
| Nuclear grade |
| G2–3 vs. G1 | 2.45
(1.06–5.64) | 0.035 | 0.80
(0.29–2.18) | 0.656 |
| Ki67 labeling index
(%) |
| ≥15 vs.
<15 | 5.29
(1.25–22.4) | 0.024 | 2.40
(0.55–10.6) | 0.245 |
| ER status |
| Positive vs.
negative | 0.11
(0.05–0.25) | <0.001 | 0.09
(0.03–0.26) | <0.001 |
| PR status |
| Positive vs.
negative | 0.14
(0.06–0.36) | <0.001 | | |
| HER2 status |
| Positive vs.
negative | 0.91
(0.31–2.63) | 0.856 | | |
| MACC1
expression |
| mRNA high vs.
low | 0.42
(0.19–0.89) | 0.024 | 0.25
(0.11–0.58) | 0.001 |
| Protein high vs.
low | 0.37
(0.17–0.79) | 0.011 | 0.37
(0.16–0.83) | 0.016 |
Relationship between MACC1 expression and
other clinicopathological factors
We also examined the correlation between MACC1
expression and clinicopathological factors, but no significant
correlations were found (Table
III). We further evaluated the correlation between MACC1
protein and c-Met mRNA expressions based on the findings that MACC1
is a transcriptional regulator of c-Met in colorectal cancer. We
found no strong positive correlation between them, with a
Spearman’s coefficient of 0.16 (P=0.0067).
| Table IIICorrelations between MACC1 expression
and clinicopathological characteristics. |
Table III
Correlations between MACC1 expression
and clinicopathological characteristics.
| No. of
patients | MACC1
mRNA
Low/high (%) | Pa | MACC1
protein
Low/high (%) | Pa |
|---|
| No. | | 76/228 (75) | | 82/218 (73) | |
| Age |
| <50 | 71 | 13/58 (82) | 0.136 | 18/53 (75) | 0.438 |
| ≥50 | 229 | 62/167 (73) | | 69/160 (70) | |
| Menopausal
status |
| Pre- | 79 | 16/63 (80) | 0.196 | 23/56 (71) | 0.997 |
| Post- | 220 | 59/161 (73) | | 64/156 (71) | |
| Tumor size |
| ≤2 cm | 152 | 42/110 (72) | 0.286 | 44/108 (71) | 0.984 |
| >2 cm | 148 | 33/115 (78) | | 43/105 (71) | |
| Nodal status |
| Negative | 179 | 41/138 (77) | 0.308 | 56/123 (69) | 0.289 |
| Positive | 121 | 34/87 (72) | | 31/90 (74) | |
| Stage |
| I | 111 | 28/83 (75) | 0.777 | 33/78 (70) | 0.819 |
| II | 159 | 38/121 (76) | | 47/112 (70) | |
| III | 30 | 9/21 (70) | | 7/23 (77) | |
| Nuclear grade |
| 1 | 152 | 38/114 (75) | 0.386 | 51/101 (66) | 0.149 |
| 2 | 74 | 15/59 (80) | | 16/58 (78) | |
| 3 | 73 | 22/51 (70) | | 19/54 (74) | |
| ER status |
| Negative | 68 | 17/51 (75) | 1.00 | 22/46 (68) | 0.488 |
| Positive
(≥1%) | 232 | 58/174 (75) | | 65/167 (72) | |
| PR status |
| Negative | 101 | 31/70 (69) | 0.105 | 34/67 (66) | 0.205 |
| Positive
(≥1%) | 199 | 44/155 (78) | | 53/146 (73) | |
| HER2 status |
| Negative | 257 | 67/190 (74) | 0.295 | 76/181 (70) | 0.594 |
| Positive | 43 | 8/35 (81) | | 11/32 (74) | |
| Ki67 labeling
index |
| <15% | 95 | 22/73 (77) | 0.616 | 33/62 (65) | 0.136 |
| ≥15% | 205 | 53/152 (74) | | 54/151 (74) | |
| Tumor subtype |
| Luminal | 219 | 52/167 (76) | 0.096 | 59/160 (73) | 0.072 |
| Luminal-HER2 | 14 | 4/10 (71) | | 5/9 (64) | |
| HER2-rich | 29 | 4/25 (86) | | 6/23 (79) | |
|
Triple-negative | 38 | 15/23 (61) | | 17/21 (55) | |
MACC1 and c-Met are differentially
expressed in breast and colorectal cancer cell lines
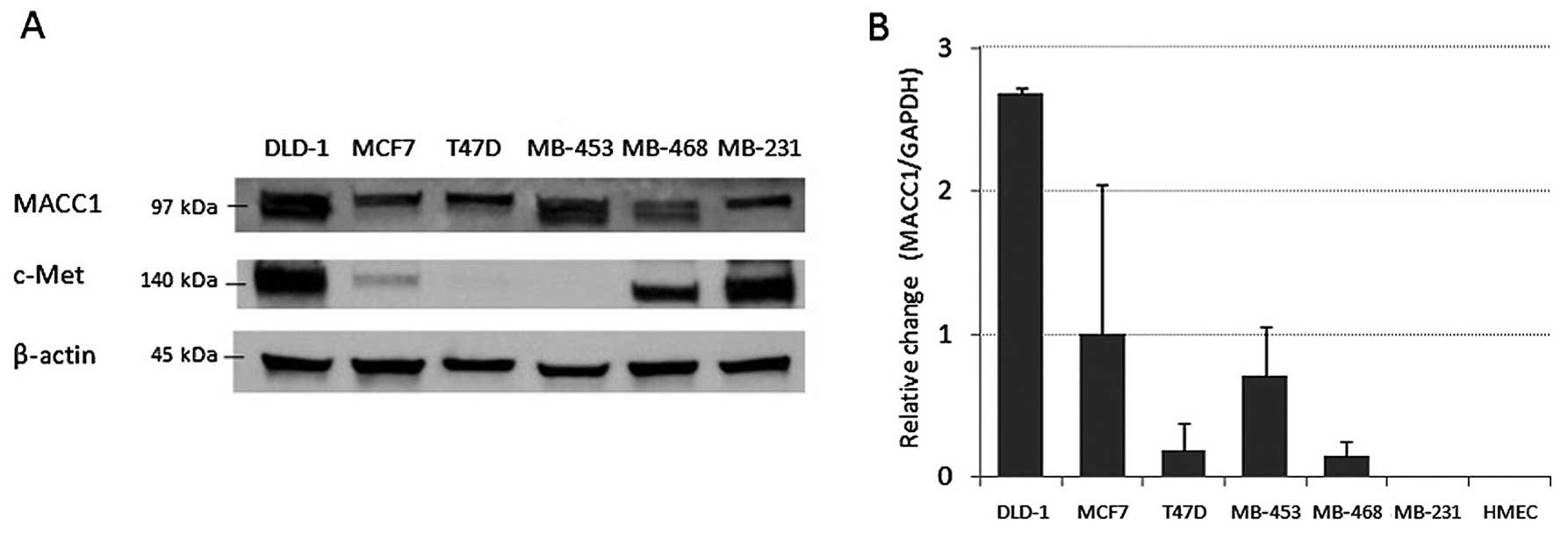
Next, we evaluated the level of MACC1 expression in
colon cancer and several breast cancer cell lines by western
blotting and RT-qPCR analyses (Fig.
4). MACC1 expression was much higher in DLD-1 colon cancer
cells than in any of the breast cancer cell lines. Further, we
found differences in its expression among breast cancer cell lines;
luminal-type cell lines including MCF7 and T47D had lower levels of
MACC1 protein than HER2-type (MDA-MB-453) or triple-negative-type
cell lines (MDA-MB-468). c-Met expression, in DLD-1, MDA-MB-231,
and MDA-MB-468 cell lines exhibited high levels.
MACC1 overexpression does not influence
the expression of c-Met protein in breast cancer cell lines
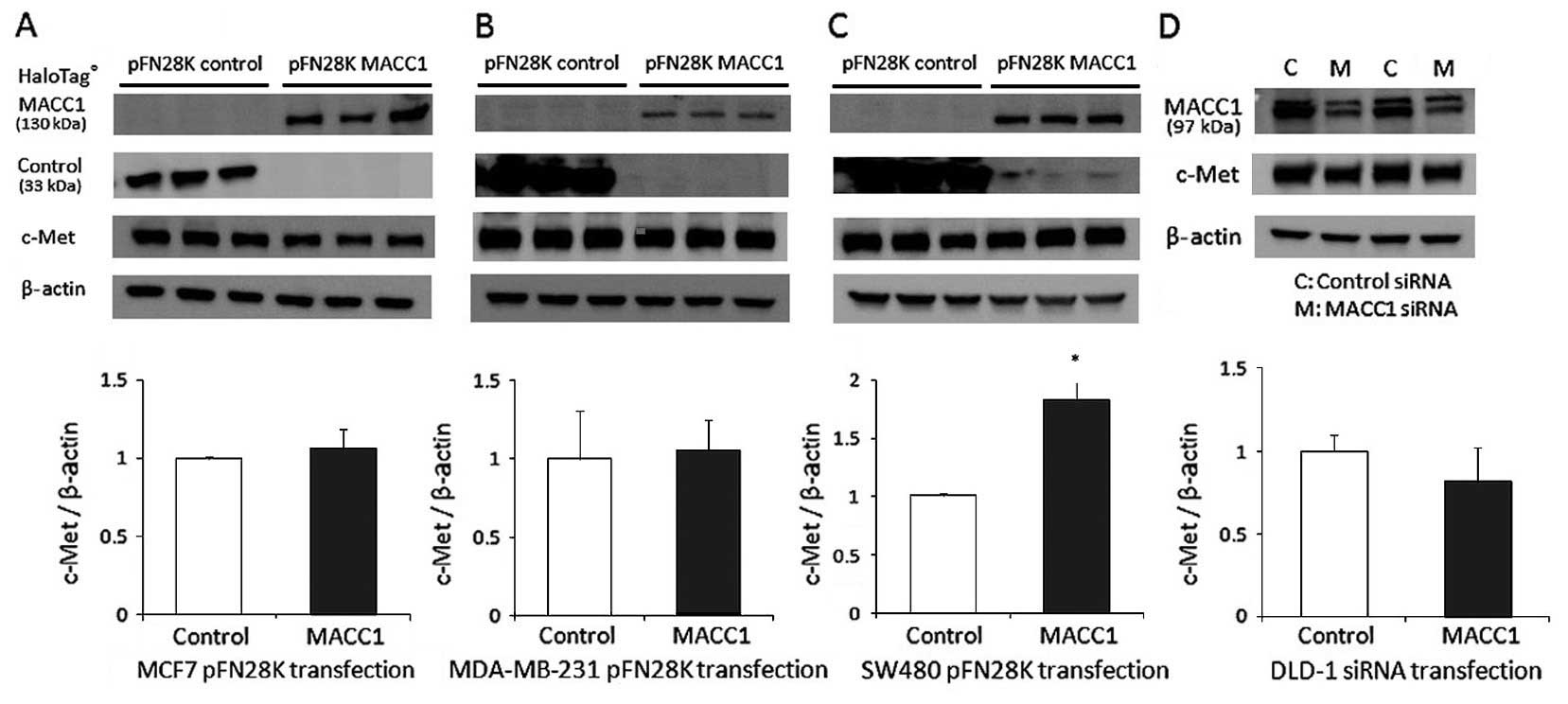
To evaluate the impact of MACC1 on the biological
function of the cells, we transfected MACC1 constructs (pFN28K
HaloTag® MACC1 and pFN28K HaloTag® control)
into the MCF7, MDA-MB-231, and SW480 cells which normally do not
express endogenous MACC1. As shown in Fig. 5, a significant increase in c-Met
expression was found only in SW480 cells transfected with MACC1 for
48 h (Fig. 5C). There was no
significant difference in MCF7 or MDA-MB-231 cells (Fig. 5A and B). In the colon cancer cell
line DLD-1 which exhibited high expression of MACC1, c-Met
expression was suppressed in the cells transfected with MACC1 siRNA
for 48 h (Fig. 5D).
Moreover, we evaluated the effect of MACC1
expression on the migratory and proliferative potential of the
transfected cells. MACC1 overexpression had no impact on the
proliferative ability of either breast or colon cancer cells (data
not shown). In contrast, SW480 cells transfected with pFN28K MACC1
showed enhanced migratory ability compared to that of pFN28K
control-transfected cells (Fig.
6B), whereas in MDA-MB-231 cells, transfection of MACC1 had no
impact on this ability (Fig.
6A).
HGF induction of c-Met expression is not
enhanced in breast cancer cells transfected with MACC1
Since it was previously reported that HGF, which is
a ligand of c-Met, induces translocation of MACC1 from the
cytoplasm to the nucleus in SW480 cells, leading to the activation
of c-Met signaling (8), we
compared the HGF-induced c-Met expression in MCF7 and SW480 cells
transfected with MACC1. As shown in Fig. 7, the SW480 cells transfected with
pFN28K MACC1 expressed more c-Met protein after HGF treatment
compared to the controls transfected with pFN28K. In contrast,
transfection of the corresponding MCF7 cells with MACC1 had no
impact on the effect of HGF-induced c-Met expression. This
indicates that the MACC1-HGF/c-Met loop has different roles in
breast and colorectal cancers.
MACC1 does not bind to the c-Met promoter
region in breast cancer cell lines
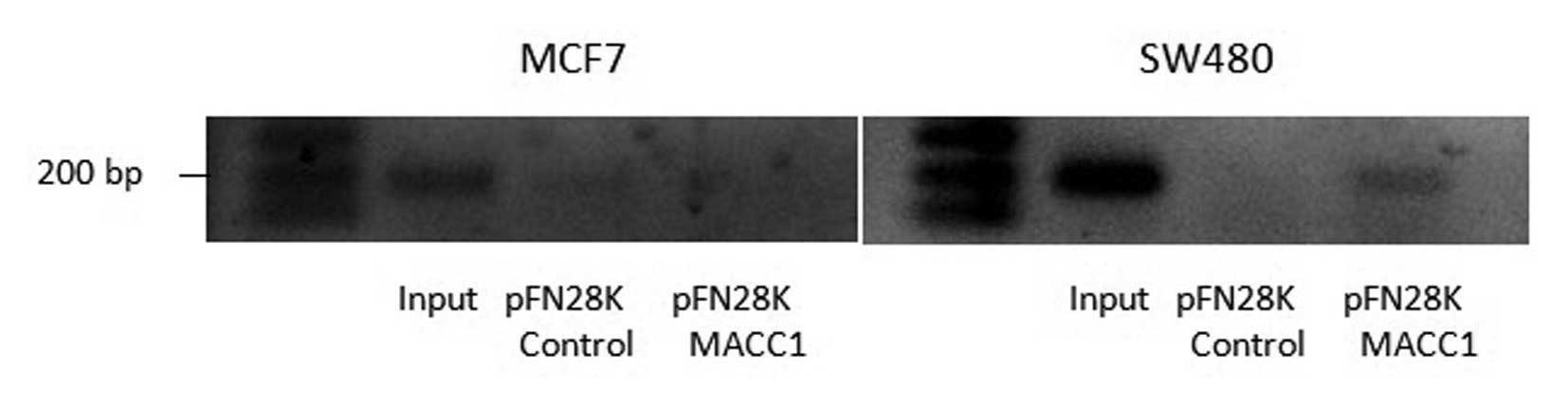
It was previously demonstrated that MACC1
specifically binds to the SP1 site of the c-Met promoter to
activate the c-Met signaling cascade in SW480 cells (21). Based on this finding, we performed
the ChIP analysis using the HaloCHIP™ System (described in
Materials and methods) to confirm that MACC1 binds to the c-Met
promoter in all the cell lines tested (Fig. 8). A visible band on amplification
of immune precipitated chromatin for the c-Met promoter was found
in SW480 cells transfected with MACC1, whereas no signal bands were
detected in MCF7 cells. This suggests that MACC1 binds to the c-Met
promoter region in SW480 cells, but not in MCF7 cells.
Discussion
The results of this study provide additional
evidence regarding the role of MACC1, indicating that it plays
different roles in breast cancer and in several other cancers. In
the present study, MACC1 expression was associated with prolonged
RFS and BCSS in breast cancer patients, which was beyond our
expectation and not in agreement with previous studies (8,10–12).
Additionally, our in vitro studies suggest that MACC1 may
not regulate c-Met expression in breast cancer cells, a theory
which was also given support by the finding that MACC1 did not bind
to the c-Met promoter in MCF7 cells when analyzed by ChIP. Further,
MACC1 overexpression did not have a functional impact on cell
migration or proliferation abilities in breast cancer cells.
Consequently MACC1 appears not to contribute to disease progression
in breast cancer via HGF/c-Met signaling.
MACC1 was first identified as a colon cancer
oncogene which promoted metastasis, and to date, a few studies have
demonstrated its role in several cancers (8–11).
However, little is known about its role in breast cancer, except
for one report by Huang et al (26). The results of their study,
suggesting that MACC1 may be an independent prognostic indicator of
a worse outcome is inconsistent with our findings. Possible reasons
for these discordant findings could be the differences in selection
of the patient cohort and in determination of adjuvant treatment.
In our study, the majority of the patients received some adjuvant
treatment, including the use of endocrine, chemotherapy, and
trastuzumab therapy; only 41 (13%) patients did not receive any
treatment. In contrast, in the study by Huang et al, most of
the patients (56.2%) received no endocrine treatment although
information of regarding other treatment was not available
(26). There is the possibility
that the effect of adjuvant treatment modified the prognostic role
of MACC1 expression. To date, many biomarkers have been
investigated and proposed as prognostic markers, nevertheless, only
a small proportion of those ultimately proved clinically useful.
This may partly be due to insufficient validation or conflicting
results across the studies. For example, IGF-1R has been identified
as an adverse prognostic factor for breast cancer in some studies
(27,28), whereas others report the favorable
prognostic role of IGF-1R (29,30).
Thus, evidence for the potential role of MACC1 as a biomarker in
breast cancer will need to be accumulated from further studies in
other large cohort.
Our results reveal that patients with low levels of
MACC1 are more likely to have a worse prognosis compared to those
with higher levels, when analyzed using an optimally-estimated
cut-off point (Fig. 3). The
biological mechanism of MACC1 which underlies improvement of breast
cancer prognosis remains unelucidated in the present study. Our
results suggest that MACC1 expression is intrinsically low in
breast cancer based on the following findings. Our in vitro
studies show that MACC1 protein expression in breast cancer cells,
especially in luminal-type cells, is generally lower than that in
colon cancer cells (DLD-1) (Fig.
4). Additionally, a comprehensive analysis of gene expression
indicates differences in MACC1 expression levels in a variety of
human tissues (31). According to
the database, the highest MACC1 expression was observed in
colorectal carcinoma, followed by stomach, gastrointestinal,
pancreatic, and ovarian carcinoma, and its expression in breast
cancer was much lower than in these carcinomas (32). These differences in the levels of
endogenous MACC1 expression might be attributable to differences in
physiological function between breast cancer and several other
types of cancers.
We further investigated the regulatory effect of
MACC1 on c-Met expression in cell lines, and demonstrated that
c-Met expression was not induced after MACC1 transfection in breast
cancer cell lines, a finding that differed from the results
observed in colon cancer cells (Fig.
5). MACC1 siRNA treatment of MDA-MB-468 cells, in which MACC1
expression was relatively high, also revealed no alteration of
c-Met expression (data not shown). c-Met is a proto-oncogene
considered essential for conferring metastatic potential in various
tumors (14,15,33,34).
c-Met activates its downstream effectors of the
Rasmitogen-activated protein kinase (MAPK) and phosphatidylinositol
3-kinase (PI3K)-Akt pathways to promote the invasive growth
characteristic of malignancies (35,36).
Also in breast cancer, disruption of the c-Met pathway has been
shown to contribute to worse prognosis (20,37)
and confer resistance to endocrine therapy or trastuzumab treatment
(38–40). In addition to c-Met, its ligand HGF
is also suggested to be an independent prognostic parameter for
breast cancer (41,42). Hence, it was of great interest to
us to understand the significance of MACC1 as a candidate regulator
of the HGF/c-Met cascade in breast cancer. A previous study has
suggested the hypothesis of a MACC1-driven positive feedback loop
after its binding to the c-Met promoter and subsequent activation
of c-Met signaling (43). In our
study, HGF-induced c-Met expression was not enhanced by MACC1
transfection in MCF7 cells in contrast to the effect in SW480 cells
(Fig. 7), indicating uncertainty
of this feedback mechanism in breast cancer. Since c-Met
transcription appears to be activated by several factors besides
MACC1, such as hypoxia-inducible factor 1 (HIF1) and AP-1 (34,44),
it is possible that MACC1 does not act as the exclusive master
regulator of the HGF/c-Met signaling involved in disease
progression in breast cancer.
The MACC1 gene is located on human chromosome
7 (7p21.1) and consists of seven exons and six introns. The MACC1
protein harbors an Src homology 3 (SH3) domain, ZU5 domain, and two
death domains (DD) (31).
Protein-protein interactions involving MACC1 are possible based on
its domain structure (45).
Further, MACC1 binds to the SP1 domain, which is found within the
promoter regions of numerous genes (46). Accordingly, MACC1 could act as a
transcriptional factor for a hitherto unknown target or interact
with other proteins leading to improved outcomes for patients with
breast cancer. Comprehensive analyses such as ChIP-seq analysis or
mass spectrometry are therefore required to confirm this
hypothesis.
In conclusion, our study reveals that MACC1
expression is associated with prolonged RFS and BCSS, and
demonstrates that its prognostic impact is independent of
established prognostic factors. In breast cancer cells, our
findings suggest that MACC1 is not a major regulator of the
transcriptional target gene, c-Met. Although MACC1 has been
suggested to be a crucial marker related to cancer development in
colorectal cancer, its role in breast cancer appears to differ.
Further evidence for any potential role of MACC1 as a biomarker in
breast cancer should be accumulated.
Acknowledgements
The authors are grateful to Y. Azakami for excellent
technical support, to A. Okabe for clinical data management, to Dr
M. Nakano (Department of Molecular Genetics, Kumamoto University)
for providing breast cancer cell lines and technical support, and
to Dr K. Kuwahara (Department of Immunology, Kumamoto University)
for providing the SW480 cell line. This study was supported by a
Grant in Aid for Scientific Research from the Ministry of
Education, Culture, Sports, Science and Technology of Japan (Y.
Yamamoto, Grant no. 24591909).
References
|
1
|
Matsuda T, Marugame T, Kamo K, Katanoda K,
Ajiki W and Sobue T; Japan Cancer Surveillance Research Group.
Cancer incidence and incidence rates in Japan in 2005: based on
data from 12 population-based cancer registries in the Monitoring
of Cancer Incidence in Japan (MCIJ) project. Jpn J Clin Oncol.
41:139–147. 2011. View Article : Google Scholar
|
|
2
|
Parker JS, Mullins M, Cheang MC, et al:
Supervised risk predictor of breast cancer based on intrinsic
subtypes. J Clin Oncol. 27:1160–1167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van’t Veer LJ, Dai H, van de Vijver MJ, et
al: Gene expression profiling predicts clinical outcome of breast
cancer. Nature. 415:530–536. 2002. View
Article : Google Scholar
|
|
4
|
Cronin M, Sangli C, Liu ML, Pho M, Dutta
D, Nguyen A, Jeong J, Wu J, Langone KC and Watson D: Analytical
validation of the Oncotype DX genomic diagnostic test for
recurrence prognosis and therapeutic response prediction in
node-negative, estrogen receptor-positive breast cancer. Clin Chem.
53:1084–1091. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sierra JR and Tsao MS: c-MET as a
potential therapeutic target and biomarker in cancer. Ther Adv Med
Oncol. 3(Suppl 1): S21–S35. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Turner N, Lambros MB, Horlings HM, et al:
Integrative molecular profiling of triple negative breast cancers
identifies amplicon drivers and potential therapeutic targets.
Oncogene. 29:2013–2023. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Turner N, Tutt A and Ashworth A: Hallmarks
of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 4:814–819. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stein U, Walther W, Arlt F, Schwabe H,
Smith J, Fichtner I, Birchmeier W and Schlag PM: MACC1, a newly
identified key regulator of HGF-MET signaling, predicts colon
cancer metastasis. Nat Med. 15:59–67. 2009. View Article : Google Scholar
|
|
9
|
Boardman LA: Overexpression of MACC1 leads
to downstream activation of HGF/MET and potentiates metastasis and
recurrence of colorectal cancer. Genome Med. 1:362009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qiu J, Huang P, Liu Q, Hong J, Li B, Lu C,
Wang L, Wang J and Yuan Y: Identification of MACC1 as a novel
prognostic marker in hepatocellular carcinoma. J Transl Med.
9:1662011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang L, Wu Y, Lin L, et al:
Metastasis-associated in colon cancer-1 upregulation predicts a
poor prognosis of gastric cancer, and promotes tumor cell
proliferation and invasion. Int J Cancer. 133:1419–1430. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang R, Shi H, Chen Z, Wu Q, Ren F and
Huang H: Effects of metastasis-associated in colon cancer 1
inhibition by small hairpin RNA on ovarian carcinoma OVCAR-3 cells.
J Exp Clin Cancer Res. 30:832011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maulik G, Shrikhande A, Kijima T, Ma PC,
Morrison PT and Salgia R: Role of the hepatocyte growth factor
receptor, c-Met, in oncogenesis and potential for therapeutic
inhibition. Cytokine Growth Factor Rev. 13:41–59. 2002. View Article : Google Scholar
|
|
14
|
Bottaro DP, Rubin JS, Faletto DL, Chan AM,
Kmiecik TE, Vande Woude GF and Aaronson SA: Identification of the
hepatocyte growth factor receptor as the c-met proto-oncogene
product. Science. 251:802–804. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Di Renzo MF, Olivero M, Giacomini A, et
al: Overexpression and amplification of the met/HGF receptor gene
during the progression of colorectal cancer. Clin Cancer Res.
1:147–154. 1995.PubMed/NCBI
|
|
16
|
Humphrey PA, Zhu X, Zarnegar R, Swanson
PE, Ratliff TL, Vollmer RT and Day ML: Hepatocyte growth factor and
its receptor (c-MET) in prostatic carcinoma. Am J Pathol.
147:386–396. 1995.PubMed/NCBI
|
|
17
|
Amemiya H, Kono K, Itakura J, Tang RF,
Takahashi A, An FQ, Kamei S, Iizuka H, Fujii H and Matsumoto Y:
c-Met expression in gastric cancer with liver metastasis. Oncology.
63:286–296. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jin L, Fuchs A, Schnitt SJ, Yao Y, Joseph
A, Lamszus K, Park M, Goldberg ID and Rosen EM: Expression of
scatter factor and c-met receptor in benign and malignant breast
tissue. Cancer. 79:749–760. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Camp RL, Rimm EB and Rimm DL: Met
expression is associated with poor outcome in patients with
axillary lymph node negative breast carcinoma. Cancer.
86:2259–2265. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lengyel E, Prechtel D, Resau JH, et al:
C-Met overexpression in node-positive breast cancer identifies
patients with poor clinical outcome independent of Her2/neu. Int J
Cancer. 113:678–682. 2005. View Article : Google Scholar
|
|
21
|
Stein U, Smith J, Walther W and Arlt F:
MACC1 controls Met: what a difference an Sp1 site makes. Cell
Cycle. 8:2467–2469. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Goldhirsch A, Wood WC, Coates AS, Gelber
RD, Thürlimann B and Senn HJ; Panel members. Strategies for
subtypes - dealing with the diversity of breast cancer: highlights
of the St. Gallen International Expert Consensus on the Primary
Therapy of Early Breast Cancer 2011. Ann Oncol. 22:1736–1747. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yamamoto S, Ibusuki M, Yamamoto Y, Fu P,
Fujiwara S, Murakami K and Iwase H: Clinical relevance of Ki67 gene
expression analysis using formalin-fixed paraffin-embedded breast
cancer specimens. Breast Cancer. 20:262–270. 2013. View Article : Google Scholar
|
|
24
|
Dowsett M, Nielsen TO, A’Hern R, et al;
International Ki-67 in Breast Cancer Working Group. Assessment of
Ki67 in breast cancer: recommendations from the International Ki67
in Breast Cancer working group. J Natl Cancer Inst. 103:1656–1664.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hartzell DD, Trinklein ND, Mendez J,
Murphy N, Aldred SF, Wood K and Urh M: A functional analysis of the
CREB signaling pathway using HaloCHIP-chip and high throughput
reporter assays. BMC Genomics. 10:4972009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang Y, Zhang H, Cai J, Fang L, Wu J, Ye
C, Zhu X and Li M: Overexpression of MACC1 and its significance in
human breast cancer progression. Cell Biosci. 3:162013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Law JH, Habibi G, Hu K, et al:
Phosphorylated insulin-like growth factor-i/insulin receptor is
present in all breast cancer subtypes and is related to poor
survival. Cancer Res. 68:10238–10246. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Railo MJ, von Smitten K and Pekonen F: The
prognostic value of insulin-like growth factor-I in breast cancer
patients. Results of a follow-up study on 126 patients. Eur J
Cancer. 30A:307–311. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shin A, Ren Z, Shu XO, Cai Q, Gao YT and
Zheng W: Expression patterns of insulin-like growth factor 1
(IGF-I) and its receptor in mammary tissues and their associations
with breast cancer survival. Breast Cancer Res Treat. 105:55–61.
2007. View Article : Google Scholar
|
|
30
|
Papa V, Gliozzo B, Clark GM, McGuire WL,
Moore D, Fujita-Yamaguchi Y, Vigneri R, Goldfine ID and Pezzino V:
Insulin-like growth factor-I receptors are overexpressed and
predict a low risk in human breast cancer. Cancer Res.
53:3736–3740. 1993.PubMed/NCBI
|
|
31
|
Stein U, Dahlmann M and Walther W: MACC1 -
more than metastasis? Facts and predictions about a novel gene. J
Mol Med Berl. 88:11–18. 2010. View Article : Google Scholar
|
|
32
|
|
|
33
|
Kang JY, Dolled-Filhart M, Ocal IT, Singh
B, Lin CY, Dickson RB, Rimm DL and Camp RL: Tissue microarray
analysis of hepatocyte growth factor/Met pathway components reveals
a role for Met, matriptase, and hepatocyte growth factor activator
inhibitor 1 in the progression of node-negative breast cancer.
Cancer Res. 63:1101–1105. 2003.PubMed/NCBI
|
|
34
|
Boccaccio C and Comoglio PM: Invasive
growth: A MET-driven genetic programme for cancer and stem cells.
Nat Rev Cancer. 6:637–645. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Peschard P and Park M: From Tpr-Met to
Met, tumorigenesis and tubes. Oncogene. 26:1276–1285. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Trusolino L, Bertotti A and Comoglio PM:
MET signalling: Principles and functions in development, organ
regeneration and cancer. Nat Rev Mol Cell Biol. 11:834–848. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ghoussoub RA, Dillon DA, D’Aquila T, Rimm
EB, Fearon ER and Rimm DL: Expression of c-met is a strong
independent prognostic factor in breast carcinoma. Cancer.
82:1513–1520. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hiscox S, Jordan NJ, Jiang W, Harper M,
McClelland R, Smith C and Nicholson RI: Chronic exposure to
fulvestrant promotes overexpression of the c-Met receptor in breast
cancer cells: Implications for tumour-stroma interactions. Endocr
Relat Cancer. 13:1085–1099. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shattuck DL, Miller JK, Carraway KL III
and Sweeney C: Met receptor contributes to trastuzumab resistance
of Her2-over-expressing breast cancer cells. Cancer Res.
68:1471–1477. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Minuti G, Cappuzzo F, Duchnowska R, et al:
Increased MET and HGF gene copy numbers are associated with
trastuzumab failure in HER2-positive metastatic breast cancer. Br J
Cancer. 107:793–799. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jiang WG, Martin TA, Parr C, Davies G,
Matsumoto K and Nakamura T: Hepatocyte growth factor, its receptor,
and their potential value in cancer therapies. Crit Rev Oncol
Hematol. 53:35–69. 2005. View Article : Google Scholar
|
|
42
|
Maemura M, Iino Y, Yokoe T, Horiguchi J,
Takei H, Koibuchi Y, Horii Y, Takeyoshi I, Ohwada S and Morishita
Y: Serum concentration of hepatocyte growth factor in patients with
metastatic breast cancer. Cancer Lett. 126:215–220. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Arlt F and Stein U: Colon cancer
metastasis: MACC1 and Met as metastatic pacemakers. Int J Biochem
Cell Biol. 41:2356–2359. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pennacchietti S, Michieli P, Galluzzo M,
Mazzone M, Giordano S and Comoglio PM: Hypoxia promotes invasive
growth by transcriptional activation of the met protooncogene.
Cancer Cell. 3:347–361. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ipsaro JJ, Huang L and Mondragón A:
Structures of the spectrin-ankyrin interaction binding domains.
Blood. 113:5385–5393. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kadonaga JT, Jones KA and Tjian R:
Promoter-specific activation of RNA polymerase II transcription by
Sp1. Trends Biochem Sci. 11:20–23. 1986. View Article : Google Scholar
|