Introduction
Hepatocellular carcinoma (HCC) is one of the most
common and lethal cancers in the human population, ranking as the
third most common cause of cancer-associated mortality worldwide
(1,2). Persistent hepatitis B virus infection
is a well-established risk factor for liver cancer, but HCC is
caused by a complex interaction between numerous factors, such as
environmental exposure, extensive alcohol intake, transgenic
oncogenes, and other causes of hepatic cirrhosis (3–6).
Despite recent progress in anticancer therapy, the efficacy of
currently available anticancer treatments is limited because of
natural or acquired resistance to conventional chemotherapy and
limited efficacy of radiotherapy (7). Patients with advanced HCC therefore
continue to show poor outcomes (8,9),
which emphasizes the need to identify and develop more potent
anticancer drugs with increased selectivity and reduced toxicity
for HCC treatment.
Apoptosis is a highly regulated process of
programmed cell death, and disruption of this process represents a
major contributing factor in the pathology of cancer (10). In general, apoptosis may be
initiated through two major pathways, the extrinsic [death receptor
(DR)-mediated] and the intrinsic (mitochondria-mediated) pathways
(11,12). The extrinsic pathway is initiated
by binding of death ligands with their cognate receptors, which
leads to the activation of caspase-8, while the intrinsic pathway
is initiated by release of cytochrome c from mitochondria
into the cytosol following loss of inner mitochondrial membrane
integrity and activation of caspase-9 (11,13).
A series of signaling cascades is activated, and both pathways
ultimately result in activation of caspase-3 and -7, the final
executioners of apoptotic cell death (14,15).
Therefore, agents that target the apoptosis pathway without
affecting normal cells have significant potential as drug targets
for cancer treatment.
Accumulating evidence now indicates that cancer
metabolism mediates cancer development and progression via its
activation of related signaling pathways, such as the AMP-activated
protein kinase (AMPK) pathway (16,17).
Under metabolic stress conditions, where decreases in intracellular
ATP and increases in AMP levels are common, AMPK is activated by
phosphorylation of a critical amino acid residue (Thr172) to
function as a major metabolic switch that maintains energy
homeostasis (18,19). Once activated, AMPK suppresses cell
proliferation in tumor as well as non-tumor cells, through
regulation of the cell cycle, apoptosis, autophagy, and inhibition
of protein synthesis (20–25). Current evidence supports the
alteration of AMPK levels in various cancers, implicating AMPK as a
potential target for prevention and/or treatment of cancer,
specifically HCC (26–28). Several studies have also
demonstrated that activation of AMPK induces apoptosis in many
human cancer cells, including HCC cells (29–31).
This finding indicates that AMPK plays a critical role in HCC and
that its loss can contribute to the progression of HCC. Indeed, a
variety of AMPK activators show anticancer effects (27,32,33).
For example, metformin, a common diabetes drug, shows anticancer
effects against HCC by activation of AMPK (27,34–36).
Similarly, a number of medicinal herb extracts exert anticancer
effects through activation of AMPK-dependent cell death pathways
(29,31,37–40).
Recently, interest has been increasing in
traditional herbs, with pharmacological activity confirmed by
traditional medicine, as new therapies for diseases like cancer
that are difficult to treat (41,42).
One of these is the deciduous tree, Kalopanax septemlobus
(Thunb.) Koidz. (Araliacieae; common name: prickly castor oil
tree), which is widely recognized in its native region of
northeastern Asia as a treatment for rheumatic arthritis, nephritis
edema, cholera, dysentery, and neurotic pain. The extracts and/or
components that K. septemlobus possess have been confirmed
recently to have pharmacological activities ranging from
anti-oxidant, anti-inflammatory, and hypoglycemic functions to
regulation of neurite outgrowth (43–46).
The molecular mechanisms underlying the effects of K.
septemlobus extracts appear to involve apoptosis, but the
precise pathways have not been elucidated to date. In the present
study, we evaluated whether an ethanol extract of K.
septemlobus leaves (EEKS) could inhibit the growth of HepG2
human HCC cells. We found that EEKS triggered caspase-dependent
apoptosis through activation of both intrinsic and extrinsic
pathways. We also investigated the potential roles for and
underlying mechanisms of AMPK signaling in mediating EEKS-induced
apoptosis in HepG2 cells.
Materials and methods
Preparation of EEKS
The leaves of K. septemlobus were obtained
from Gurye Wild Flower Institute (Gurye, Korea) and authenticated
by Professor S.H. Hong, Department of Biochemistry, Dongeui
University College of Korean Medicine (Busan, Korea). The dried
leaves (50 μg) were cut into small pieces, ground into a fine
powder, and then soaked with 500 ml 70% ethanol (500 ml) for 2
days. The extracted liquid was filtered twice through Whatman No. 3
filter paper to remove any insoluble materials and was then
concentrated using a rotary evaporator (Rikakikai Co., Tokyo,
Japan). The resulting extract (EEKS) was dissolved in
dimethylsulfoxide (DMSO; Sigma-Aldrich Co., St. Louis, MO, USA) to
a final concentration of 200 mg/ml (extract stock solution) and was
subsequently diluted with medium to the desired concentration prior
to use.
Cell culture
HepG2 HCC cells and Chang liver cells (an
immortalized non-tumor cell line derived from normal liver tissue)
were purchased from the American Type Culture Collection (Manassas,
MD, USA). The cells were cultured at 37°C in humidified air with 5%
CO2 in RPMI-1640 medium (Gibco-BRL, Gaithersburg, MD,
USA), supplemented with 10% fetal bovine serum (FBS, Gibco-BRL),
100 U/ml of penicillin, and 100 mg/ml of streptomycin
(Sigma-Aldrich Co.), with or without added EEKS.
Cell viability assay
Cells were incubated with EEKS, with or without
z-Val-Ala-Asp-fluoromethylketone (z-VAD-fmk, a caspase-3 inhibitor;
Calbiochem, San Diego, CA, USA) or dorsomorphin dihydrochloride
(compound C, an AMPK inhibitor; Tocris Bioscience, Bristol, UK) at
serial concentrations. The culture medium was then aspirated and
the cells were washed with phosphate-buffered saline (PBS),
followed by incubation with 0.5 mg/ml of
3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT,
Sigma-Aldrich Co.) at 37°C for 2 h. After incubation, the
supernatant was removed and the cells were treated with DMSO to
dissolve the formazan reaction product. The concentration of
formazan was determined by measuring the absorbance at 540 nm using
an enzyme-linked immunosorbent assay (ELISA) reader (Molecular
Devices, Sunnyvale, CA, USA).
Fluorescence microscopy examination of
apoptosis
Apoptosis was assessed by washing the cells with
PBS, followed by fixation in 3.7% paraformaldehyde (Sigma-Aldrich
Co.) in PBS for 10 min at room temperature. The fixed cells were
washed, permeabilized with 0.2% Triton X-100 in PBS for 10 min at
room temperature, and then incubated with 2.5 μg/ml
4,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich Co.) for 10 min
at room temperature. The cells were washed twice with PBS and
images were then captured using a fluorescence microscope (Carl
Zeiss, Jena, Germany).
Detection of apoptosis by DNA laddering
assay
The control and EEKS-treated cells were harvested
and washed with PBS, and the pellets were lysed for 1 h on ice in
lysis buffer containing 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM
trypsin-ethylenediaminetetraacetic acid (EDTA), and 0.5% Triton
X-100. Following centrifugation at 19,000 x g for 30 min at 4°C,
DNA in the supernatant was extracted in an equal volume of neutral
phenol : chloroform : isoamylalcohol (25:24:1, v/v/v, Sigma-Aldrich
Co.). Equal volumes of DNA samples were then separated by
electrophoresis in 1.5% agarose gel at 40 V, stained with 0.1 μg/ml
ethidium bromide (EtBr, Sigma-Aldrich Co.), and visualized under
ultraviolet light.
DNA flow cytometric detection of
apoptosis
After treatment with EEKS, the cells were harvested,
washed twice with ice-cold PBS, and fixed with 75% ethanol at 4°C
for 30 min. The cells were then stained with 5 μl Annexin
V-fluorescein isothiocyanate (FITC) (R&D Systems, Minneapolis,
MN, USA) and 5 μl propidium iodide (PI, Sigma-Aldrich Co.). The
samples were incubated for 15 min at room temperature in the dark,
and then the degree of apoptosis was quantified by flow cytometry
as a percentage of the Annexin V-positive and PI-negative (Annexin
V+/PI− cells) cells (Becton-Dickinson, San
Jose, CA, USA).
Protein extraction and western blot
analysis
After treatment with different concentrations of
EEKS, the cells were lysed in buffer containing 40 mM Tris (pH
8.0), 120 mM NaCl, 0.5% Nonidet P-40, 0.1 mM sodium orthovanadate,
2 μg/ml aprotinin, 2 μg/ml leupeptin and 100 μg/ml
phenymethylsulfonyl fluoride. In a parallel experiment, the
mitochondrial and cytosolic fractions were isolated using a
mitochondrial fractionation kit (Active Motif, Carlsbad, CA, USA)
according to the manufacturer's instructions. The Bio-Rad protein
assay (Bio-Rad, Hercules, CA, USA) was used according to the
manufacturer's instructions to determine the protein
concentrations. For western blot assays, equal amounts of protein
(30–50 μg/lane) were subjected to electrophoresis on sodium dodecyl
sulfate (SDS)-polyacrylamide gels, and then transferred onto
nitrocellulose membranes (Schleicher and Schuell, Keene, NH, USA).
The membranes were blocked with Tris-buffered saline (10 mM
Tris-Cl, pH 7.4) containing 0.5% Tween-20 and 5% non-fat dry milk
for 1 h at room temperature and incubated with the primary
antibodies overnight. Membranes were then washed with PBS and
incubated with the secondary antibody conjugated to horseradish
peroxidase (Amersham Co., Arlington Heights, IL, USA) for 1 h at
room temperature. Immunoreactivity was detected by using the
enhanced chemiluminescence (ECL) western blotting detection system
(Amersham Co.) according to the manufacturer's instructions. The
primary antibodies used in this study were as follows: actin
(1:1,000, sc-7120; rabbit polyclonal, Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA), caspase-3 (1:1,000, sc-7272; mouse
monoclonal, Santa Cruz Biotechnology, Inc.), caspase-8 (1:1,000,
sc-7890; rabbit polyclonal, Santa Cruz Biotechnology, Inc.),
caspase-9 (1:1,000, sc-7885; rabbit polyclonal, Santa Cruz
Biotechnology, Inc.), PARP (1:1,000, sc-7150; rabbit polyclonal,
Santa Cruz Biotechnology, Inc.), XIAP (1:1,000, sc-11426; rabbit
polyclonal, Santa Cruz Biotechnology, Inc.), cIAP-1 (1:1,000,
sc-7943; rabbit polyclonal, Santa Cruz Biotechnology, Inc.), cIAP-2
(1:1,000, sc-7944; rabbit polyclonal, Santa Cruz Biotechnology,
Inc.), TRAIL (1:500, sc-7877; rabbit polyclonal, Santa Cruz
Biotechnology, Inc.), DR4 (1:1,000, sc-7863; rabbit polyclonal,
Santa Cruz Biotechnology, Inc.), DR5 (1:1,000, sc-65314; mouse
monoclonal, Santa Cruz Biotechnology, Inc.), Fas (1:1,000, sc-715;
rabbit polyclonal, Santa Cruz Biotechnology, Inc.), FasL (1:1,000,
sc-957; rabbit polyclonal, Santa Cruz Biotechnology, Inc.), Bcl-2
(1:1000, sc-509; mouse monoclonal, Santa Cruz Biotechnology, Inc.),
Bax (1:1,000, sc-493; rabbit polyclonal, Santa Cruz Biotechnology,
Inc.), Bid (1:500, sc-11423; rabbit polyclonal, Santa Cruz
Biotechnology, Inc.), cytochrome c (1:500, sc-7159; rabbit
polyclonal, Santa Cruz Biotechnology, Inc.), COX IV (1:1,000,
sc-376731; mouse monoclonal, Santa Cruz Biotechnology, Inc.), AMPK
(1:1,000, sc-25792; rabbit polyclonal, Santa Cruz Biotechnology,
Inc.), p-AMPK (1:500, 2535; rabbit polyclonal, Cell Signaling
Technology, Inc., Boston, MA, USA), ACC (1:1,000, sc-30212; rabbit
polyclonal, Santa Cruz Biotechnology, Inc.) and p-ACC (1:500, 3661;
rabbit polyclonal, Cell Signaling Technology, Inc.)
Measurement of mitochondrial membrane
potential (MMP, Δψm)
The MMP values were determined using the
dual-emission potential-sensitive probe,
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide
(JC-1, Sigma-Aldrich Co.), which is internalized and concentrated
by respiring mitochondria and can therefore reflect MMP changes in
cells. Briefly, cells were fixed and permeabilized with 0.2% Triton
X-100 in PBS for 10 min at room temperature, and then incubated
with 10 μM JC-1 for 30 min at 37°C in the dark. Subsequently, the
cells were washed with PBS to remove unbound dye, and the amount of
JC-1 retained by 10,000 cells per sample was measured at 488 and
575 nm using a flow cytometer.
Caspase activity assay
The activities of the caspases were assessed by
marking cells with colorimetric assay kits (R&D Systems), which
utilize synthetic tetrapeptides (Asp-Glu-Val-Asp (DEAD) for
caspase-3; Ile-Glu-Thr-Asp (IETD) for caspase-8; and
Leu-Glu-His-Asp (LEHD) for caspase-9) labeled with p-nitroaniline
(pNA) that is linked to the end of the caspase-specific substrate.
Briefly, the cells were washed with PBS and lysed in the supplied
lysis buffer, according to the manufacturer's instructions. The
supernatants were collected and incubated at 37°C for 2 h in the
dark with the supplied reaction buffer containing dithiothreitol
and DEAD-pNA, IETD-pNA, or LEHD-pNA as substrates. The reactions
were measured by changes in absorbance at 405 nm using an ELISA
reader.
Statistical analysis of data
The experiments were repeated three times, and the
results were expressed as means ± standard deviation (SD). A
one-way analysis of variance (ANOVA), followed by Dunnett's t-test,
was applied to assess the statistical significance of the
difference among the study groups. A value of p<0.05 was
considered to be statistically significant.
Results
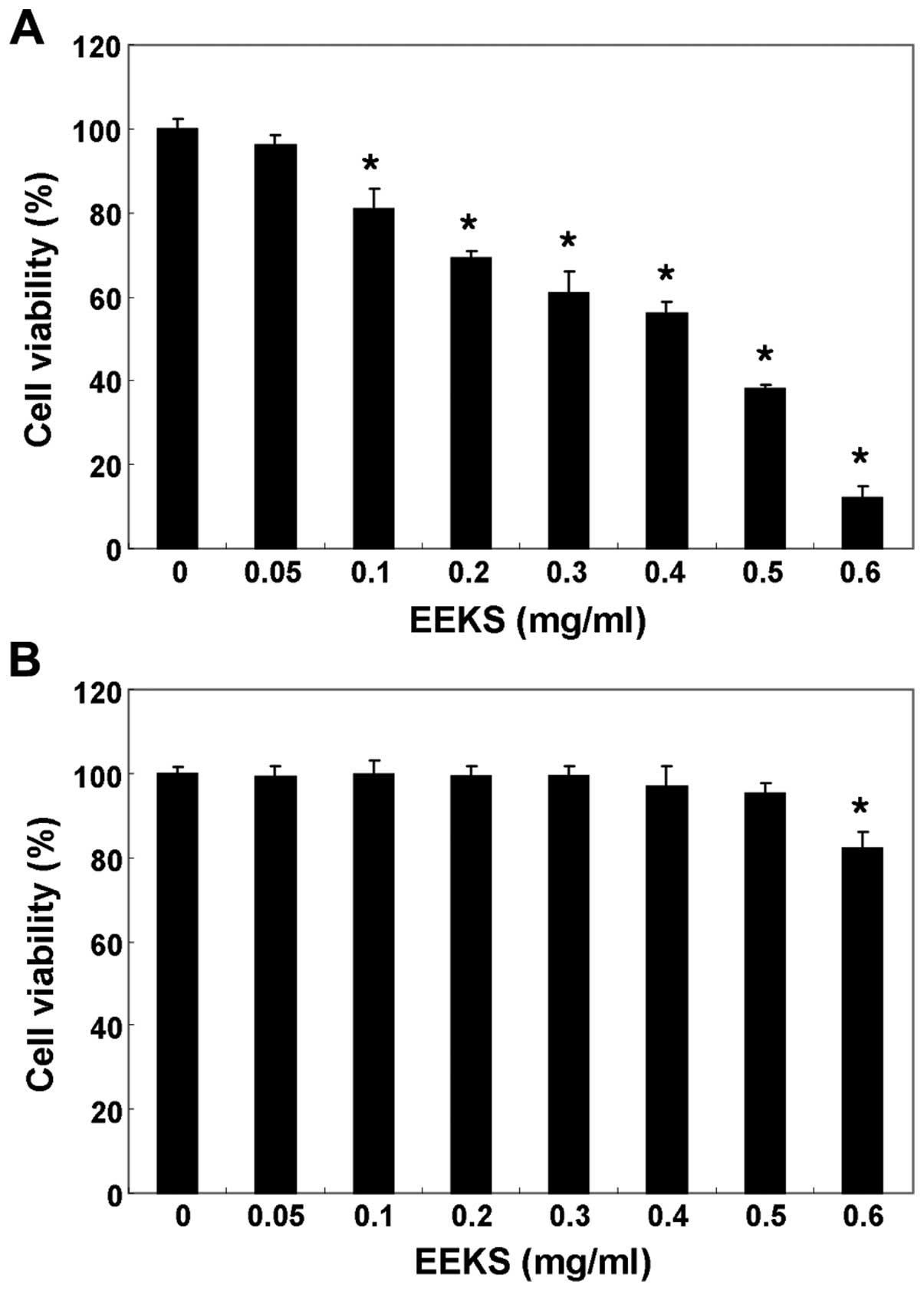
EEKS treatment suppresses the viability
of HepG2 cells
The effects of EEKS on cell viability were evaluated
by stimulating the HepG2 cells with various concentrations of EEKS
for 24 h, and performing an MTT assay. As shown in Fig. 1A, EEKS caused significant
concentration-dependent decreases in cell viability in HepG2 cells.
The results of an additional experiment using Chang liver cells,
conducted to examine the effect of EEKS on the proliferation of
normal cells, are shown in Fig.
1B. EEKS concentrations ≤500 μmg/ml did not induce
cytotoxicity, whereas 600 μg/ml EEKS significantly reduced cell
viability. Therefore, in this study, conditions for subsequent
experiments were chosen that induced no cytotoxicity in Chang liver
cells.
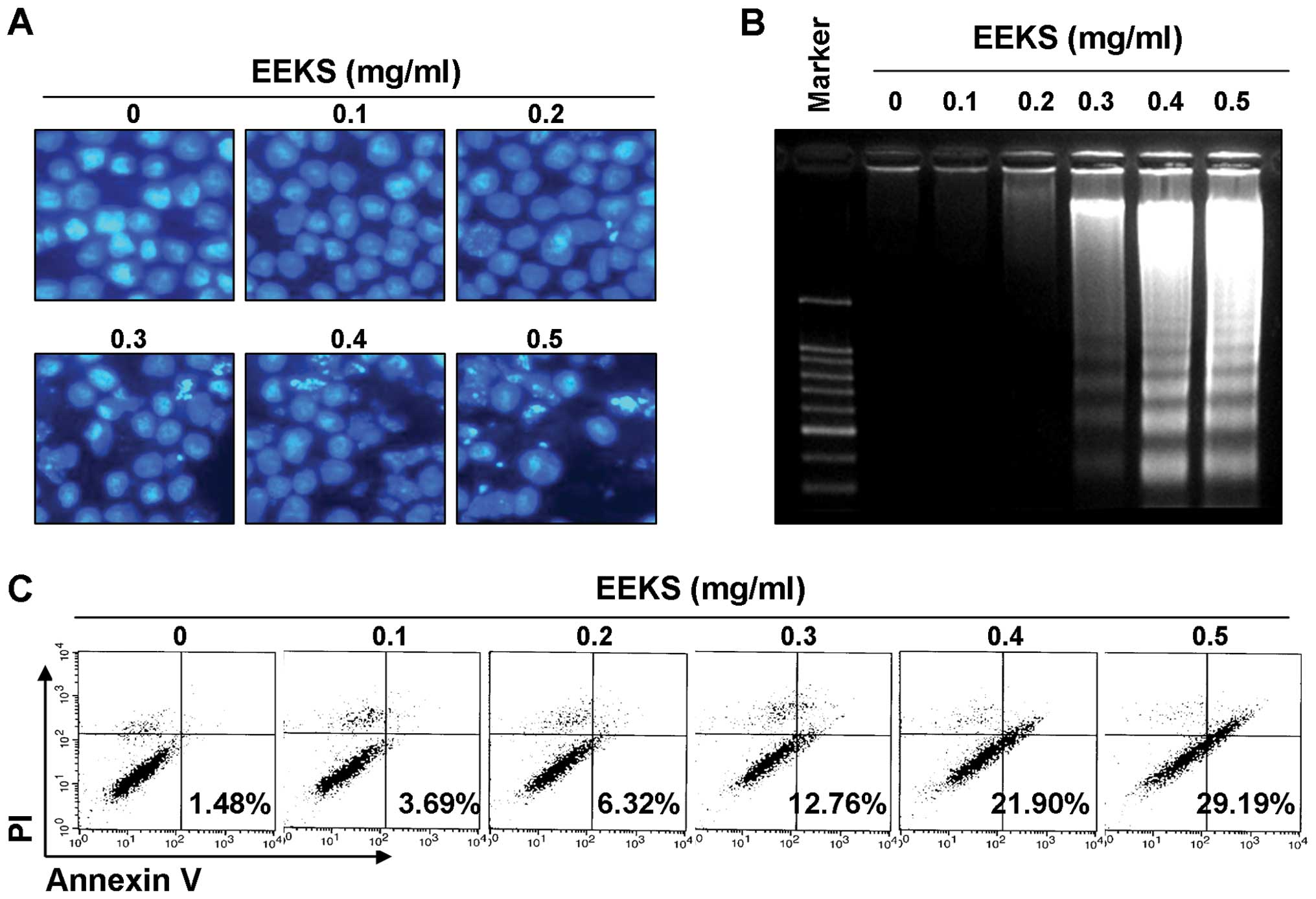
EEKS treatment enhances apoptosis in
HepG2 cells
The possibility that the growth inhibitory activity
of EEKS was caused by an induction of apoptosis was examined based
on three established criteria for assessment of apoptosis. First,
morphological changes in HepG2 cells were determined using DAPI
staining, as shown in Fig. 2A. A
significant number of cells showed chromatin condensation, loss of
nuclear construction, and formation of apoptotic bodies following
EEKS treatment, whereas these features were not observed in control
cells. Second, treatment with EEKS induced progressive accumulation
of fragmented DNA in a concentration-dependent manner, which
appeared as a typical ladder pattern of DNA fragmentation, another
hallmark of apoptosis (Fig. 2B).
In addition, flow cytometry analysis confirmed an increased
accumulation of Annexin V-positive cells following EEKS treatment,
indicating a greater degree of apoptosis in cells treated with EEKS
(Fig. 2C). These results suggested
that EEKS-induced apoptosis in HepG cells contributed to its
anti-proliferative and cytotoxic effects.
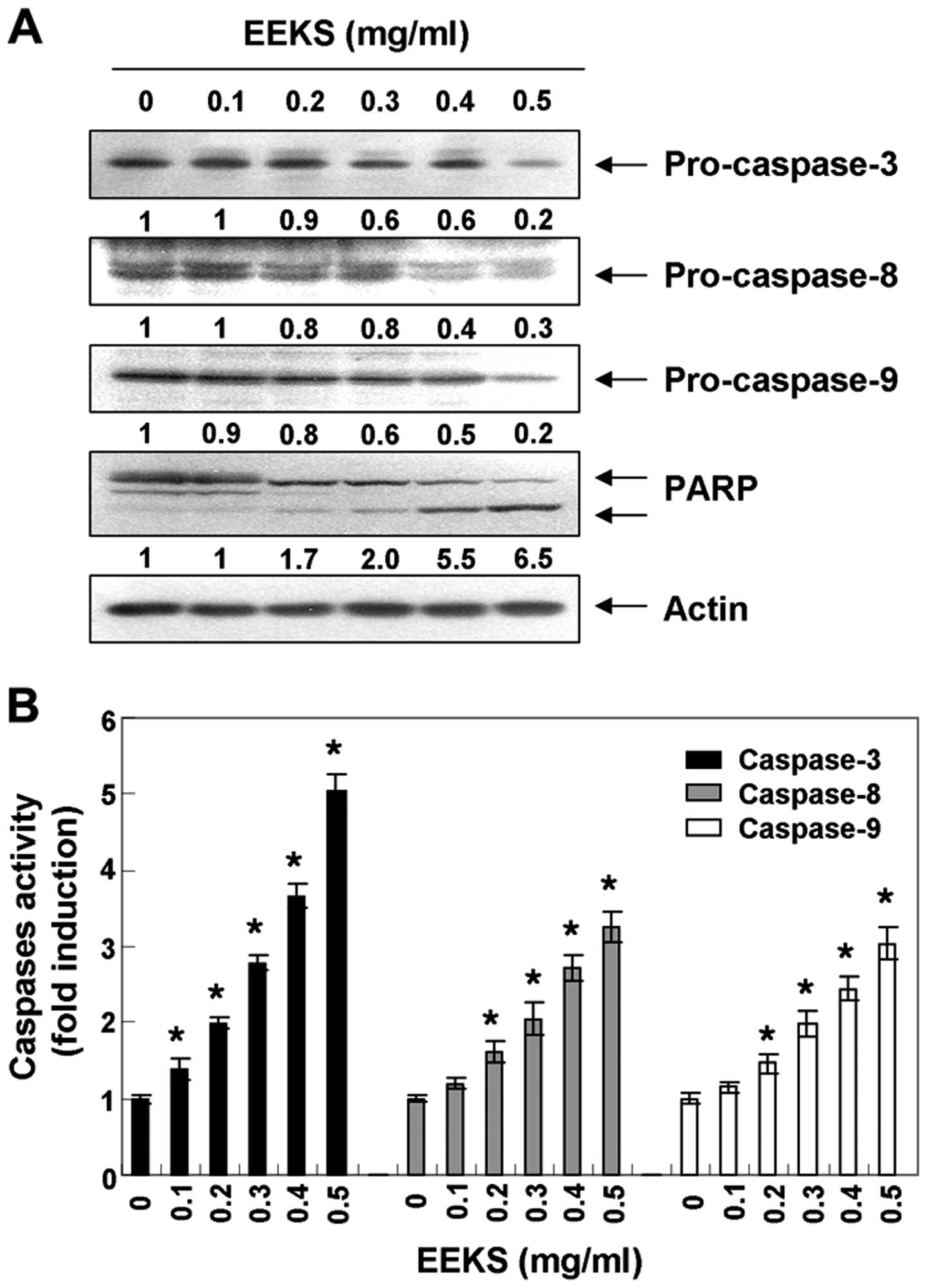
EEKS-induced apoptosis is
caspase-dependent in HepG2 cells
The molecular mechanism of EEKS-induced apoptosis in
HepG2 cells was examined by determining whether EEKS affected
activation of caspase cascades-key executioners of apoptosis.
Western blot analyses revealed that the expression levels of
pro-caspase-8 and -9, upstream activators of caspase-3/-7 in the
extrinsic and intrinsic pathways, respectively, were downregulated
in a concentration-dependent manner (Fig. 3A). In addition to the reduced
expression of pro-caspase-8 and -9, the level of pro-caspase-3 was
also concentration-dependently reduced in response to EEKS
treatment, and correspondingly, EEKS treatment led to progressive
proteolytic cleavage of PARP (116-kDa), a known caspase-3 substrate
(47), to yield an 85-kDa cleaved
fragment. In addition, equal amounts of proteins from lysates from
cells treated with EEKS were assayed to assess in vitro
caspase activity. As shown in Fig.
3B, treatment with EEKS significantly increased the activity of
caspase-3, -8 and -9.
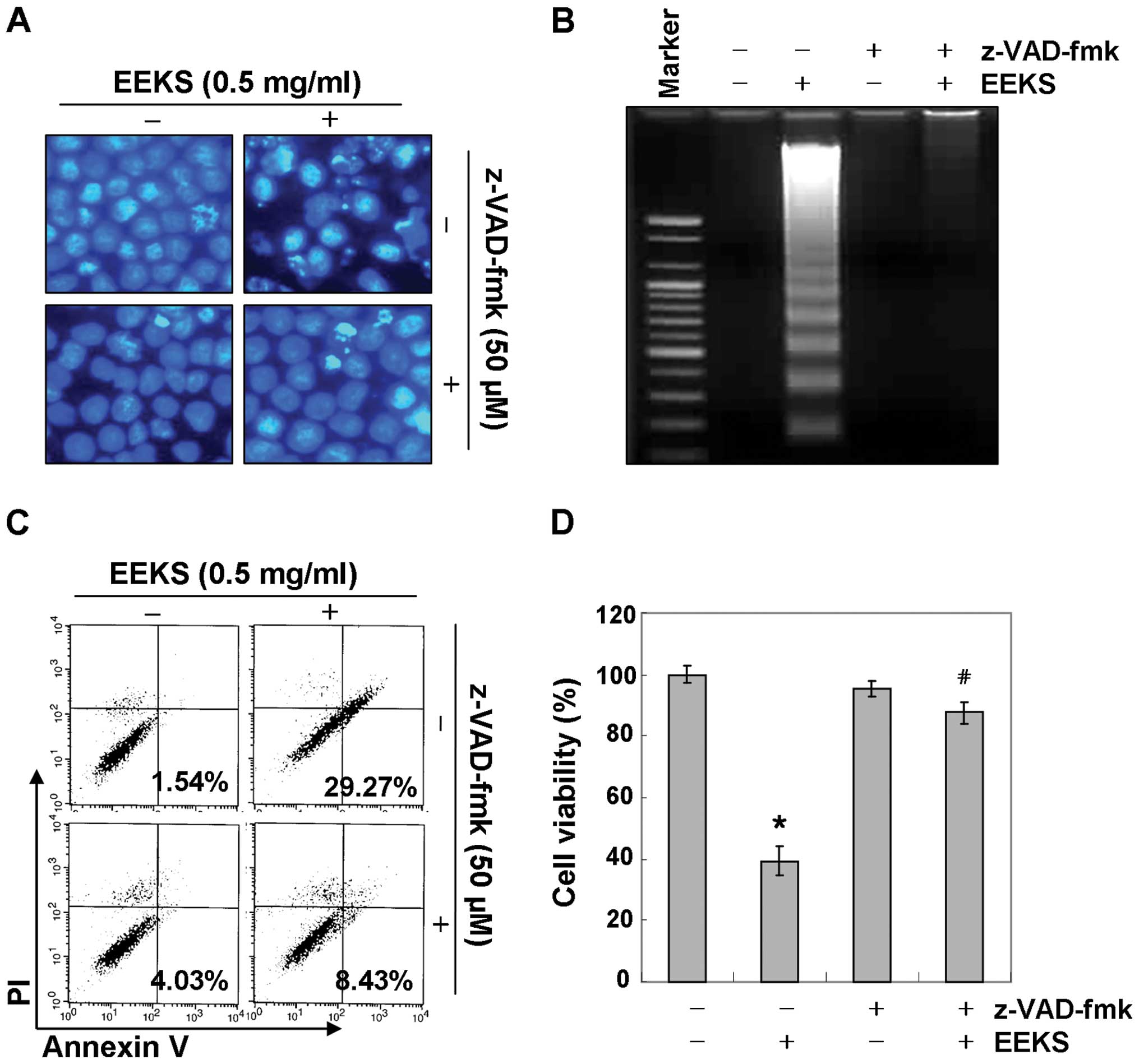
We further confirmed the involvement of activation
of caspases in the apoptosis induced by EEKS by pretreating the
cells with a pan-caspase inhibitor, z-VAD-fmk, for 1 h, followed by
EEKS treatment. As shown in Fig. 4A
and B, pre-treatment with z-VED-fmk significantly prevented the
appearance of apoptotic features such as chromatin condensation and
DNA fragmentation. Flow cytometry analysis and MTT assay also
revealed that the pan-caspase inhibitor suppressed EEKS-induced
apoptosis and reduction in viability in HepG2 cells (Fig. 4C and D). These results clearly
indicate that EEKS-triggered apoptosis may be mediated through
caspase-dependent extrinsic and intrinsic pathways in HepG2 cells.
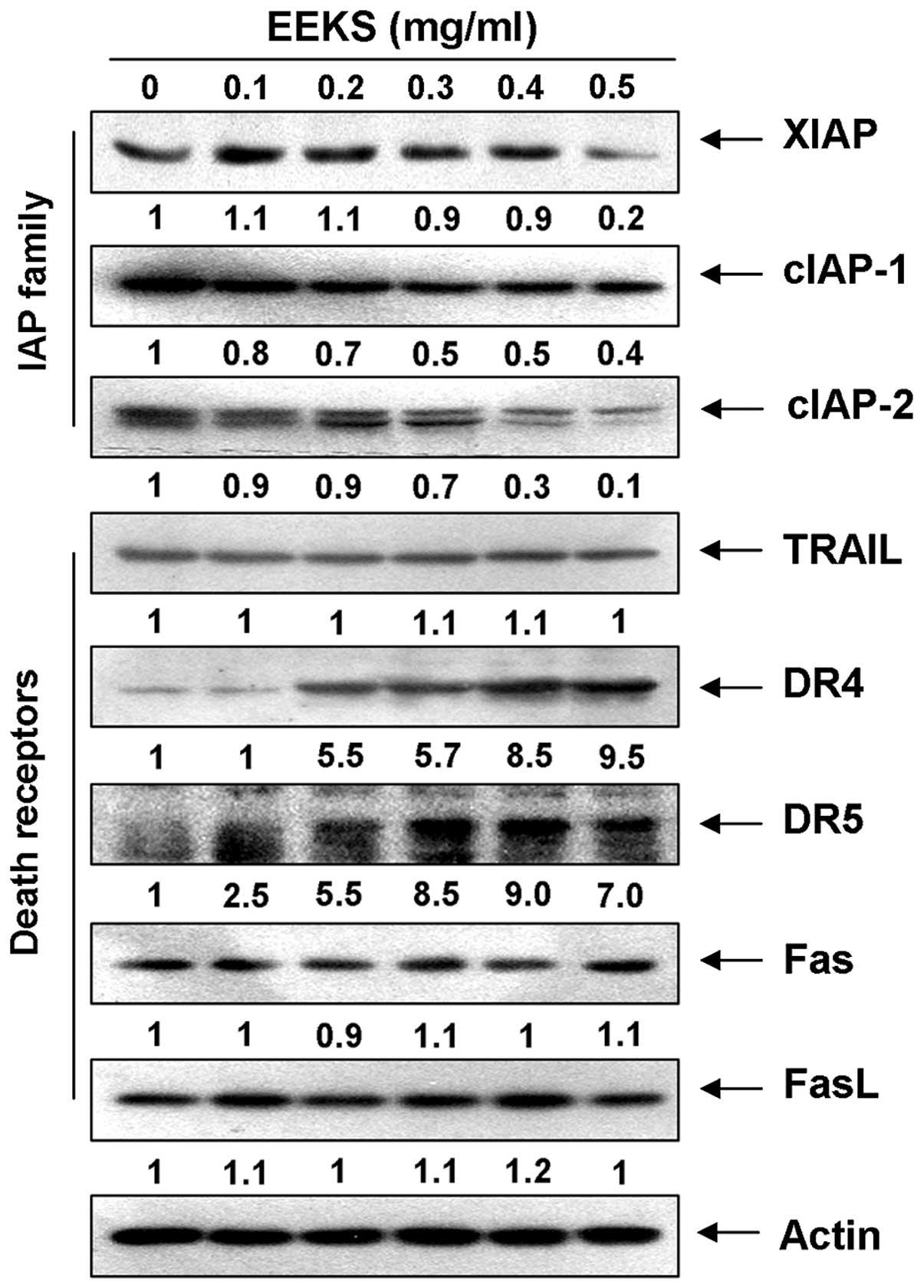
In addition, EEKS treatment reduced the expression of members of
the inhibitor of the apoptosis proteins (IAP) family, such as XIAP,
cIAP-1, and cIAP-2 (Fig. 5), which
bind to caspases and lead to their inactivation (48,49).
This finding indicated that EEKS-induced activation of caspases was
associated with a reduction in IAP family proteins.
EEKS treatment induces the expression of
death receptor (DR)-related proteins and truncation of Bid in HepG2
cells
We further explored the induction of apoptosis of
HepG2 cells by EEKS by detecting the expression levels of
DR-mediated apoptosis regulators and Bid, a BH3 pro-apoptotic
protein, which might be involved in the regulation of extrinsic
apoptosis pathway. Western blot analysis revealed that the levels
of DR ligands, such as the tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL) and Fas ligand (FasL), were
relatively unchanged in response to EEKS treatment; however, the
levels of DR4 and DR5, but not Fas, increased markedly in a
concentration-dependent manner (Fig.
3). A subsequent immunoblotting analysis revealed that exposure
of HepG2 cells to EEKS caused a progressive accumulation of
truncated Bid (tBid) (Fig. 4A),
presumably resulting from truncation by activated caspase-8. These
results indicate that the cytotoxic effects induced by EEKS could
be mediated through DR-mediated apoptosis, thereby accentuating
cross-talk between the intrinsic and extrinsic apoptosis
pathways.
EEKS treatment activates the
mitochondria-mediated intrinsic apoptosis pathway in HepG2
cells
We next evaluated the effect of EEKS on the
mitochondrial apoptosis signaling pathway. Notably, the total
levels of pro-apoptotic Bax and anti-apoptotic Bcl-2 proteins
remained unchanged in response to EEKS treatment (Fig. 6A); however, EEKS treatment
decreased the cytosolic levels of Bax, whereas its mitochondrial
levels were increased significantly after treatment with increasing
concentrations of EEKS (Fig. 6B).
An indicative stage in the intrinsic apoptotic pathway is the
depolarization of the mitochondrial membrane and the subsequent
increase in permeability of the outer membrane, followed by pore
formation and the release of proapoptotic molecules including
cytochrome c into cytosol (50,51).
The present results demonstrated that treatment with increasing
concentrations of EEKS significantly reduced the MMP levels
(Fig. 6C), suggesting
depolarization of the mitochondria by EEKS. Further
characterization of the mitochondrial-mediated apoptotic effect of
EEKS, by analyzing the release of cytochrome c, revealed
that EEKS treatment promoted a concentration-dependent increase in
the release of cytochrome c from mitochondria into the
cytosol (Fig. 4B), suggesting that
EEKS also activated the mitochondria-mediated intrinsic apoptosis
pathway in HepG2 cells.
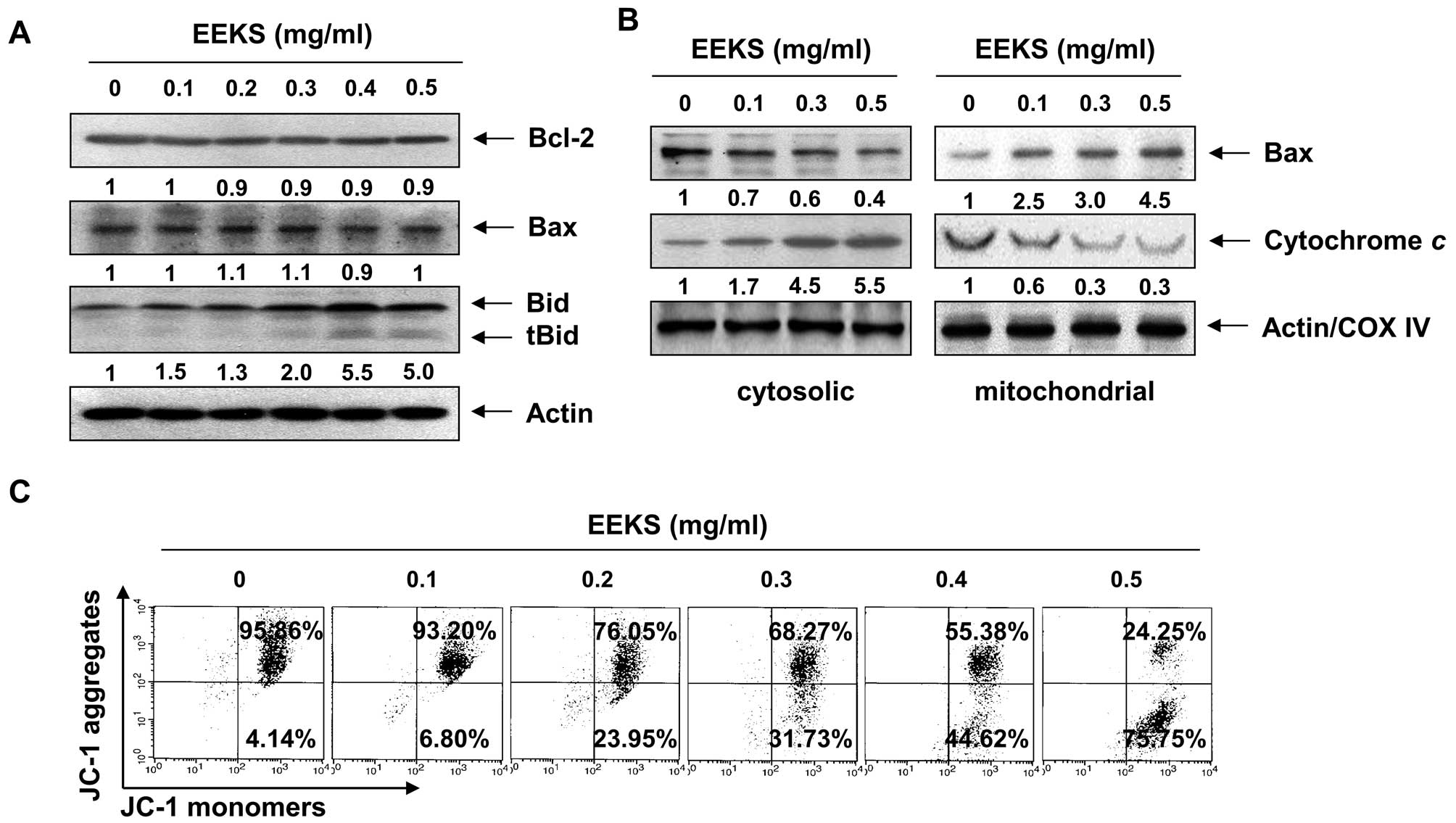 | Figure 6Effects of EEKS treatment on the
levels of Bcl-2 family proteins and cytochrome c, and on the
MMP values in HepG2 cells. The cells were treated with the
indicated concentrations of EEKS for 24 h. (A and B) The total
cellular proteins (A) or cytosolic and mitochondrial proteins (B)
were extracted and separated by SDS-polyacrylamide gel
electrophoresis, followed by western blot analysis using the
indicated antibodies and an enhanced chemiluminescence (ECL)
detection system. Actin and cytochrome c oxidase subunit 4
(COX IV) were used as internal controls for the cytosolic and
mitochondrial fractions, respectively. The relative ratios of
expression in the results of western blotting are presented at the
bottom of each result as relative value of actin or COX IV
expression. (C) The cells were collected and incubated with 10 μM
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl-imidacarbocyanine iodide
(JC-1) for 20 min at 37°C in the dark. The cells were then washed
with PBS, and the mean JC-1 fluorescence intensity was detected
using a flow cytometer. The data represent the means of two
independent experiments. |
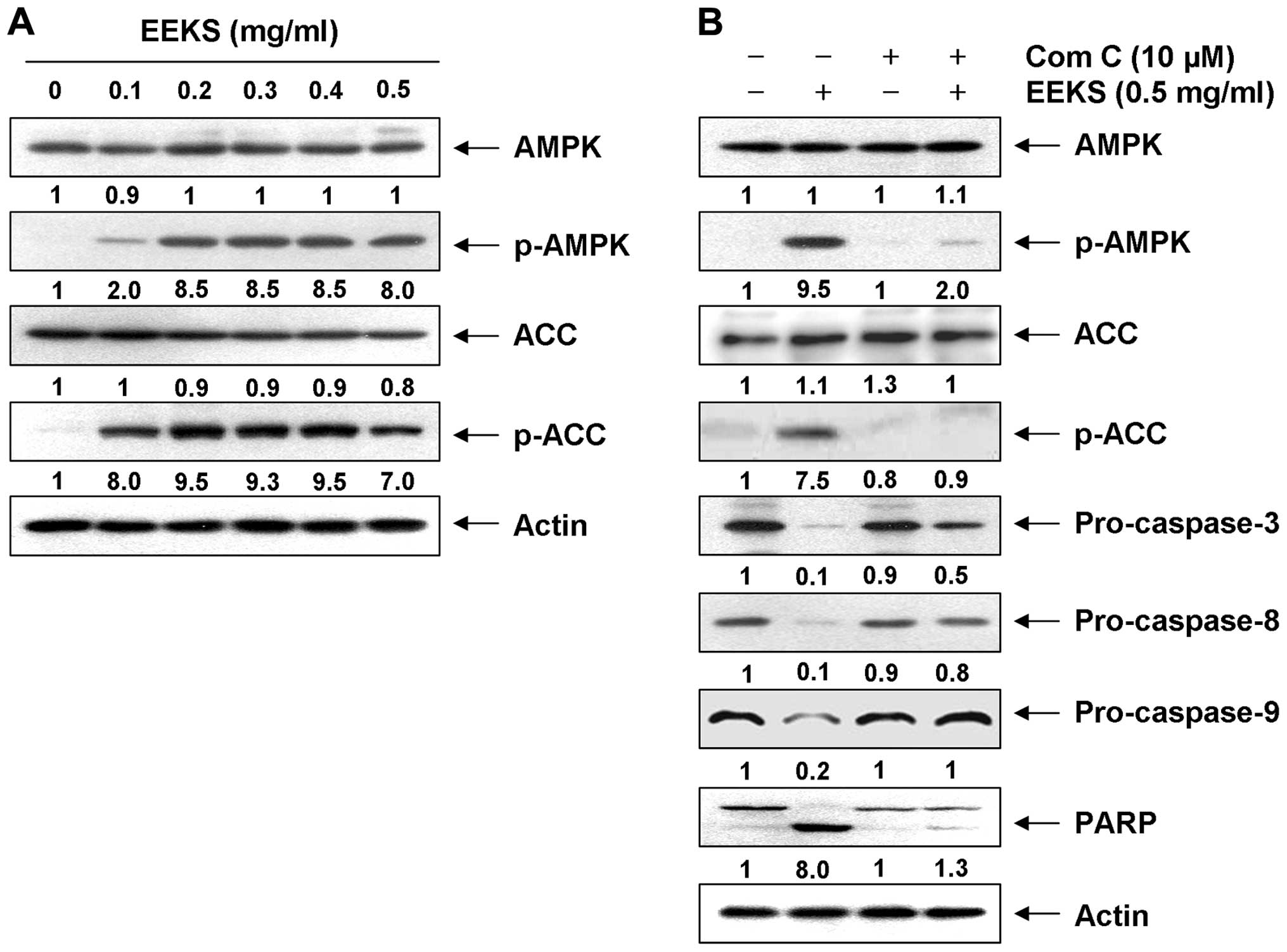
EEKS treatment induces apoptosis and
lowers cell viability of HepG2 cells via AMPK activation
Several reports have demonstrated that activation of
AMPK leads to the induction of apoptosis in numerous human cancer
cell types (29–31). We therefore investigated whether
the phosphorylation of AMPK is induced by EEKS. As demonstrated in
Fig. 7A, when compared with the
basal level, EEKS treatment led to increased levels of
phosphorylation of AMPK (Thr172) in a concentration-dependent
manner, without inducing significant changes in the total protein
levels. Consistent with the activation of AMPK, the phosphorylation
of acetyl-CoA carboxylase (ACC) at Ser79 (the best-characterized
site phosphorylated by AMPK) (52)
also increased after EEKS administration, indicating that the AMPK
pathway in HepG2 cells was activated in the presence of EEKS.
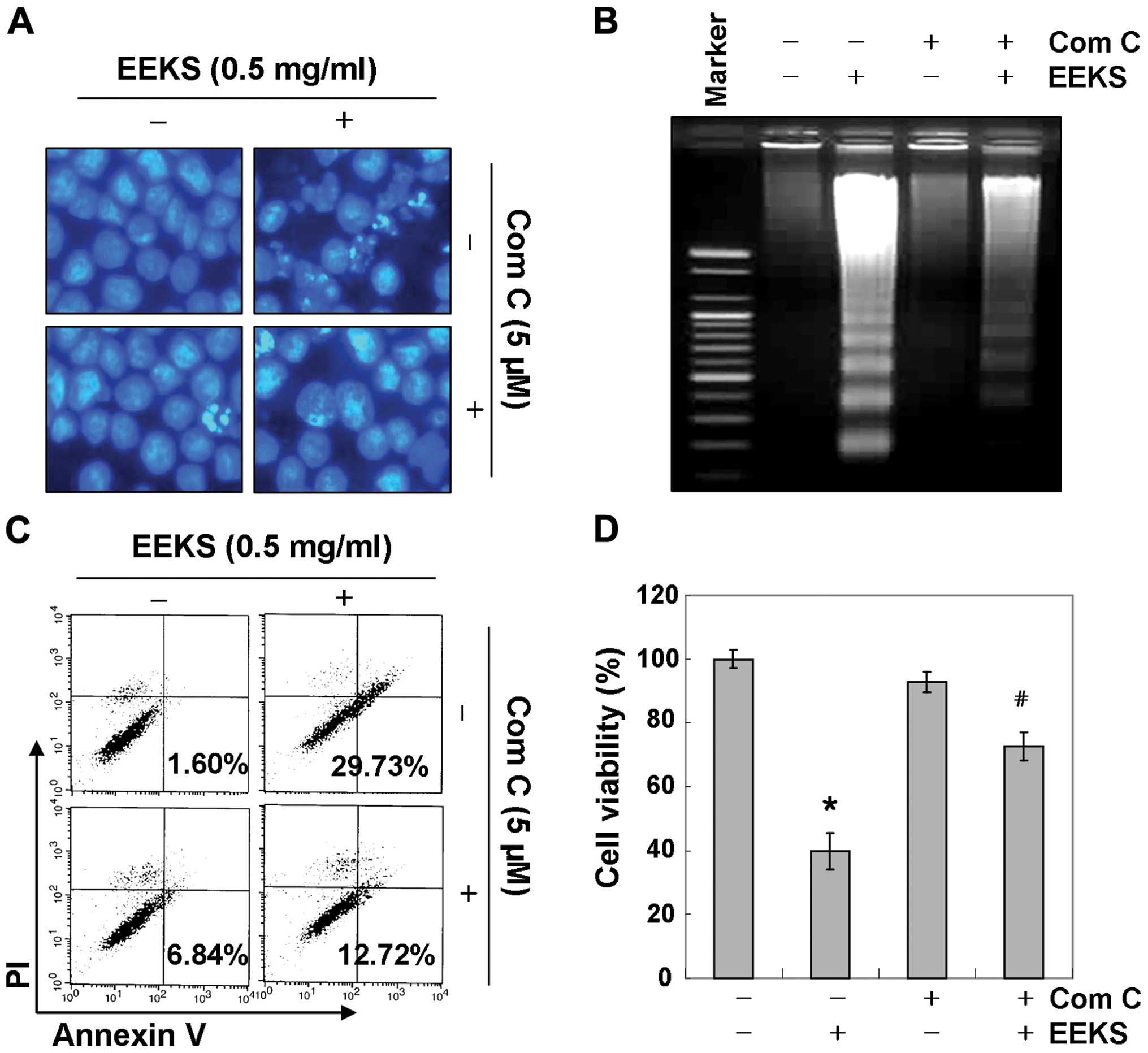
We further confirmed the relationship between
apoptosis induction and AMPK activation in EEKS-stimulated HepG2
cells by examining the effects of compound C, a specific AMPK
inhibitor. As shown in Fig. 7B,
treatment with compound C blocked phosphorylation of AMPK as well
as ACC and, interestingly, prevented cleavage of caspases
(caspase-3, -8 and -9) and degradation of PARP in EEKS-treated
HepG2 cells, implying a linkage between caspase activation and AMPK
activation. In line with these observations, results in Fig. 8 demonstrated that AMPK inhibition
significantly reduced the increase in apoptosis induced by EEKS
(Fig. 8A–C) and the cell viability
loss (Fig. 8D). These results
suggested that EEKS-induced apoptosis and loss of viability were
mediated by activation of AMPK, and that AMPK was probably upstream
of caspase activation in the signaling pathway involved in this
process.
Discussion
Apoptosis is an essential biological process for
maintaining cell stability, structure, and function, and for proper
development. Therefore, induction of apoptosis in target cells is
an ideal strategy for anticancer therapy. In this process, caspase-
and mitochondria-mediated apoptosis coordinately occur in response
to a wide range of death stimuli (15,53).
The caspase-8-mediated extrinsic apoptotic pathway and the
tBid-mediated intrinsic apoptotic pathway can both contribute, in
widely varying degrees, to death receptor-induced apoptosis
(11,54). Our data revealed that EEKS promoted
the expression of DR4 and DR5, activation of caspase-8 and -3, and
concomitant PARP cleavage, together with a rise in tBid. Therefore,
the data indicate that the extrinsic pathway contributes, at least
in part, to the observed EEKS-induced apoptosis in HepG2 cells.
In addition to the extrinsic pathway, we also found
that EEKS activated critical regulatory elements of the intrinsic
mitochondrial executioner pathway, indicated by the loss of the
MMP, translocation of Bax from the cytoplasm to mitochondria,
release of cytochrome c from mitochondria into the cytosol,
and activation of caspase-9. These data suggest that Bax was
oligomerized and anchored onto the outer mitochondrial membrane,
thereby forming mitochondrial permeability transition pores that
disrupted the MMP. Bax translocation could therefore be related to
the mitochondrial response to the generation of tBid, which led to
mitochondrial disturbance, release of cytochrome c, and
activation of caspase-9 to intensify the initial apoptotic
response. However, pretreatment with the pan-caspase inhibitor,
z-VAD-fmk, significantly attenuated EEKS-induced cell death and
growth inhibition. Although the existence of a direct link between
effects of EEKS on the intrinsic and extrinsic apoptosis pathways
is unclear, our results indicate that EEKS appears to induce
apoptosis in HepG2 cells primarily via a caspase-dependent action
on both intrinsic and extrinsic apoptotic pathways.
Recently, activation of the AMPK signaling pathway
has been shown to play a critical role in regulation of energy
metabolism under both physiological and pathological conditions via
downstream pathways relevant to the control of cellular
proliferation (16,17). AMPK is known to function as an
intracellular energy sensor, being activated in response to an
increased AMP/ATP ratio, a condition of energetic stress, to
promote the catabolic pathway, thereby inhibiting cell
proliferation (18,19). Growing evidence suggests that AMPK
is dysregulated in most cancer tissues, including HCC, when
compared to normal tissues (26–28),
and several studies have shown that AMPK activators exhibit
inhibitory effects on HCC growth (21,26,27,29–31,36).
Scientific interest continues to grow in the search for molecular
pathways and novel compounds that target AMPK signaling as new
potential therapeutic options for HCC. The results of the present
study demonstrated that EEKS treatment of HepG2 cells promoted a
concentration-dependent phosphorylation of AMPK and of ACC its
downstream target.
We examined the action of EEKS on AMPK activation
using the synthetic AMPK inhibitor compound C. As expected, the
ability of EEKS to stimulate the phosphorylation of both AMPK and
ACC was diminished markedly by compound C treatment, suggesting
that EEKS could be an AMPK activator. Moreover, the inhibition of
AMPK in HepG2 cells attenuated the EEKS-induced activation of
caspases and prevented PARP cleavage. In addition, the induction of
apoptosis and reduction in cell viability by EEKS was prevented by
compound C pretreatment, strongly suggesting that EEKS activated
AMPK, which led to caspase-dependent cell apoptosis in HepG2
cells.
In conclusion, the present study revealed that EEKS
triggers apoptosis of human HCC HepG2 cells through activation of
both the intrinsic caspase pathway and the extrinsic pathway. The
resulting activation of caspases is responsible for the mediation
of EEKS-induced apoptosis. In addition, we propose that
EEKS-mediated apoptosis is caused by activation of AMPK, which may
contribute to the induction of caspase-dependent HepG2 cell
apoptosis. Our findings indicate that AMPK activation plays a
pivotal role in EEKS-induced apoptosis of HepG2 cells, but further
studies are needed to pinpoint the active compounds in the EEKS.
Identification of the underlying molecular mechanisms and studies
confirming the biological efficacy of EEKS using in vivo
models are also required.
Acknowledgements
This study was supported by Basic Science Research
Program through the National Research Foundation of Korea (NRF)
grant funded by the Korea government (2013 041811,
2015R1A2A2A01004633 and 2014R1A1A1008460).
References
|
1
|
Kao JH and Chen DS: Changing disease
burden of hepatocellular carcinoma in the Far East and Southeast
Asia. Liver Int. 25:696–703. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Venook AP, Papandreou C, Furuse J and de
Guevara LL: The incidence and epidemiology of hepatocellular
carcinoma: A global and regional perspective. Oncologist. 15(Suppl
4): 5–13. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen CJ, Wang LY, Lu SN, Wu MH, You SL,
Zhang YJ, Wang LW and Santella RM: Elevated aflatoxin exposure and
increased risk of hepatocellular carcinoma. Hepatology. 24:38–42.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Feitelson MA, Sun B, Satiroglu Tufan NL,
Liu J, Pan J and Lian Z: Genetic mechanisms of
hepatocarcinogenesis. Oncogene. 21:2593–2604. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thorgeirsson SS and Grisham JW: Molecular
pathogenesis of human hepatocellular carcinoma. Nat Genet.
31:339–346. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ben Ari Z, Weitzman E and Safran M:
Oncogenic viruses and hepatocellular carcinoma. Clin Liver Dis.
19:341–360. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Avila MA, Berasain C, Sangro B and Prieto
J: New therapies for hepatocellular carcinoma. Oncogene.
25:3866–3884. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bruix J and Sherman M; American
Association for the Study of Liver Diseases. Management of
hepatocellular carcinoma: An update. Hepatology. 53:1020–1022.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deng GL, Zeng S and Shen H: Chemotherapy
and target therapy for hepatocellular carcinoma: New advances and
challenges. World J Hepatol. 7:787–798. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fadeel B and Orrenius S: Apoptosis: A
basic biological phenomenon with wide-ranging implications in human
disease. J Intern Med. 258:479–517. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fulda S and Debatin KM: Extrinsic versus
intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene.
25:4798–4811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lavrik IN: Systems biology of apoptosis
signaling networks. Curr Opin Biotechnol. 21:551–555. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Walczak H and Krammer PH: The CD95
(APO-1/Fas) and the TRAIL (APO-2L) apoptosis systems. Exp Cell Res.
256:58–66. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ola MS, Nawaz M and Ahsan H: Role of Bcl-2
family proteins and caspases in the regulation of apoptosis. Mol
Cell Biochem. 351:41–58. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hensley P, Mishra M and Kyprianou N:
Targeting caspases in cancer therapeutics. Biol Chem. 394:831–843.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mantovani J and Roy R: Re-evaluating the
general(ized) roles of AMPK in cellular metabolism. FEBS Lett.
585:967–972. 2011. View Article : Google Scholar
|
|
17
|
Li W, Saud SM, Young MR, Chen G and Hua B:
Targeting AMPK for cancer prevention and treatment. Oncotarget.
6:7365–7378. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Marley AE, Sullivan JE, Carling D, Abbott
WM, Smith GJ, Taylor IW, Carey F and Beri RK: Biochemical
characterization and deletion analysis of recombinant human protein
phosphatase 2C alpha. Biochem J. 320:801–806. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Oakhill JS, Scott JW and Kemp BE: AMPK
functions as an adenylate charge-regulated protein kinase. Trends
Endocrinol Metab. 23:125–132. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hardie DG: AMPK: A key regulator of energy
balance in the single cell and the whole organism. Int J Obes.
32(Suppl 4): S7–S12. 2008. View Article : Google Scholar
|
|
21
|
Lee CW, Wong LL, Tse EY, Liu HF, Leong VY,
Lee JM, Hardie DG, Ng IO and Ching YP: AMPK promotes p53
acetylation via phosphorylation and inactivation of SIRT1 in liver
cancer cells. Cancer Res. 72:4394–4404. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Han JY, Park SH, Yang JH, Kim MG, Cho SS,
Yoon G, Cheon SH and Ki SH: Licochalcone suppresses LXRα-induced
hepatic lipogenic gene expression through AMPK/Sirt1 pathway
activation. Toxicol Res. 30:19–25. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hwang WC, Kim MK, Song JH, Choi KY and Min
S: Inhibition of phospholipase D2 induces autophagy in colorectal
cancer cells. Exp Mol Med. 46:e1242014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Min B, Lee H, Song JH, Han MJ and Chung J:
Arctiin inhibits adipogenesis in 3T3-L1 cells and decreases
adiposity and body weight in mice fed a high-fat diet. Nutr Res
Pract. 8:655–661. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rehman G, Shehzad A, Khan AL and Hamayun
M: Role of AMP-activated protein kinase in cancer therapy. Arch
Pharm (Weinheim). 347:457–468. 2014. View Article : Google Scholar
|
|
26
|
Martelli AM, Chiarini F, Evangelisti C,
Ognibene A, Bressanin D, Billi AM, Manzoli L, Cappellini A and
McCubrey JA: Targeting the liver kinase B1/AMP-activated protein
kinase pathway as a therapeutic strategy for hematological
malignancies. Expert Opin Ther Targets. 16:729–742. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zheng L, Yang W, Wu F, Wang C, Yu L, Tang
L, Qiu B, Li Y, Guo L, Wu M, et al: Prognostic significance of AMPK
activation and therapeutic effects of metformin in hepatocellular
carcinoma. Clin Cancer Res. 19:5372–5380. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lv Q, Zhen Q, Liu L, Gao R, Yang S, Zhou
H, Goswami R and Li Q: AMP-kinase pathway is involved in tumor
necrosis factor alpha-induced lipid accumulation in human hepatoma
cells. Life Sci. 131:23–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu R, Zhang ZQ, Wang B, Jiang HX, Cheng L
and Shen LM: Berberine-induced apoptotic and autophagic death of
HepG2 cells requires AMPK activation. Cancer Cell Int. 14:492014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ishijima N, Kanki K, Shimizu H and Shiota
G: Activation of AMP-activated protein kinase by retinoic acid
sensitizes hepatocellular carcinoma cells to apoptosis induced by
sorafenib. Cancer Sci. 106:567–575. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu JJ, Omar HA, Lee YR, Teng YN, Chen PS,
Chen YC, Huang HS, Lee KH and Hung JH: 6-Shogaol induces cell cycle
arrest and apoptosis in human hepatoma cells through pleiotropic
mechanisms. Eur J Pharmacol. 762:449–458. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rattan R, Giri S, Singh AK and Singh I:
5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside inhibits
cancer cell proliferation in vitro and in vivo via AMP-activated
protein kinase. J Biol Chem. 280:39582–39593. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao H, Zhu H, Lin Z, Lin G and Lv G:
Compound 13, an α1-selective small molecule activator of AMPK,
inhibits Helicobacter pylori-induced oxidative stresses and gastric
epithelial cell apoptosis. Biochem Biophys Res Commun. 463:510–517.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qu Z, Zhang Y, Liao M, Chen Y, Zhao J and
Pan Y: In vitro and in vivo antitumoral action of metformin on
hepatocellular carcinoma. Hepatol Res. 42:922–933. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Saito T, Chiba T, Yuki K, Zen Y, Oshima M,
Koide S, Motoyama T, Ogasawara S, Suzuki E, Ooka Y, et al:
Metformin, a diabetes drug, eliminates tumor-initiating
hepatocellular carcinoma cells. PLoS One. 8:e700102013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang X, Sun D, Tian Y, Ling S and Wang L:
Metformin sensitizes hepatocellular carcinoma to arsenic
trioxide-induced apoptosis by downregulating Bcl2 expression.
Tumour Biol. 36:2957–2964. 2015. View Article : Google Scholar
|
|
37
|
Chou CW, Cheng YW and Tsai CH:
Phyllostachys edulis extract induces apoptosis signaling in
osteosarcoma cells, associated with AMPK activation. Drug Des Devel
Ther. 8:1577–1584. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee HW, Jang KS, Choi HJ, Jo A, Cheong JH
and Chun KH: Celastrol inhibits gastric cancer growth by induction
of apoptosis and autophagy. BMB Rep. 47:697–702. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shin JA, Kwon KH and Cho SD:
AMPK-activated protein kinase activation by Impatiens balsamina L.
is related to apoptosis in HSC-2 human oral cancer cells.
Pharmacogn Mag. 11:136–142. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang TP, Lee HJ, Ou TT, Chang YJ and Wang
CJ: Mulberry leaf polyphenol extract induced apoptosis involving
regulation of adenosine monophosphate-activated protein
kinase/fatty acid synthase in a p53-negative hepatocellular
carcinoma cell. J Agric Food Chem. 60:6891–6898. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang G, Li X, Li X, Wang L, Li J, Song X,
Chen J, Guo Y, Sun X, Wang S, et al: Traditional Chinese medicine
in cancer care: A review of case series published in the chinese
literature. Evid Based Complement Alternat Med.
2012:7510462012.PubMed/NCBI
|
|
42
|
Wang X, Feng Y, Wang N, Cheung F, Tan HY,
Zhong S, Li C and Kobayashi S: Chinese medicines induce cell death:
The molecular and cellular mechanisms for cancer therapy. Biomed
Res Int. 2014:5303422014.PubMed/NCBI
|
|
43
|
Shin JS, Kim YM, Hong SS, Kang HS, Yang
YJ, Lee DK, Hwang BY, Ro JS and Lee MK: Induction of neurite
outgrowth by (-)-(7R, 8S)-dihydrodehydrodiconiferyl alcohol from
PC12 cells. Arch Pharm Res. 28:1337–1340. 2005. View Article : Google Scholar
|
|
44
|
Jun DH, Lee JT, Cheon SJ, Lee CE, Kim TH,
Lee DH, Han J and Kim SH: Polyphenol and anti-oxidant effects of
Kalopanax septemlobus Koidz. leaf extracts Korean. J Plant Res.
22:343–348. 2009.
|
|
45
|
Wang LS, Zhao DQ, Xu TH, Zhou XF, Yang XW
and Liu YH: A new triterpene hexaglycoside from the bark of
Kalopanax septemlobus (Thunb.) Koidz. Molecules. 14:4497–4504.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kim HJ, Kim MJ, Oh SI, Hwangbo MH, Jang
SJ, Kim HI and Lee IS: Antioxidant activity of Kalopanax pictus
leaf extract and its effects on the quality characteristics of
fried pork skin. Korean J Food Sci Technol. 44:185–190. 2012.
View Article : Google Scholar
|
|
47
|
Duriez PJ and Shah GM: Cleavage of
poly(ADP-ribose) polymerase: A sensitive parameter to study cell
death. Biochem Cell Biol. 75:337–349. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
de Graaf AO, de Witte T and Jansen JH:
Inhibitor of apoptosis proteins: New therapeutic targets in
hematological cancer? Leukemia. 18:1751–1759. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Danson S, Dean E, Dive C and Ranson M:
IAPs as a target for anticancer therapy. Curr Cancer Drug Targets.
7:785–794. 2007. View Article : Google Scholar
|
|
50
|
Scorrano L and Korsmeyer SJ: Mechanisms of
cytochrome c release by proapoptotic BCL-2 family members. Biochem
Biophys Res Commun. 304:437–444. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jourdain A and Martinou JC: Mitochondrial
outer-membrane permeabilization and remodelling in apoptosis. Int J
Biochem Cell Biol. 41:1884–1889. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Scott JW, Norman DG, Hawley SA,
Kontogiannis L and Hardie DG: Protein kinase substrate recognition
studied using the recombinant catalytic domain of AMP-activated
protein kinase and a model substrate. J Mol Biol. 317:309–323.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hassan M, Watari H, AbuAlmaaty A, Ohba Y
and Sakuragi N: Apoptosis and molecular targeting therapy in
cancer. BioMed Res Int. 2014:1508452014. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kantari C and Walczak H: Caspase-8 and
bid: Caught in the act between death receptors and mitochondria.
Biochim Biophys Acta. 1813:558–563. 2011. View Article : Google Scholar : PubMed/NCBI
|