Introduction
Liver cancer is the fifth most common malignancy and
a major cause of mortality and morbidity worldwide (1). Although treatments directed at liver
cancer patients have advanced and the molecular mechanisms of
carcinogenesis have been elucidated, the prognosis of liver cancer
patients has remained poor (2,3).
Therefore, the molecular characteristics of liver cancer need to be
examined in more detail and new therapeutic methods developed.
Hyperthermia (HT) is a cancer therapy that has been
used since ancient times. Although HT may be administered alone, it
is frequently used in combination with other therapies including
chemotherapy and radiation therapy. HT has been distinguished into
local, regional, and whole body HT. Among currently available liver
cancer therapies, ablation therapies, which involve local HT
including radiofrequency and microwave ablation, have been widely
used and are regarded as standard treatments (4). Moreover, the effectiveness of
combination therapy of chemotherapy and loco-regional HT has been
investigated in clinical trials (5). HT is expected to become a therapeutic
method that improves the prognosis of liver cancer patients.
Water channels (aquaporin: AQP) are a family of
transmembrane proteins responsible for water transport, and 13 of
these subtypes are known to be expressed in mammals (6). AQPs regulate transcellular and
trans-epithelial water movement. They have been suggested to play a
key role in cell volume regulation and cell migration through water
transport (7,8). The expression of AQPs has been
detected in several types of tumors, and AQPs are receiving
increasing attention as new biomarkers or therapeutic targets
(9,10). Previous studies reported that the
expression of AQP5 was altered in tumors of various organs such as
the esophagus, lung, prostate, liver, stomach, breast, and
gallbladder (11–17). The strong expression of AQP5 has
been identified as a prognostic factor in human cancer samples
(11–15). In an in vitro study, the
down-regulated expression of AQP5 was shown to suppress cancer cell
proliferation and survival via the regulation of p21 (11), while the upregulated expression of
AQP5 promoted cancer proliferation (15). Moreover, the downregulated
expression of AQP5 was found to inhibit cancer cell migration
(14–17). These findings demonstrated that
AQP5 is not only involved in cancer cell migration via
physiological water transport, but it also plays a role in cancer
proliferation and survival via the molecular mechanisms responsible
for cancer. Therefore, targeted therapy for AQP5 may be effective
in tumors that strongly express AQP5.
In the present study, we investigated changes in
AQP5 protein expression in HCC cell lines strongly expressing AQP5
that were exposed to heat shock, and demonstrated that heat shock
decreased the expression of AQP5 on cellular membranes and in the
cytoplasm using western blotting (WB) and immunofluorescent
staining (IF). Furthermore, heat shock induced similar changes in
cell volume and the suppression of cancer proliferation and cancer
cell migration/invasion to those induced by the downregulated
expression of AQP5 using siRNA knockdown. We also demonstrated that
the mechanism underlying the downregulated expression of AQP5
induced by heat shock was related to autophagic degradation. These
results suggest that heat shock is an effective treatment against
liver cancer that strongly expresses AQP5, which was reported as a
negative prognostic factor in histopathological features of liver
cancer (14), via the anticancer
effects induced by the autophagic degradation of AQP5.
Materials and methods
Cell lines, antibodies, and other
reagents
The human liver cancer cell lines, HLE and Alexander
cells, were obtained from the Japanese Collection of Research
Bioresources Cell Bank. The human HCC cell line, Hep-G2, was
obtained from the Riken Cell Bank. These cells, with less than
thirty passages, were used in all analyses, and were grown in
plastic culture flasks (Corning Inc., NY, USA). HLE and Hep-G2
cells were maintained in DMEM medium (Nacalai Tesque, Kyoto, Japan)
and Alexander cells were maintained in RPMI-1640 medium (Nacalai
Tesque). Each medium was supplemented with 10% fetal bovine serum
(FBS), 100 U/ml of penicillin, and 100 µg/ml of
streptomycin.
The following antibodies were used in our study: a
mouse monoclonal AQP1 antibody (Santa Cruz Biotechnology, CA, USA),
rabbit polyclonal AQP3 antibody (Santa Cruz Biotechnology), rabbit
monoclonal AQP5 antibody (Abcam, Cambridge, MA, USA), rabbit
monoclonal p21 antibody (Cell Signaling Technology, Beverly, MA,
USA), mouse monoclonal LC3B antibody (Medical and Biological
Laboratories, Nagoya, Japan), rabbit polyclonal E-cadherin antibody
(Santa Cruz Biotechnology), rabbit monoclonal histone H3 antibody
(Cell Signaling Technology), mouse monoclonal β-actin (ACTB)
antibody (Sigma-Aldrich, St. Louis, MO, USA), horseradish
peroxidase (HRP)-conjugated anti-rabbit secondary antibody (Cell
Signaling Technology), and HRP-conjugated anti-mouse secondary
antibody (Cell Signaling Technology). The following reagents were
used in our study: the protein synthesis inhibitor (CHX)
(Sigma-Aldrich), proteasome inhibitor (EPX) (Peptide Institute,
Osaka, Japan), and autophagy inhibitor (BafA1) (Tocris Bioscience,
Ellisville, MO, USA).
Protein isolation
Cells were lysed with M-PER lysis buffer
supplemented with Halt protease and phosphatase inhibitor cocktail
(Thermo Fisher Scientific, Rockford, IL, USA), sonicated, and
centrifuged at 15,000 rpm at 4°C for 10 min in order to obtain
supernatants, which contained total protein. The Pierce cell
surface protein isolation kit (Pierce, Rockford, IL, USA) was used
to isolate cell surface proteins according to the manufacturer's
protocol. NE-PER (Pierce) was used for the isolation of nuclear
protein and cytoplasm protein according to the manufacturer's
protocol.
Western blotting
Protein concentrations were measured using a Protein
Assay Rapid kit (Wako, Osaka, Japan). Cell lysates containing equal
amounts of protein were separated by SDS-PAGE, and then transferred
onto PVDF membranes (Merck Millipore, Billerica, MA, USA). The
membranes were probed with the indicated antibodies, and proteins
were detected by the ECL Plus Western Blotting Detection system (GE
Healthcare). The primary ACTB antibody was used as the loading
control of whole lysates, the primary E-cadherin antibody was used
as that of cell membrane proteins, and the primary histone H3
antibody was used as that of nuclear proteins.
Immunofluorescence staining
Cells were stained according to a standard cell
staining protocol. Briefly, Alexander cells were cultured on SPL
cell culture slides, which are 8-chamber slides (SPL Life Science,
Pocheon, Korea) for 24 h. Subsequently, for IF to compare protein
expression between cells under normal conditions and those that
were thermally stimulated, cells were heated at 37 or 42°C for 1 h
in 5% CO2. Cells were subsequently fixed with 4%
paraformaldehyde at room temperature for 20 min, permeabilized in
0.25% Triton X-100 in phosphate-buffered saline (PBS), and
incubated in blocking buffer containing 1% bovine serum albumin.
Cells were then incubated with the anti-AQP5 or anti-AQP1 antibody
at room temperature for 1 h. After three washes in PBS, cells were
incubated with Alexa Fluor 488-labeled goat anti-rabbit secondary
antibodies at room temperature for 1 h. After three washes in PBS,
cells were incubated with rhodamine phalloidin and
40,6-diamidino-2-phenylindole (DAPI) for 30 min. Slides were then
mounted with Vectashield Mounting Medium (Vector Laboratories,
Burlingame, CA, USA). In IF on cells heated with BafA1, cells were
pre-incubated with 50 nM BafA1 for 30 min, and then warmed at 37 or
42°C for 1 h in 5% CO2. After fixing, permeabilizing,
and blocking, cells were incubated with the anti-AQP5 antibody for
1 h. After three washes in PBS, cells were incubated with Alexa
Fluor 594-labeled goat anti-rabbit secondary antibodies at room
temperature for 1 h. Thereafter, cells were incubated with the
anti-LC3B antibody and Alexa Fluor 488-labeled goat anti-mouse
secondary antibodies. DAPI staining and mounting were performed.
The distribution of AQP5 and LC3B proteins was examined using
BZ-X700 (Keyence, Tokyo, Japan).
Small interfering RNA (siRNA)
transfection
Cells were transfected with 12 nM AQP5 siRNA
(Stealth RNAi™ siRNA #1: HSS100611, #2: HSS179941, #3: HSS179942;
Invitrogen, Carlsbad, CA, USA) using the Lipofectamine RNAiMAX
reagent (Invitrogen) according to the manufacturer's instructions.
Medium containing siRNA was replaced with fresh medium after 24 h.
The provided control siRNA (Stealth RNAi™ siRNA negative control;
Invitrogen) was used as the negative control.
Real-time quantitative RT-PCR
Total RNA was extracted using an RNeasy kit (Qiagen,
Valencia, CA, USA). Messenger RNA (mRNA) expression was measured by
quantitative real-time PCR (7300 Real-Time PCR system; Applied
Biosystems, Foster City, CA, USA) using TaqMan gene expression
assays (Applied Biosystems) according to the manufacturer's
instructions. Expression levels were measured for AQP5
(Hs00387048_m1). The expression of each gene was normalized against
the housekeeping gene ACTB (Hs01060665_g1; Applied Biosystems).
Each assay was performed in triplicate.
Measurement of cell volume changes using
a high resolution flow cytometer
Cell volume measurements were performed using a high
resolution flow cytometer, the Cell Lab Quanta (Beckman Coulter,
Fullerton, CA, USA), according to a previously described procedure
(18–21). This flow cytometer was designed to
measure the electronic volume (EV) of a cell, and the EV data of
>10,000 cells were collected and analyzed using the Quanta
control software. A total of 1.0×106 pelleted Alexander
cells, which had been heated at 37 or 42°C using a water bath for 1
h or transfected with control/AQP5 siRNA, were suspended in 1 ml of
RPMI-1640. These suspensions were subsequently displaced into a
Vi-CELL™ Sample Cup (Beckman Coulter), and cell volume was
measured.
Cell proliferation assay
In AQP5 knockdown experiments, cells were seeded
onto 6-well plates at a density of 5×104 cells per well
and incubated at 37°C with 5% CO2. Twenty-four hours
after cell seeding, siRNA transfection was performed and 48 and 72
h after siRNA transfection, cells were detached from flasks using
trypsin-EDTA, and a viable cell count was then performed using
trypan blue and the Countess Automated Cell Counter (Invitrogen,
Tokyo, Japan). In heat shock experiments, 5×104 cell
pellets were heated at 37 or 42°C using a water bath for 1 h, and
then re-seeded onto 6-well plates and incubated at 37°C with 5%
CO2. A viable cell count was performed 48 and 72 h after
the heat shock treatment.
Cell cycle analysis
In AQP5 knockdown experiments, cell cycle
progression was evaluated 48 h after siRNA transfection using
fluorescence-activated cell scoring (FACS). In heat shock
experiments, cell cycle progression was evaluated 24 h after the
heat shock treatment for 2 h. Briefly, cells were treated with
Triton X-100, and their nuclei were stained with PI RNase staining
buffer (Becton-Dickinson Biosciences, San Jose, CA, USA). The DNA
content was then measured using a Becton-Dickinson Accuri C6 FACS
(Becton-Dickinson Biosciences). At least 10,000 cells were counted,
and BD Accuri C6 software was used to analyze the cell cycle
distribution.
Invasion and migration assay
The migration assay was conducted using a Cell
Culture Insert with a pore size of 8 µm (BD Biosciences,
Bedford, MA, USA). Biocoat Matrigel (BD Biosciences) was used to
evaluate cell invasion potential. Briefly, cells
(2.0×105 cells per well), which had been heated at
37/42°C or transfected with AQP5/control siRNA, were seeded on the
upper chamber in serum-free medium. The lower chamber contained
medium with 10% FBS. The chambers were incubated for 24 h at 37°C
in 5% CO2, and non-migrating or non-invading cells were
then removed from the upper side of the membrane by scrubbing with
cotton swabs. Migrated or invaded cells were fixed on the membrane
and stained with Diff-Quick staining reagents (Sysmex, Kobe,
Japan). The migrated or invaded cells on the lower side of the
membrane were counted in four independent fields of view at ×100 or
×200 magnification of each insert. Each assay was performed in
triplicate.
Analysis of apoptotic cells
Cells were harvested 24 h after siRNA transfection
or heat shock for 1 h, and then stained with fluorescein
isothiocyanate-conjugated Annexin V and phosphatidylinositol using
the Annexin V kit (Beckman Coulter, Brea, CA, USA) according to the
manufacturer's protocols. Becton-Dickinson Accuri C6 FACS was used
to analyze the proportion of apoptotic cells.
CHX chase experiments
Alexander cells were plated at a density of
1.0×105 on 6-well plates and incubated for 24 h at 37°C
with 5% CO2. Medium was changed to antibiotic-free
medium containing 1.0 µg/ml CHX with/without 1 µM EPX
or 50 nM BafA1, and cells were then incubated at 37°C or 42°C with
5% CO2 for 1, 2, 3,6, 12, and 24 h. After this
treatments, total protein isolation and WB was performed.
Statistical analysis
Results were expressed as means ± SEM. Statistical
analyses were carried out using the Student's t-test. Differences
were considered significant when the P-value was <0.05.
Statistical analyses were performed using JMP version 10.
Results
Protein expression of AQP in liver cancer
cells, and the suppression of proliferation by heat shock in
accordance with AQP expression
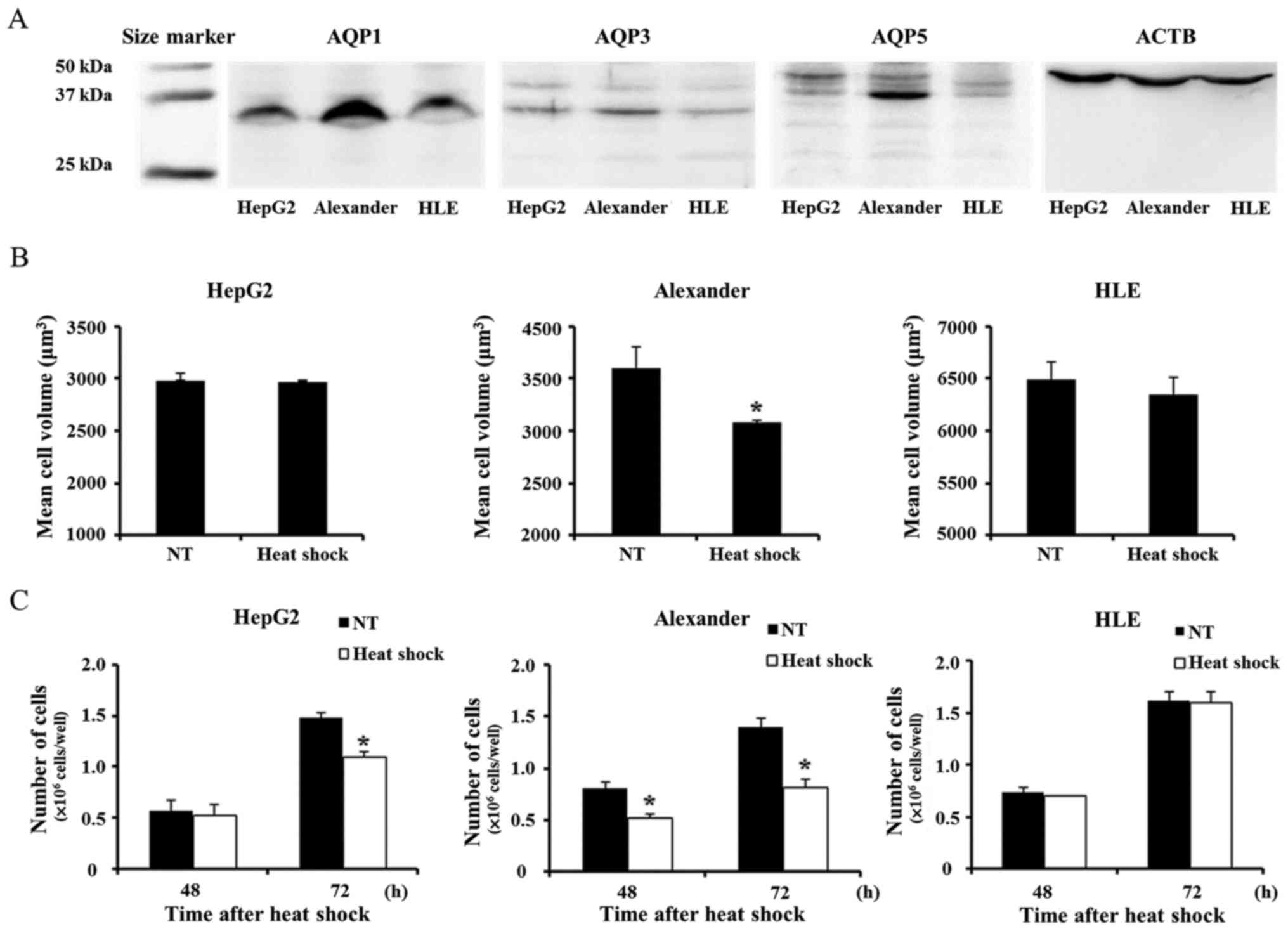
In order to investigate AQP expression in HCC cells,
we first evaluated AQP1, AQP3, and AQP5 protein expression in the
liver cancer cell lines, HepG2 and HLE, and Alexander cells. The
expression of these AQP isoforms in liver cancer was described
previously (14,22). WB revealed that AQP was weakly
expressed in HepG2 and HLE. In contrast, AQP1 and AQP5 were more
strongly expressed in Alexander cells than in the other cells
tested (Fig. 1A). Heat shock
induced cell volume shrinkage only in Alexander cells in accordance
with the expression of AQP1 and AQP5 by each cell line (Fig. 1B). Similarly, heat shock more
strongly suppressed proliferation only in Alexander cells (Fig. 1C).
Changes induced in the distribution of
AQP5 in Alexander cells by heat shock
In order to determine whether heat shock regulates
the expression of AQP1 and AQP5 in Alexander cells strongly
expressing these AQPs, the distribution of AQP1 and AQP5 in
Alexander cells treated with heat shock was examined using IF. The
cytoplasm and nuclei of non-treated Alexander cells were diffusely
stained by AQP1 and AQP5. The distribution of AQP1 in cells treated
with heat shock was similar to that in non-treated cells (Fig. 2B). On the other hand, the staining
intensity of AQP5 in the cytoplasm of cells treated with heat shock
was weaker than that in non-treated cells (Fig. 2A), suggesting that heat shock
regulated the expression of AQP5.
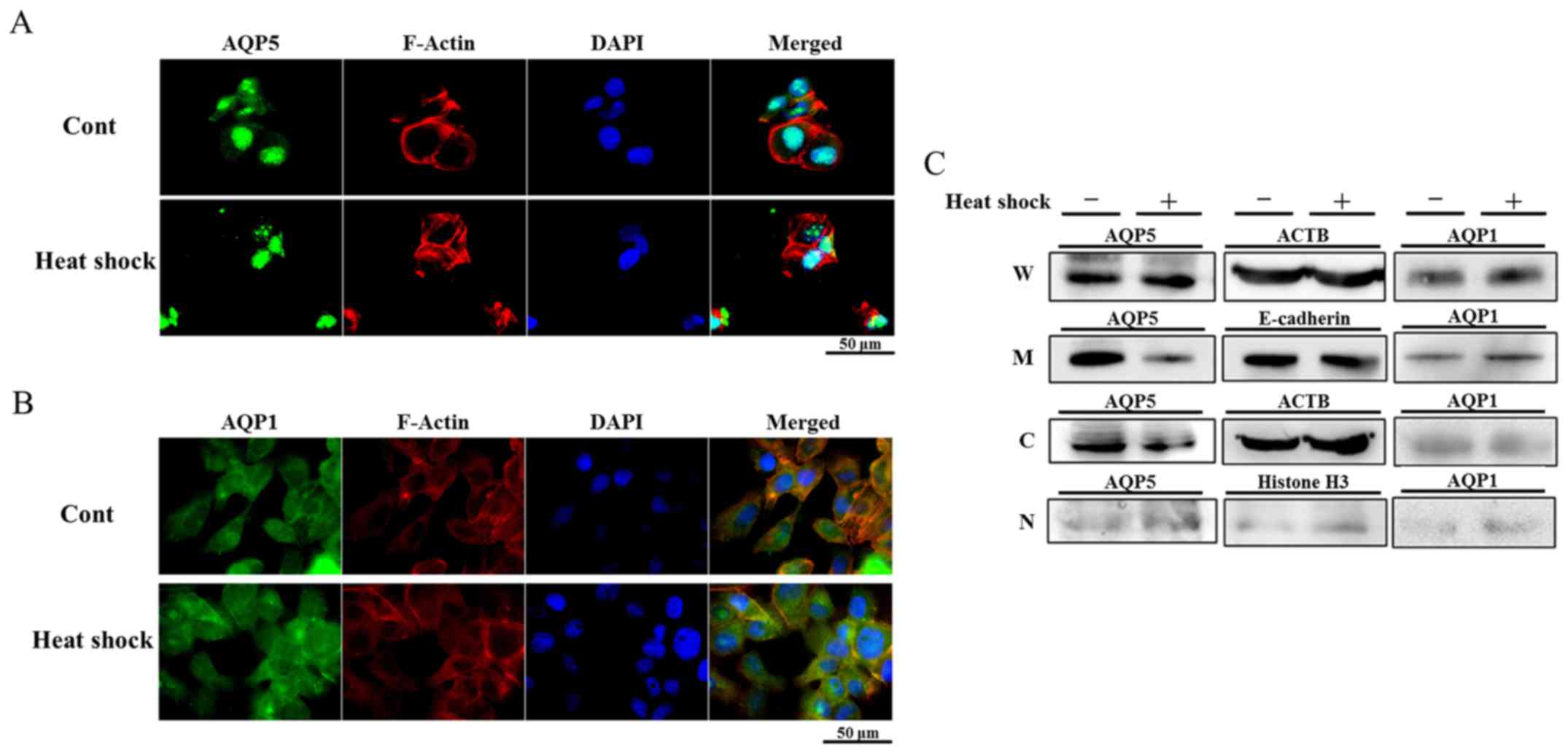 | Figure 2Heat shock decreases AQP5 protein
expression on cell membranes and in the cytoplasm. (A and B)
Immunofluorescent staining of AQP5 (A) and AQP1 (B) on heated
Alexander cells and non-treated cells (control). Heat shock
decreased AQP5 expression in the cytoplasm of Alexander cells, but
did not affect that of AQP1. (C) Western blotting of protein
fractions for the detection of AQP1 and AQP5 in heated Alexander
cells and non-treated cells (W, whole lysates, M, cellular membrane
proteins, C, cytoplasmic proteins, and N, nuclear proteins). AQP5
expression on cellular membranes and in cytoplasmic proteins of
heated cells was weaker than that in non-treated cells, and no
significant differences were observed in the expression of AQP5 in
nuclear proteins. In contrast, heat shock did not affect AQP1
expression in membrane and cytoplasmic protein. |
Heat shock decreased AQP5 protein
expression on cell membranes and in the cytoplasm of Alexander
cells
In order to examine changes induced in the
distribution of proteins by heat shock in more detail, the protein
fractions of non-treated cells and cells heated for 1 h were
isolated and WB was performed. The expression of AQP5 in the cell
membrane fraction of heated cells was weaker than that in
non-treated cells, and that in the cytoplasm was also decreased by
heat shock, which is consistent with the results of IF. On the
other hand, the expression of AQP5 in nuclei was similar between
each cell line examined (Fig. 2C).
These results suggest that heat shock decreases the expression of
AQP5 on cell membranes and in the cytoplasm by regulating membrane
trafficking or degradation in Alexander cells.
Morphological changes in Alexander cells
treated with heat shock or transfected with aquaporin 5 small
interfering RNA (siRNA)
Previous studies reported that AQP5 plays an
important role in cell migration by regulating morphological
changes induced by water transport through the cellular membrane
(7,8). Therefore, we hypothesized that heat
shock inhibits cancer cell migration by downregulating the
expression of AQP5 on cellular membranes. In order to validate this
hypothesis, we compared cell volumes between cells treated with
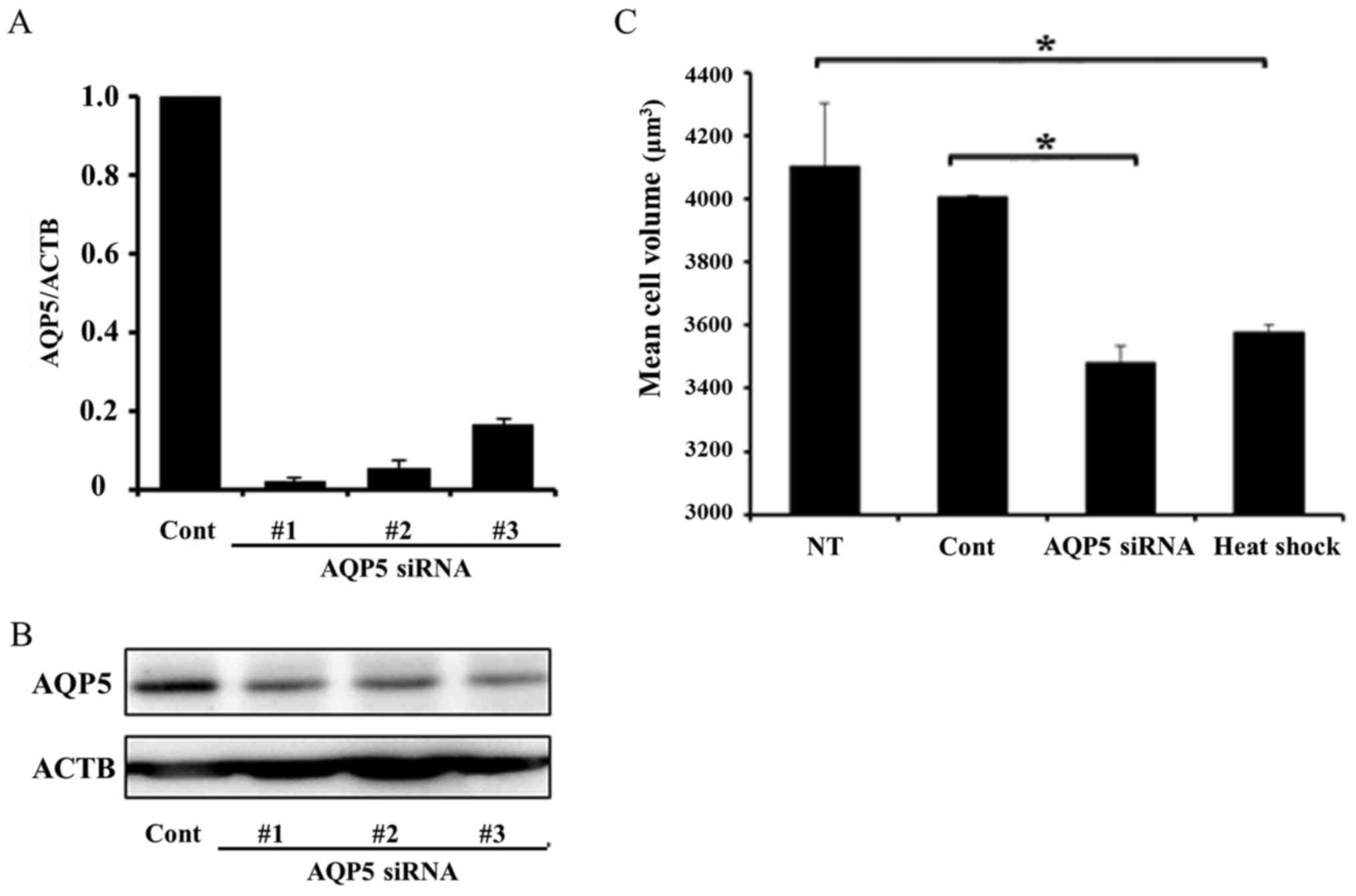
heat shock and transfected with AQP5 siRNA. Fig. 3A and B shows AQP5 mRNA and protein
expression in cells transfected with control siRNA and three types
of AQP5 siRNA (#1–3). All AQP5 siRNAs effectively decreased AQP5
mRNA and protein expression, and we used AQP5 siRNA #3 in
subsequent AQP5 knockdown experiments. Cell volumes were measured
in non-treated, heated, and transfected cells using high resolution
flow cytometry (Cell Lab Quanta). The mean cell volume of heated
cells was significantly smaller than that of non-treated cells.
Similarly, the mean cell volume of cells transfected with AQP5
siRNA was significantly smaller than those transfected with control
siRNA. These results demonstrated that heat shock affected the cell
volume of Alexander cells in a similar manner to the downregulated
expression of AQP5 (Fig. 3C).
Suppression of cell migration and
invasion induced by heat shock or AQP5 knockdown in Alexander
cells
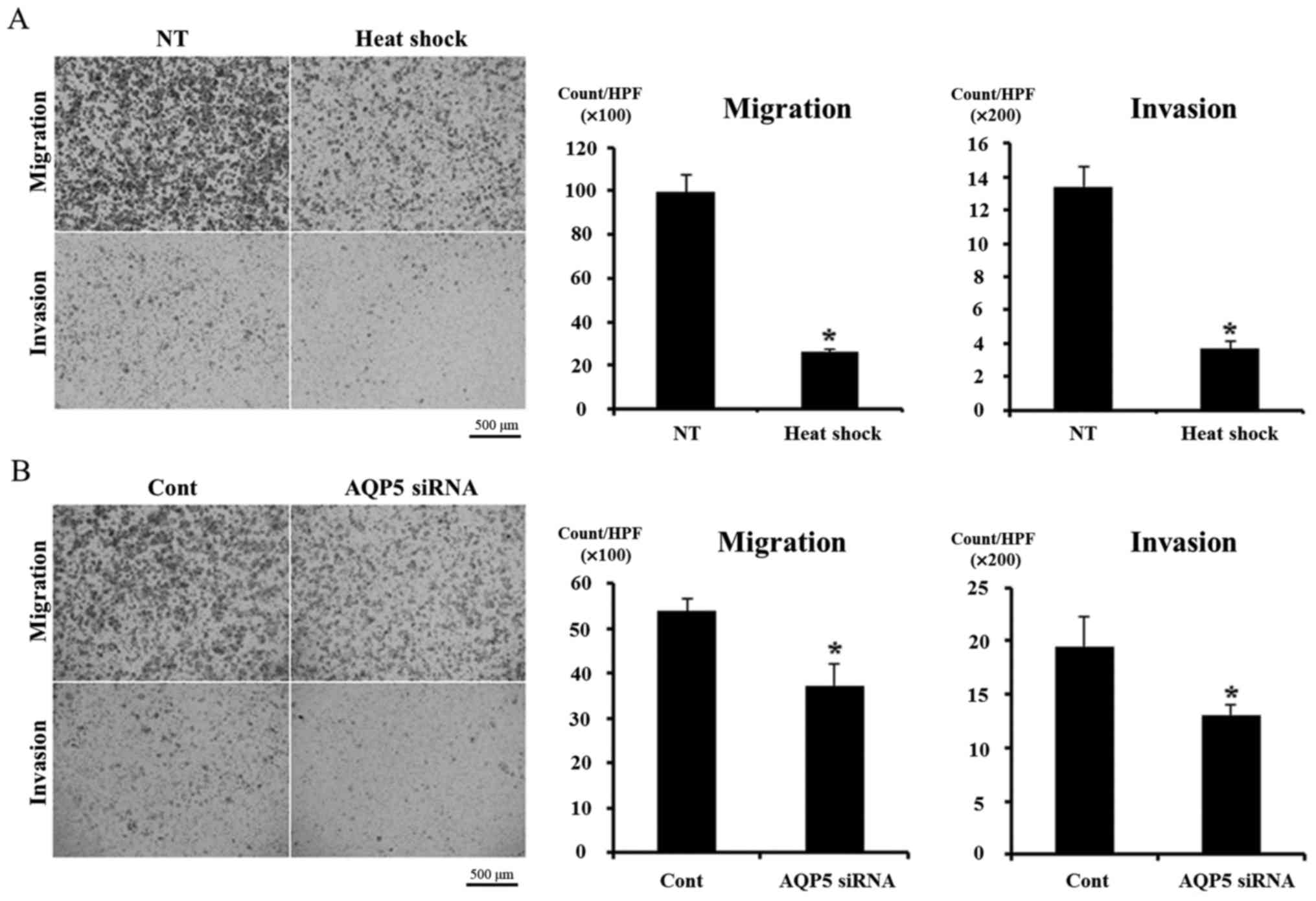
We analyzed the effects of heat shock and AQP5
knockdown on cell migration and invasion in order to investigate
our hypothesis using a Boyden chamber assay. Heat shock
significantly inhibited cell migration and invasion in Alexander
cells (Fig. 4A), and similar
results were obtained in cells transfected with AQP5 siRNA
(Fig. 4B). These results support
our hypothesis.
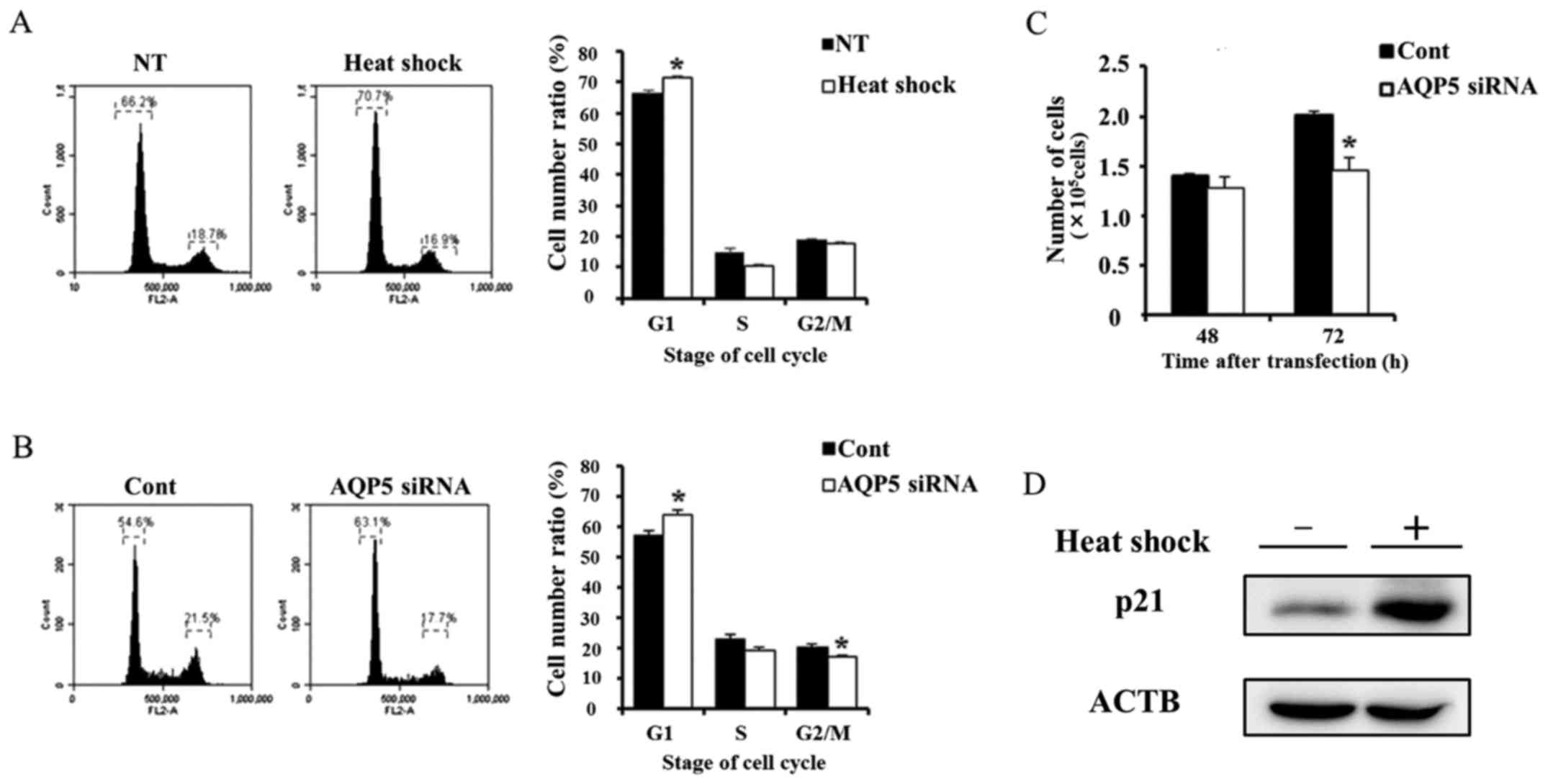
Suppression of cell cycle progression
from the G1 to S phase and proliferation in Alexander cells by heat
shock or AQP5 knockdown
A previous study reported that AQP5 controlled
cancer cell cycle progression and proliferation (11). Therefore, we investigated cancer
cell cycle progression and proliferation in cells treated with heat
shock and transfected with AQP5 siRNA. Heat shock and AQP5
knockdown partially reduced cell cycle progression from the G1 to S
phase (Fig. 5A and B). We also
performed a proliferation assay on Alexander cells subjected to
these treatments. The number of viable cells transfected with AQP5
siRNA 72 h after transfection was lower than that in cells
transfected with control siRNA (Fig.
5C), which is consistent with the results obtained in heated
cells (Fig. 1B). These results
indicate that heat shock may suppress cell cycle progression and
proliferation in Alexander cells by regulating AQP5. Moreover,
previous study reported that AQP5 was related to cancer cell
proliferation and survival via the regulation of p21 (11). Therefore, we evaluated change of
p21 protein expression induced by heat shock. Western blotting of
p21 on whole lysate of non-treated and heated cells revealed that
heat shock upregulated the p21 expression.
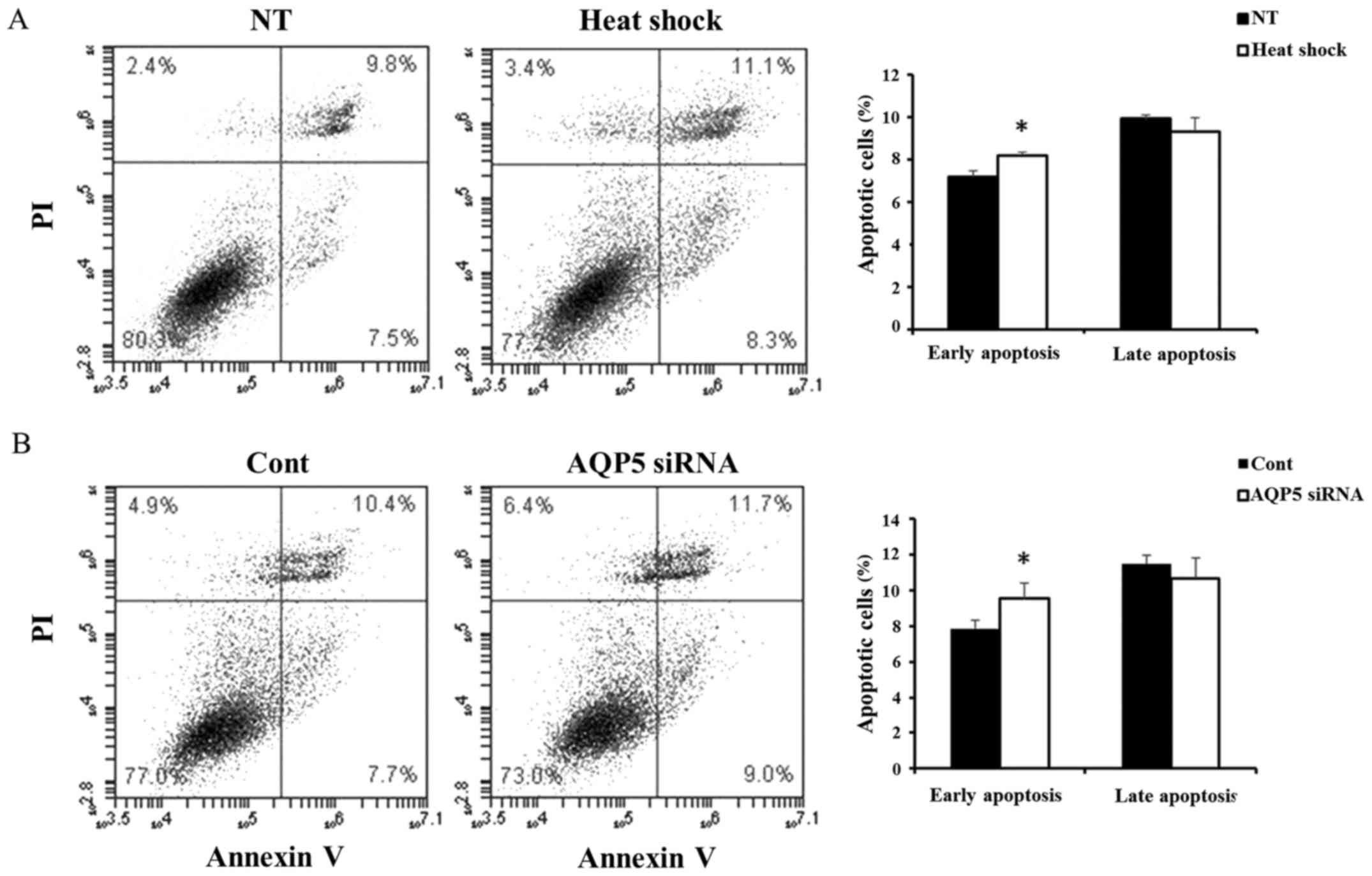
The activity of apoptosis in Alexander
cells treated with heat shock and AQP5 siRNA
Next, we compared the activity of apoptosis in
Alexander cells treated with heat shock and AQP5 siRNA. Heat shock
significantly induced early apoptosis (Fig. 6A), and similar results were
obtained in cells transfected with AQP5 siRNA (Fig. 6B). These results suggested that
heat shock induces early apoptosis in Alexander cells by regulating
AQP5.
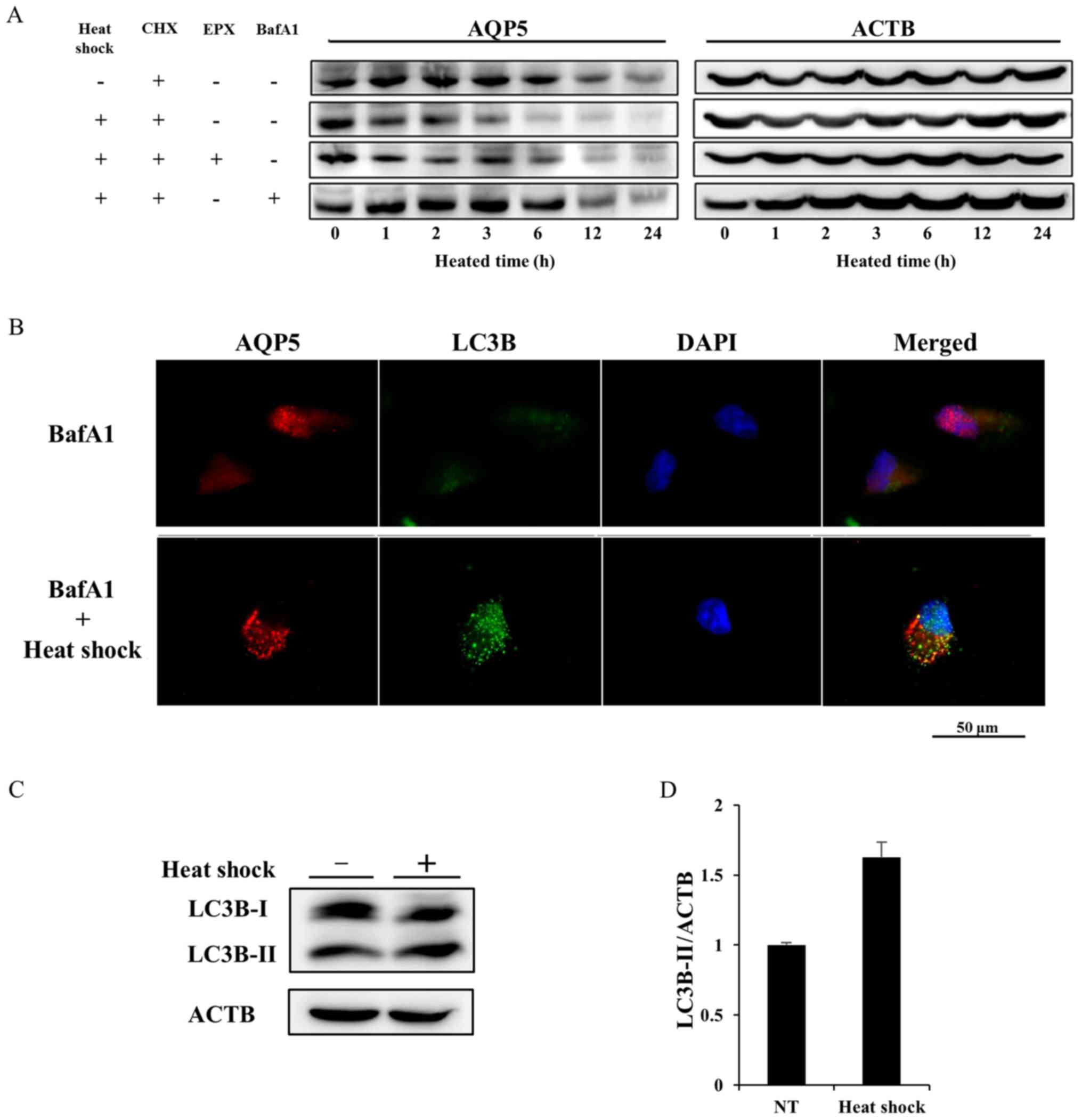
Heat shock decreases AQP5 expression in
Alexander cells by activating autophagic degradation
We determined whether the half-life of the AQP5
protein was affected by heat shock using cycloheximide (CHX), which
inhibits protein synthesis, chase experiments to elucidate the
mechanisms responsible for the downregulated expression of AQP5. In
contrast to cells treated with CHX alone (the control group), heat
shock accelerated the degradation of AQP5 in the presence of CHX
(the heat shock group) (Fig. 7A).
A CHX chase assay on heated cells treated with epoxomicin (EPX),
which inhibits protea-some (EPX group), or bafilomycin A1 (BafA1),
an autophagy inhibitor, was also performed. The degradation of AQP5
in the EPX group was similar to that in the heat shock group. In
contrast, the additional treatment of BafA1 extended the half-life
of AQP5 to the same extent as that in the control group, and
rescued heated cells from the acceleration of AQP5 degradation.
These results indicated that heat shock induced the downregulation
of AQP5 expression by activating autophagic degradation.
We then performed double IF of AQP5 and light chain
3B (LC3B) on non-treated cells or heated cells under accumulating
autophagosomes by inhibiting autolysis using bafA1 in order to
define the mechanisms underlying the heat shock-induced degradation
of AQP5. LC3B is the main biological marker for autophagy, which
has two subtypes, cytosolic-associated protein LC3B-I and the
membrane-bound LC3B-II. Autophagosome formation is associated with
the conversion of LC3B-I into LC3B-II. The double IF finding under
the treatment of bafA1 showed that heat shock induced the
accumulation of small vesicles stained by AQP5 or LC3B. Some
vesicles of AQP5 overlapped those of LC3B on merge of AQP5 and LC3B
image (Fig. 7B). Furthermore,
protein expression of LC3B-I and LC3B-II on non-treated cell and
heated cells was evaluated to confirm the activation of autophagy
induced by heat shock. Western blotting of LC3B on whole lysates of
non-treated and heated cells revealed the upregulation of LC3B-II
expression induced by heat shock (Fig.
7C and D). These results suggested that autophagy was related
to the heat shock-induced degradation of AQP5.
Discussion
Hyperthermia (HT) has been used as a cancer therapy
since ancient times. In recent years, the combination of regional
or whole body HT with radiation and chemotherapy has been employed
clinically, and has achieved positive clinical outcomes in patients
with various solid tumors (23,24).
However, the effects of these therapies are weaker than those of
other curative treatments used as standard therapies for some solid
tumors, and HT is rarely used as a primary cancer therapy. On the
other hand, a previous study reported a few cancer cases in which
tumors exhibited high sensitivity to HT (25). Therefore, further studies are
needed in order to establish methods to detect tumors with high
sensitivity to HT.
A selective tumor killing effect has been observed
between 40 and 44°C in vitro and in vivo (26). A large number of studies have
examined the mechanisms underlying this anticancer effect. Heat
shock has been shown to induce G0/G1 arrest through the
accumulation of p16 and p53 (27),
and also activated signal transduction pathways for anti-apoptosis
and/or cellular proliferation, such as Akt, p38, extracellular
signal-regulated kinase, and heat shock proteins (28). Heat shock was previously reported
to induce apoptosis through reactive oxygen species generation and
increases in intracellular calcium ion concentration (29). Furthermore, heat shock has been
shown to change water and ion permeability through cellular
membranes as well as cell volume regulation (30,31).
A relationship has been found between cell volume and apoptosis,
and apoptotic volume decrease (AVD), which is isosmotic cell
shrinkage induced by a loss in KCl via potassium chloride channels
and the concomitant loss of water through water channels or the
lipid bilayer, was shown to occur in the early phase of programmed
cell death (8,31). A relationship between cell volume
and cell proliferation was also previously reported (8). Based on the findings of relationships
between heat shock, cell volume, apoptosis, and proliferation,
molecules related to cell volume regulation, e.g., potassium,
chloride channels, the potassium and chloride co-transporter (KCC),
and AQPs, may play an important role in the anticancer effects of
HT. These molecules, e.g., KCC3, and AQP5, were found to be
overexpressed in human cancer samples, and were identified as
prognostic factors (11–15,32,33).
If HT regulates these molecules, targeted therapy to tumors
overexpressing these molecules may become possible.
A previous study reported that heat shock activated
autophagy (34). Autophagy is a
catabolic process that leads to the sequestration and degradation
of intracellular material within lysosomes. Autophagy is known to
protect against various human diseases (35). However, the role of autophagy in
tumors is more complex. Autophagy serves as an oncogenic mechanism
to promote tumor survival. On the other hand, it has a
tumor-suppressive role. Autophagy has been suggested to have
paradoxical functions in cancer (36, 37). Chemotherapeutic drugs that activate
autophagy, e.g., rapamycin, everolimus, and temsirolimus, have
already been administered to cancer patients clinically (38). The mechanisms underlying the
anticancer effects of the activation of autophagy have not yet been
elucidated in detail. However, it is clear that the activation of
autophagy induces anticancer effects. Therefore, heat shock may be
similarly effective in tumors through the activation of autophagy.
Furthermore, previous studies reported that the degradation of AQP5
occurred via selective autophagy. Hosoi described that the
selective autophagic degradation of AQP5 in submandibular grand
regulated water secretion (39).
In the present study, we selected Alexander cells
strongly expressing AQP1 and AQP5, in which heat shock induced cell
volume shrinkage and effectively suppressed cell proliferation,
from some HCC cancer cell lines depending on the results of WB and
proliferation assays. We then performed IF and WB on fractionated
samples in order to investigate heat shock-induced changes in AQP1
and AQP5 protein expression in Alexander cells. IF revealed that
the expression of AQP1 was similar between control and heated
cells, whereas that of AQP5 in the cytoplasm was decreased by heat
shock. WB on fractionated samples indicated that heat shock
decreased the expression of AQP5 on cellular membranes and in the
cytoplasm. These results prompted us to propose two hypotheses. In
the first hypothesis, we suggest that heat shock suppressed cancer
migration/invasion via water balance or cell volume regulation
induced by the decrease in AQP5 expression on cellular membranes in
liver cancer cell lines strongly expressing AQP5. The second
hypothesis proposes that heat shock inhibits cancer proliferation
through the regulation of cancer signals induced by a decrease in
AQP5 expression on cellular membranes or in the cytoplasm. In order
to validate these hypotheses, we compared changes in cell volume,
migration/invasion ability, proliferation, the cell cycle, and the
activity of apoptosis induced by heat shock between cells exposed
to heat shock and those in which the expression of AQP5 was
down-regulated using siRNA transfection. Cell volume shrinkage
similarly occurred with heat shock and the downregulation of AQP5.
Heat shock suppressed cancer migration/invasion and proliferation
to the same extent as the downregulation of AQP5. Both treatments
partially induced G0/G1 arrest, and early apoptosis. Moreover, heat
shock upregulated protein expression of p21, which was previously
reported as related protein to AQP5 (11). These results indicate that heat
shock exerts similar anticancer effects to the downregulation of
AQP5 in liver cancer cell lines strongly expressing AQP5, which
supports our hypotheses. Furthermore, we performed a CHX chase
assay using a proteasome inhibitor or autophagy inhibitor
concurrently in order to investigate the mechanisms responsible for
heat shock-induced decreases in the expression of AQP5. Heat shock
accelerated the degradation of AQP5 under the CHX treatment only,
and this was rescued under the concurrent treatment of CHX and the
autophagy inhibitor, BafA1. Moreover, double IF using the AQP5
antibody and the autophagy marker, the LC3B antibody revealed the
overlap of AQP5 and LC3B vesicles on heated cells treated with
BafA1, and western blotting of LC3B revealed that heat shock
upregulated LC3B-II expression. This result suggests that the
decreases induced in AQP5 by heat shock were related to
autophagy.
Moreover, AQP5 expression in nuclear was observed in
the present study. Although this function of AQP5 in nuclear was
not clear, previous study reported that AQP5 was involved with
nuclear protein, e.g., p21 or cyclin D1, in cancer cells (11,40).
AQP5 in nuclear may interact with these nuclear protein to maintain
cancer function. Moreover, previous study described that AQP5 is
related to water transport through osmotic gradient across the cell
membrane (7). Therefore, it is not
expected that the downregulated function of only AQP5 can induce
cell volume shrinkage. There are two mechanisms to consider when
assessing that AQP5 downregulation induces cell volume decrease.
The downregulation of AQP5 may affect mRNA or protein expression of
the other molecules, which contained ion channels and transporter.
Although these changes of expression could not be investigated in
present study, it affected osmotic gradient across the cell
membrane via the change of ion transport, as a result; cell volume
shrinkage might occur. Furthermore, AQP5 downregulation and heat
shock induced early apoptosis (Fig.
6). This finding suggested that AQP5 downregulation induced
cell volume decrease via the mechanism of AVD.
Morbidity and mortality rates are continuously
increasing in cancer. Therefore, we need to develop more therapies
related to the molecular characteristics of individual tumors in
order to overcome cancer. The effectiveness of heat shock for
cancer strongly expressing AQP5, which was reported as a poor
prognostic factor in histopathological features of liver cancer
(14), via the activation of
autophagic degradation has not yet been reported, and we herein
demonstrated this for the first time. This result may be useful for
the development of liver cancer therapy.
In conclusion, we showed that heat shock decreased
the expression of AQP5 on cellular membranes and in the cytoplasm
of HCC cell lines strongly expressing AQP5 by activating autophagic
degradation. Moreover, we found that heat shock and the
downregulation of AQP5 exerted similar anticancer effects. This
result suggests that heat shock exerts anticancer effects via the
autophagic degradation of AQP5 in liver cancer.
Acknowledgments
This study was supported by a Grant-in-Aid for
Scientific Research (C) (26461988) and Grants-in-Aid for Young
Scientists (B) (15K19903 and 15K19904) from the Japan Society for
the Promotion of Science.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Llovet JM, Burroughs A and Bruix J:
Hepatocellular carcinoma. Lancet. 362:1907–1917. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Slotta JE, Kollmar O, Ellenrieder V,
Ghadimi BM and Homayounfar K: Hepatocellular carcinoma: Surgeon's
view on latest findings and future perspectives. World J Hepatol.
7:1168–1183. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kang TW and Rhim H: Recent advances in
tumor ablation for hepatocellular carcinoma. Liver Cancer.
4:176–187. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gadaleta-Caldarola G, Infusino S, Galise
I, Ranieri G, Vinciarelli G, Fazio V, Divella R, Daniele A,
Filippelli G and Gadaleta CD: Sorafenib and locoregional deep
electro-hyperthermia in advanced hepatocellular carcinoma: A phase
II study. Oncol Lett. 8:1783–1787. 2014.PubMed/NCBI
|
|
6
|
Ishibashi K, Hara S and Kondo S: Aquaporin
water channels in mammals. Clin Exp Nephrol. 13:107–117. 2009.
View Article : Google Scholar
|
|
7
|
Day RE, Kitchen P, Owen DS, Bland C,
Marshall L, Conner AC, Bill RM and Conner MT: Human aquaporins:
Regulators of transcellular water flow. Biochim Biophys Acta.
1840:1492–1506. 2014. View Article : Google Scholar
|
|
8
|
Hoffmann EK, Lambert IH and Pedersen SF:
Physiology of cell volume regulation in vertebrates. Physiol Rev.
89:193–277. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ribatti D, Ranieri G, Annese T and Nico B:
Aquaporins in cancer. Biochim Biophys Acta. 1840:1550–1553. 2014.
View Article : Google Scholar
|
|
10
|
Papadopoulos MC and Saadoun S: Key roles
of aquaporins in tumor biology. Biochim Biophys Acta. 1848(10 Pt
B): 2576–2583. 2015. View Article : Google Scholar
|
|
11
|
Shimizu H, Shiozaki A, Ichikawa D,
Fujiwara H, Konishi H, Ishii H, Komatsu S, Kubota T, Okamoto K,
Kishimoto M, et al: The expression and role of Aquaporin 5 in
esophageal squamous cell carcinoma. J Gastroenterol. 49:655–666.
2014. View Article : Google Scholar
|
|
12
|
Song T, Yang H, Ho JC, Tang SC, Sze SC,
Lao L, Wang Y and Zhang KY: Expression of aquaporin 5 in primary
carcinoma and lymph node metastatic carcinoma of non-small cell
lung cancer. Oncol Lett. 9:2799–2804. 2015.PubMed/NCBI
|
|
13
|
Li J, Wang Z, Chong T, Chen H, Li H, Li G,
Zhai X and Li Y: Over-expression of a poor prognostic marker in
prostate cancer: AQP5 promotes cells growth and local invasion.
World J Surg Oncol. 12:2842014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guo X, Sun T, Yang M, Li Z, Li Z and Gao
Y: Prognostic value of combined aquaporin 3 and aquaporin 5
overexpression in hepatocellular carcinoma. BioMed Res Int.
2013:2065252013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang YH, Zhou XY, Wang HM, Xu H, Chen J
and Lv NH: Aquaporin 5 promotes the proliferation and migration of
human gastric carcinoma cells. Tumour Biol. 34:1743–1751. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jung HJ, Park JY, Jeon HS and Kwon TH:
Aquaporin-5: A marker protein for proliferation and migration of
human breast cancer cells. PLoS One. 6:e284922011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sekine S, Shimada Y, Nagata T, Sawada S,
Yoshioka I, Matsui K, Moriyama M, Omura T, Osawa S, Shibuya K, et
al: Role of aquaporin-5 in gallbladder carcinoma. Eur Surg Res.
51:108–117. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kosuga T, Shiozaki A, Ichikawa D, Fujiwara
H, Komatsu S, Iitaka D, Tsujiura M, Morimura R, Takeshita H, Nagata
H, et al: Pleural lavage with distilled water during surgery for
esophageal squamous cell carcinoma. Oncol Rep. 26:577–586.
2011.PubMed/NCBI
|
|
19
|
Iitaka D, Shiozaki A, Ichikawa D, Kosuga
T, Komatsu S, Okamoto K, Fujiwara H, Ishii H, Nakahari T, Marunaka
Y, et al: Blockade of chloride ion transport enhances the cytocidal
effect of hypotonic solution in gastric cancer cells. J Surg Res.
176:524–534. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nako Y, Shiozaki A, Ichikawa D, Komatsu S,
Konishi H, Iitaka D, Ishii H, Ikoma H, Kubota T, Fujiwara H, et al:
Enhancement of the cytocidal effects of hypotonic solution using a
chloride channel blocker in pancreatic cancer cells. Pancreatology.
12:440–448. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Takemoto K, Shiozaki A, Ichikawa D,
Komatsu S, Konishi H, Nako Y, Murayama Y, Kuriu Y, Nakanishi M,
Fujiwara H, et al: Evaluation of the efficacy of peritoneal lavage
with distilled water in colorectal cancer surgery: In vitro and in
vivo study. J Gastroenterol. 50:287–297. 2015. View Article : Google Scholar
|
|
22
|
Mazal PR, Susani M, Wrba F and Haitel A:
Diagnostic significance of aquaporin-1 in liver tumors. Hum Pathol.
36:1226–1231. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
van der Zee J: Heating the patient: A
promising approach? Ann Oncol. 13:1173–1184. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mallory M, Gogineni E, Jones GC, Greer L
and Simone CB II: Therapeutic hyperthermia: The old, the new, and
the upcoming. Crit Rev Oncol Hematol. 97:56–64. 2016. View Article : Google Scholar
|
|
25
|
Takeda T, Takahashi T, Yamamoto I,
Hasegawa T, Takeda T and Takeda H: Hyperthermia enhances
immunotherapy in cancer patients : Clinical and experimental
analyses. Therm Med. 28:11–16. 2012. View Article : Google Scholar
|
|
26
|
Ahmed K, Tabuchi Y and Kondo T:
Hyperthermia: An effective strategy to induce apoptosis in cancer
cells. Apoptosis. 20:1411–1419. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Valenzuela MT, Nunez MI, Villalobos M,
Siles E, McMillan TJ, Pedraza V and Ruiz de Almodóvar JM: A
comparison of p53 and p16 expression in human tumor cells treated
with hyperthermia or ionizing radiation. Int J Cancer. 72:307–312.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ohnishi K and Ohnishi T: Hyperthermic
sensitizers targeting heat-induced signal transductions. Ann Cancer
Res Therap. 15:35–40. 2007. View Article : Google Scholar
|
|
29
|
Ahmed K and Zaidi SF: Treating cancer with
heat: Hyperthermia as promising strategy to enhance apoptosis. J
Pak Med Assoc. 63:504–508. 2013.PubMed/NCBI
|
|
30
|
Platonova A, Boudreault F, Kapilevich LV,
Maksimov GV, Ponomarchuk O, Grygorczyk R and Orlov SN:
Temperature-induced inactivation of cytoplasmic biogel osmosensing
properties is associated with suppression of regulatory volume
decrease in A549 cells. J Membr Biol. 247:571–579. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bortner CD and Cidlowski JA: A necessary
role for cell shrinkage in apoptosis. Biochem Pharmacol.
56:1549–1559. 1998. View Article : Google Scholar
|
|
32
|
Shiozaki A, Takemoto K, Ichikawa D,
Fujiwara H, Konishi H, Kosuga T, Komatsu S, Okamoto K, Kishimoto M,
Marunaka Y, et al: The K-Cl cotransporter KCC3 as an independent
prognostic factor in human esophageal squamous cell carcinoma.
BioMed Res Int. 2014:9364012014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shiozaki A, Nako Y, Ichikawa D, Konishi H,
Komatsu S, Kubota T, Fujiwara H, Okamoto K, Kishimoto M, Marunaka
Y, et al: Role of the Na+/K+/2Cl−
cotransporter NKCC1 in cell cycle progression in human esophageal
squamous cell carcinoma. World J Gastroenterol. 20:6844–6859. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Han J, Xu X, Qin H, Liu A, Fan Z, Kang L,
Fu J, Liu J and Ye Q: The molecular mechanism and potential role of
heat shock-induced p53 protein accumulation. Mol Cell Biochem.
378:161–169. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rosenfeldt MT and Ryan KM: The multiple
roles of autophagy in cancer. Carcinogenesis. 32:955–963. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rosenfeldt MT and Ryan KM: The role of
autophagy in tumour development and cancer therapy. Expert Rev Mol
Med. 11:e362009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Levy JM and Thorburn A: Targeting
autophagy during cancer therapy to improve clinical outcomes.
Pharmacol Ther. 131:130–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hosoi K: Physiological role of aquaporin 5
in salivary glands. Pflugers Arch. 468:519–539. 2016. View Article : Google Scholar
|
|
40
|
Kang SK, Chae YK, Woo J, Kim MS, Park JC,
Lee J, Soria JC, Jang SJ, Sidransky D and Moon C: Role of human
aquaporin 5 in colorectal carcinogenesis. Am J Pathol. 173:518–525.
2008. View Article : Google Scholar : PubMed/NCBI
|