Introduction
Inflammation is a fundamental physiological process
for survival. However, excessive expression of pro-inflammatory
cytokines can cause a lethal systemic inflammation (1,2).
'Cholinergic anti-inflammatory pathway' is well known for its role
in autonomic nervous system (3).
Nicotine, a selective cholinergic agonist, inhibits
pro-inflammatory cytokine release in α7 nicotinic acetylcholine
receptor (α7 nAchR)-dependent manner (4). Substantial evidence points to a
critical role of nicotine in preventing nuclear translocation of
the NF-κB complex (5,6), which demonstrated the inhibitory
effects of nicotine on NF-κB activity (7,8).
Although therapeutic use of nicotine has been documented in
depression, Tourette's syndrome, Parkinson's disease, Crohn's
disease and ulcerative colitis (9–11),
the mechanisms of cholinergic anti-inflammation are still
uncertain.
Stat3 is critical for physiological regulation of
cell differentiation, growth, apoptosis and immune response
(12,13). Mice with stat3 knockout in
macrophages and neutrophils had enhanced inflammatory activity
(14). Deletion of stat3 results
in myeloid cell abnormalities and causes Crohn's disease-like
pathogenesis (15), indicating
that stat3 is a potential negative regulator of inflammation.
Nicotine exerts its anti-inflammatory effect via Jak2 and Stat3
signaling (15,16), however, little is known about the
role of stat3 phosphorylation in nicotine-inhibited NF-κB
activation.
Tumor necrosis factor-α induced protein-8-like 2
(TIPE2), a member of tumor necrosis factor-α-induced protein-8
family, inhibited TCR-mediated T cell activation, NF-κB activation
(17), and is involved in the
pathogenesis of stroke (18).
However, up to now, the role of TIPE2 in cholinergic
anti-inflammatory pathway is still unknown, which is definitely
important for controlling a variety of inflammation-associated
diseases.
Here we provide evidence that TIPE2 and stat3
mediated the anti-inflammation effect of nicotine, which is
ultimately involved in the inhibition of p65 activity. Firstly, it
was supported by the fact that both nicotine and TIPE2 inhibit
pro-inflammatory cytokine release with NF-κB inactivation.
Secondly, nicotine upregulates TIPE2 expression via Erk1/2 and
PI3K/Akt pathways. Moreover, the treatment with nicotine increase
stat3 phosphorylation and inhibits nuclear translocation of p65.
Importantly, co-immunoprecipitation assay reveals that the
treatment with nicotine augments the interaction of phosphorylated
stat3 and p65. Hence, this study reveals that TIPE2 upregulation
and stat3 phosphorylation might contribute to nicotine-mediated
anti-inflammation effect, indicating that TIPE2 and stat3 might be
potential molecules for dealing with inflammation-associated
diseases.
Materials and methods
Reagents
Nicotine, α-bungarotoxin and lipopolysaccharides
(LPS) were purchased from Sigma-Aldrich (MO, USA). PMA and
ionomycin were purchased from Beyotime (Shanghai, China).
Recombinant murine M-CSF and IL-4 were obtained from R&D
(Minneapolis, MN, USA). Fetal bovine serum was obtained from
Hyclone (Logan, UT, USA). Brefeldin A Solution, Fluorescene
conjugated antibodies and mouse IL-12/IL-4 ELISA kits were from
eBioscience (San Diego, CA, USA). NE-PER Nuclear and Cytoplasmic
Extraction reagents were purchased from Pierce (Rockford, IL, USA).
AG490, LY294002 and Wortmannin were purchased from Cayman Chemical
(Ann Arbor, MI, USA). U0126, PD98059, antibodies to total or
phosphorylated kinases, β-actin and histone H3 were purchased from
Cell Signaling (Beverly, MA, USA). TIPE2 antibody was purchased
from Abnova (Taipei, Taiwan). Protein A/G Plus-Agarose was
purchased from Santa Cruz Biotechnology (CA, USA). SYBR®
Premix Ex Taq™, TRIzol and Prime-Script Reverse Transcriptase were
purchased from Takara (Dalian, China).
Animals
Pathogen-free Balb/c mice (female, 3-week-old) were
purchased from Shanghai Laboratory Animal Center of the Chinese
Academy of Sciences (China) and kept at the Animal Center of Xiamen
University. All animal studies were approved by the Review Board of
Medical College of Xiamen University. Mice were subcutaneously
injected with nicotine (0.5 µg/10 g, twice per day) for 9
days. Control mice were subcutaneously injected with PBS. At the
end of treatment, mice were sacrificed and further investigation
was performed.
Cell lines
Murine macrophage cell lines Raw 264.7 cells were
obtained from Shanghai Cell Bank (Shanghai, China). Cells were
cultured in DMEM medium with 10% fetal bovine serum at 37°C in 5%
CO2. Cells were synchronized by serum starvation for at
least 12 h before the treatment of nicotine or LPS.
TIPE2 transfection
TIPE2 overexpressed Raw 264.7 cells were established
according to the method described previously (17). Briefly, 6×105 cells/well
were seeded in 6-well plates and transfected with pcDNA3.1-TIPE2
constructs and empty vector using PEI (Polyplus, AFAQ) according to
the manufacturer's instructions. TIPE2 overexpressed cells were
selected in 500 µg/ml G418 condition and confirmed by RT-PCR
and western blotting, respectively.
Bone marrow-derived murine
macrophage
Bone marrow-derived macrophage was prepared as
previous description (19).
Briefly, bone marrow mononuclear cells were prepared from bone
marrow suspensions by depletion of red cells and then cultured at a
density of 1×106 cells/ml in RPMI-1640 medium with 10
ng/ml of M-CSF and 1 ng/ml of IL-4. Non-adherent cells were gently
washed out with PBS on day 4 of culture; the remaining loosely
adherent clusters were used as macrophages. To explore the effect
of nicotine on α7 nAchR and TIPE2 expressions, macrophage was
conferred 0.1–10 µM nicotine 24-h stimulation. To determine
the effect of nicotine on TNF-α expression, macrophage pretreated
with 1 µg/ml α-bungarotoxin prior to nicotine treatment was
further conferred 100 ng/ml LPS stimulation.
ELISA
To investigate the effect of nicotine on cytokine
secretion, purified splenic T cells from nicotine-administered mice
were cultured at a density of 2×105/well in U-bottom
96-well plates in the presence of PMA (50 ng/ml) and ionomycin (1
µM) for 12 h. Culture supernatants were collected and the
concentrations of IL-12, IL-4 were determined by ELISA (21).
Flow cytometric measurement
Expressions of cell surface molecules and
pro-inflammatory cytokine release were determined by flow cytometry
according to the methods described previously (20).
Cytoplasmic and nuclear extracts
isolation
Cytoplasmic and nuclear extract was prepared as
previously described (21).
Briefly, TIPE2 overexpressed and control Raw 264.7 cells were
pretreated with nicotine (1 µM) prior to 100 ng/ml LPS 6-h
stimulation. Then, the cells were collected and suspended in
ice-cold CER buffer (cytoplasmic extraction reagent), vortexed for
10 min and ice-cold CER was added. The cytosolic fraction
(supernatant) was separated by centrifugation (16,000 × g, 5 min,
4°C) and the nuclear protein was separated by incubating insoluble
fraction with ice-cold NER (nuclear extraction reagent) for 40 min
and centrifuged at 16,000 × g for 10 min, 4°C. Protein
concentration was estimated using the Bio-Rad protein assay reagent
and an equal amount of proteins per sample of nuclear extract was
further analyzed by western blotting.
Western blotting
Proteins were obtained in lysis buffer as previously
described (22). Proteins were
loaded onto SDS-PAGE gels for electrophoresis and transferred to
PVDF membranes. After blocking in 5% fat-free milk in TBST for 1.5
h, the membranes were incubated with primary antibodies at 4°C
overnight. After that, the membranes were incubated with
corresponding HRP-conjugated secondary antibodies at room
temperature for 1.5 h. After washing six times with TBST (for 10
min each), bound antibodies were visualized using chemiluminescence
ECL. β-actin or histone H3 were used as loading control.
Reverse transcriptase polymerase chain
reaction
TIPE2 overexpressed Raw 264.7 and control cells were
seeded in 6-well plate (1×105 cells/well) and total RNA
was isolated using TRIzol and reverse-transcription was performed
according to the standard procedure (20). Subsequent PCR amplification was
performed using 2 µg cDNA in the following condition: 95°C
for 30 sec, 35 cycles of (95°C for 5 sec, 58°C for 30 sec, and 72°C
for 30 sec). β-actin was used as internal control. PCR products
were run on 2% agarose gels and analyzed under ultraviolet (UV)
light after ethidium bromide staining. β-actin: sense,
5′-ACCGTGGAGAAGAGC TACGA-3′; antisense, 5′-GTACTTGCGCTCAGAAGGAG-3′.
TIPE2 sense, 5′-CACCGCAATGGCTCCTTT-3′; antisense,
5′-CACCAACTCTAGCAGCACATC-3′.
Real-time PCR
Total RNA was extracted from mouse spleen and thymus
according to the manufacturer's instructions (17). Reverse transcription was performed
with oligo dT primers. Real-time PCR was carried out in Applied
Biosystems 7500 system with Power SYBR Green PCR Master Mix
(Applied Biosystems). Relative TIPE2 expression was determined with
β-actin as the control.
Immunoprecipitation
Immunoprecipitation was performed as previously
described (23). Briefly, Raw
264.7 cells pretreated with nicotine (1 µM) prior to 100
ng/ml LPS stimulation were harvested and lysed in RIPA buffer (PBS
containing 0.1% SDS, 0.5% sodium deoxycholate, 1% Nonidet P-40, 1
mM sodium orthovanadate, 1 mM PMSF and 3% protease inhibitor
cocktail). These lysates were then pre-cleared by incubation with
20 µl/ml Protein A/G agarose beads for 1 h at 4°C. After
brief centrifugation, the supernatant was added to the indicated
primary antibody or control IgG in RIPA buffer overnight at 4°C,
followed by the addition of 20 µl/ml Protein A/G agarose
beads. Immunoprecipitates were washed in RIPA buffer, re-suspended
in SDS sample buffer, boiled for 5 min and analysed by SDS/PAGE.
Proteins were electrophoretically transferred to PVDF membranes and
subjected to western blot analysis using the indicated
antibodies.
Statistical analysis
Each experiment was repeated at least 3 times and
confirmed that similar data were obtained. All data were presented
as mean with standard error means. Statistical significance was
tested using Student's t-test, one-way ANOVA with post Newman-Keuls
test. Statistical differences were considered to be significant at
p<0.05.
Results
α7 nAChR is involved in nicotine-mediated
inhibitory effect on inflammation
Although α7 nAChR is constitutive expressed on both
DCs and monocytes, the roles of α7 nAChR in nicotine-mediated
anti-inflammation are still uncertain. Toward this end, Balb/c mice
were subcutaneously administered with nicotine and pro-inflammatory
cytokines was determined. A statistically significant reduction in
LPS inducing pro-inflammatory cytokines of TNF-α, IL-4, IL-12 and
IL-2 occurred in both splenic CD4+ T cells (Fig. 1A and B). Subsequently, we tested if
such nicotine treatment affects α7 nAChR expression. To this end,
bone marrow-derived macrophage was treated with nicotine
(10−5–10−7 mol/l) and α7 nAChR expression was
determined. Importantly, a clear upregulation of α7 nAChR was
achieved by the treatment with nicotine (Fig. 1C), indicating that α7 nAChR is
involved in nicotine-mediated anti-inflammation effect. To test
this hypothesis, we pre-incubated macrophage with the selective α7
nAChR antagonist α-bungarotoxin. Interestingly, α-bungarotoxin
pre-incubation abolished the nicotine effect on TNF-α expression
(Fig. 1D), indicating that the
anti-inflammation effect of nicotine is α7 nAChR-dependent.
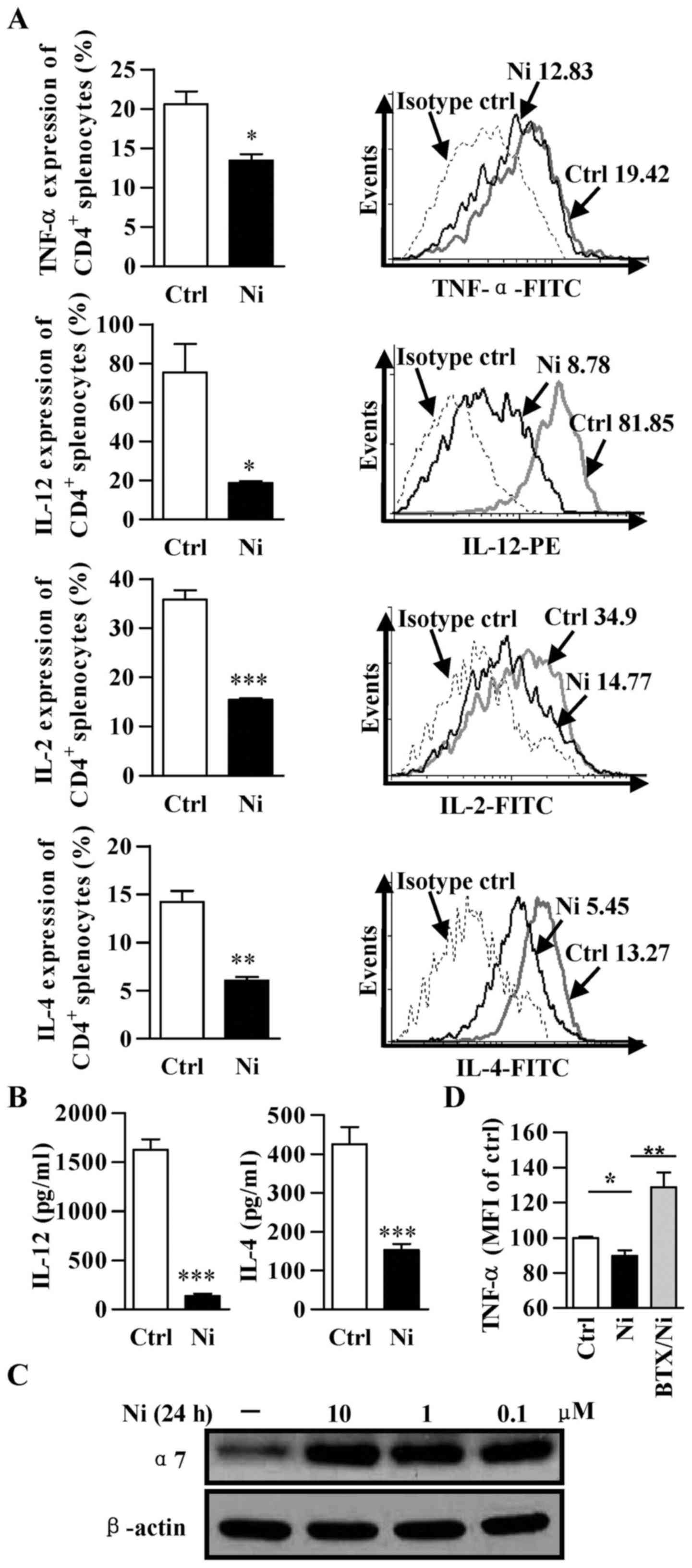 | Figure 1Nicotine treatment inhibits
pro-inflammatory cytokine production. Balb/c mice were
subcutaneously injected with nicotine (0.5 µg/10 g, twice
per day) for 9 days. Then, splenic T cells were stimulated with PMA
(50 ng/ml), ionomycin (1 µM) and LPS (100 ng/ml) for 6 h and
pro-inflammatory cytokines were determined by flow cytometry (A)
and ELISA (B) respectively. Numbers in (A) represent mean
fluorescence intensity (MFI) and positive cell percentage in each
gated area. Murine macrophages derived from bone marrow were
treated with nicotine for 24 h and α7 nAchR expression was
determined by western blotting (C). Murine macrophage pretreated
with 1 µg/ml α-bungarotoxin prior to nicotine (1 µM)
stimulation were further conferred LPS treatment (100 ng/ml) and
TNF-α expression was determined by flow cytometry (D). Data are
shown as mean ± SEM, n=3, *p<0.05, **p<0.01,
***p<0.001, Student's t-test or one-way ANOVA with
post Newman-Keuls test. A representative out of three independent
experiments is shown. Ni, nicotine; BTX, α-bungarotoxin. |
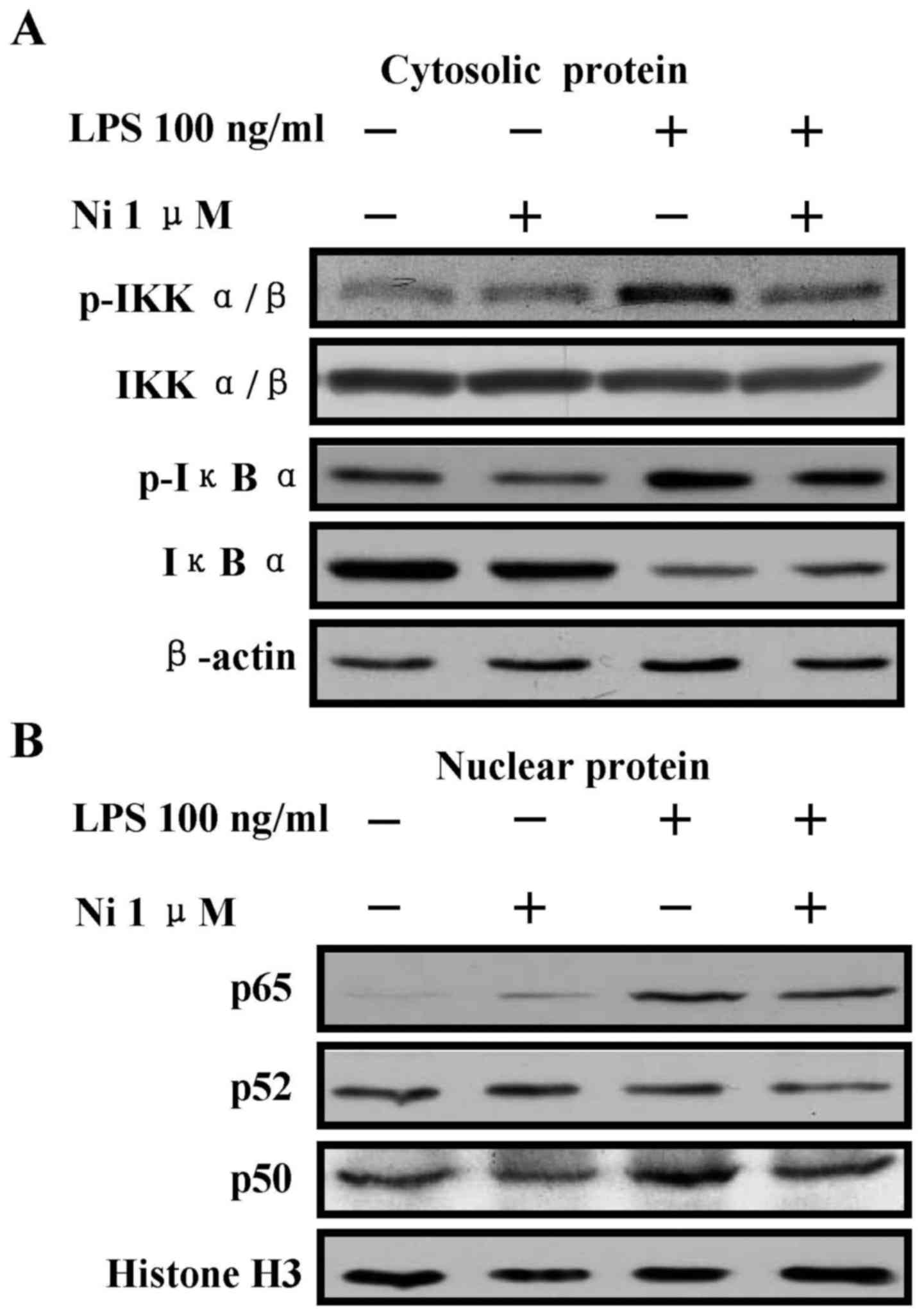
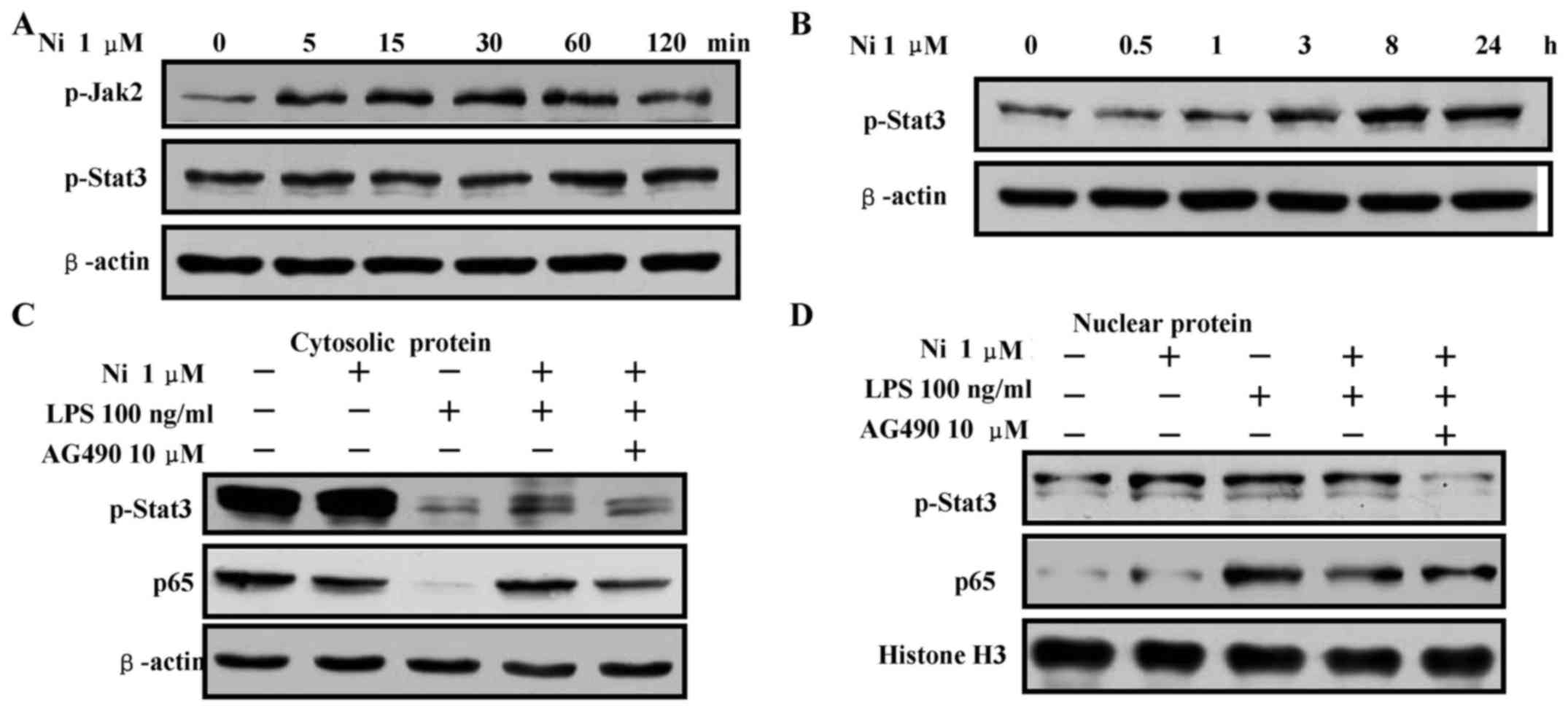
The treatment with nicotine inhibits
LPS-induced NF-κB activation
NF-κB pathway is crucial for pro-inflammatory
cytokine production (24), which
is regulated by IκBα phosphorylation and degradation (25). LPS stimulation augmented the
phosphorylation of IκBα, IKKα/β in cytoplasm (Fig. 2A) and increased the translocation
of p65 and p50 from cytosol to nucleus (Fig. 2B). The nicotine pretreatment
abrogated the LPS effect on the phosphorylation of IκBα, IKKα/β
(Fig. 2A) and the translocation of
p65 and p50 (Fig. 2B). These
observations were consistent to the studies previously reported
(6,26). All these findings indicate that
nicotine exhibits anti-inflammation effect by achieving NF-κB
inactivation.
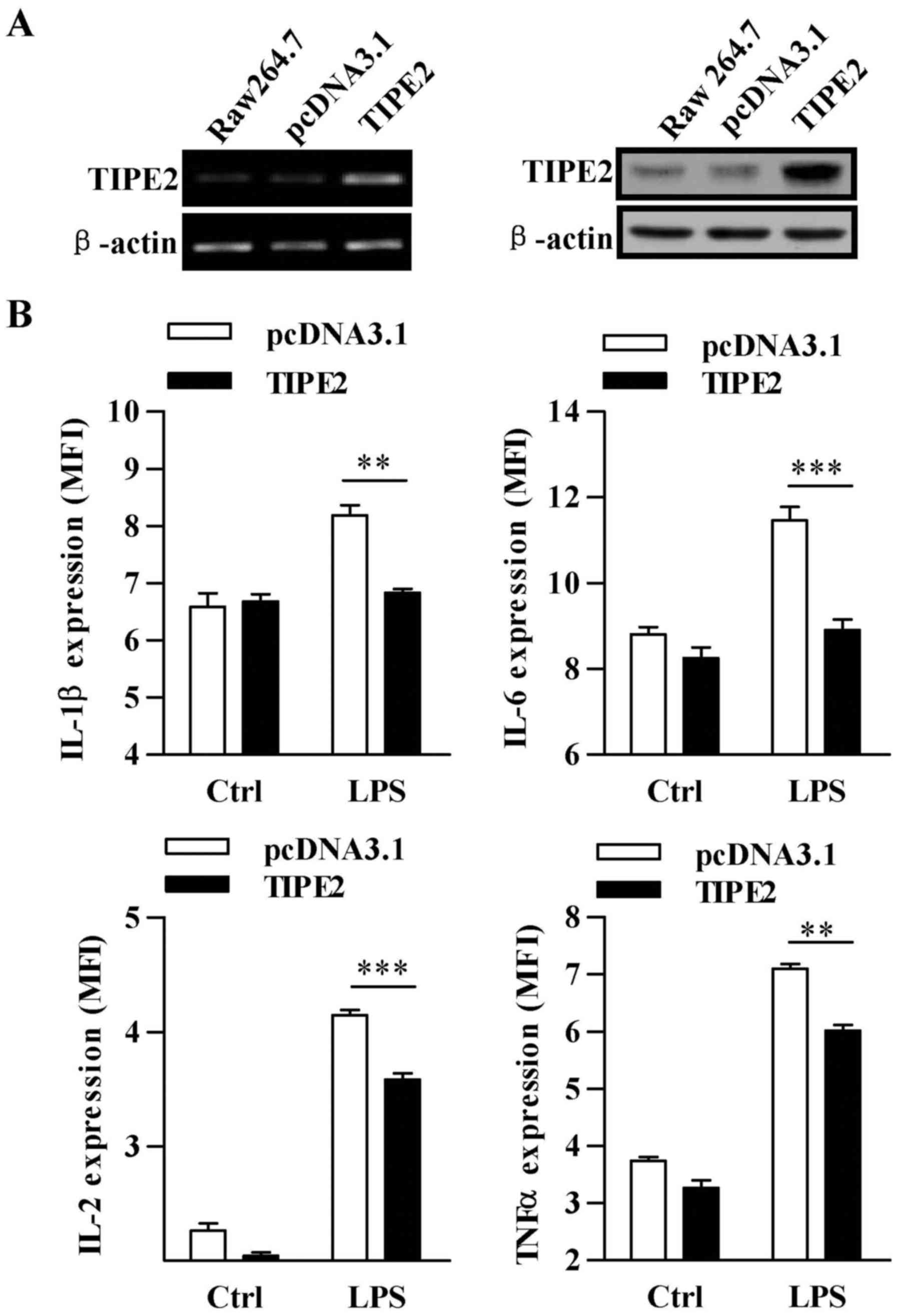
TIPE2 abrogates LPS-induced
pro-inflammatory cytokine expression
TIPE2 overexpressed Raw 264.7 cells were established
(Fig. 3A) and then the effect of
TIPE2 on pro-inflammatory cytokine expression was determined.
Without LPS stimulation, TIPE2 overexpression had no effect on
pro-inflammatory cytokine secretion. Although LPS increased the
expression of IL-1β, IL-6, IL-2 and TNF-α, TIPE2 overexpression
abrogated the effect of LPS on these pro-inflammatory cytokine
release with the inhibitory rates of 16.5, 22.3, 13.6 and 15.2%,
respectively (Fig. 3B), indicating
that TIPE2 is a negative regulator of inflammation.
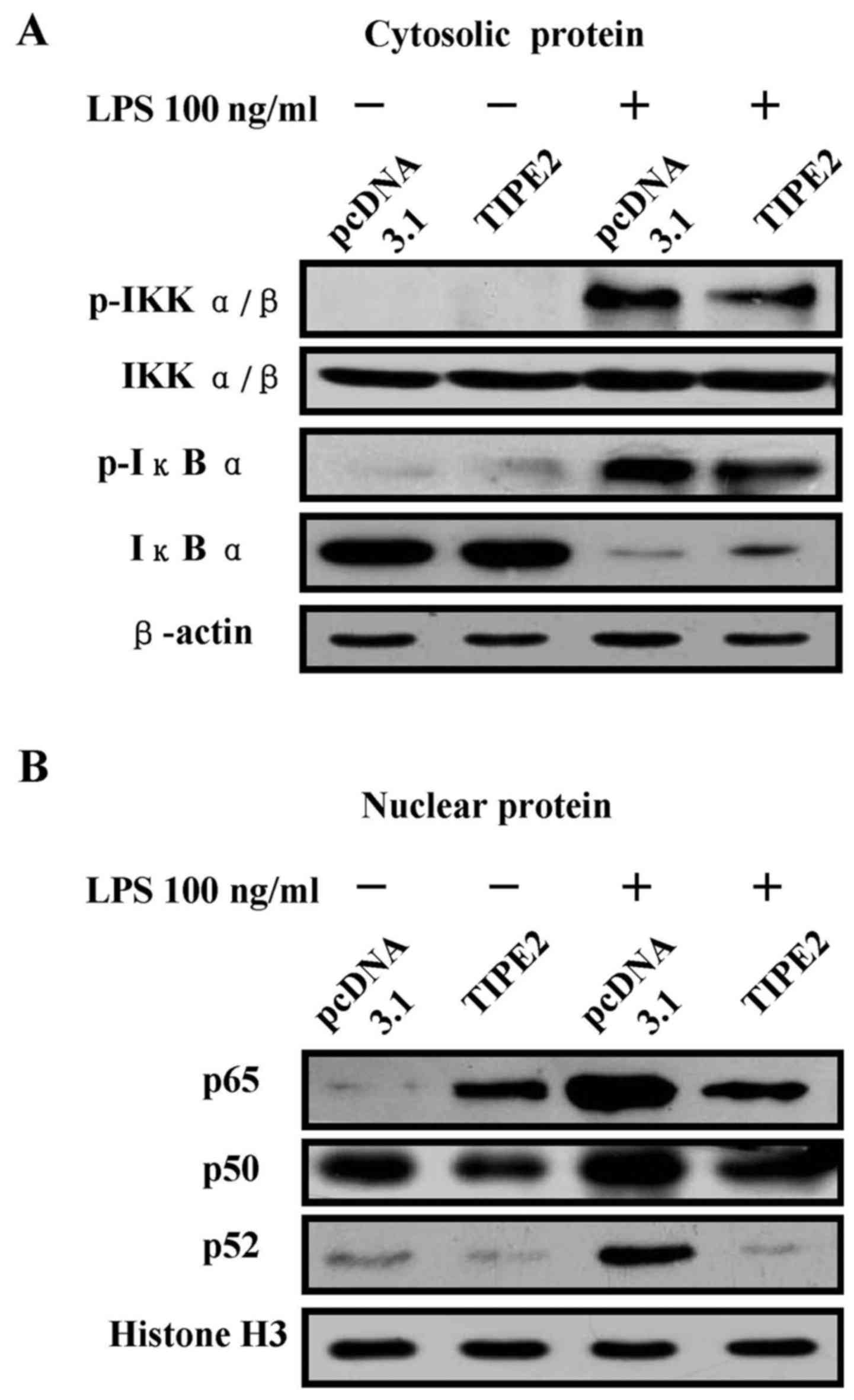
TIPE2 abolishes LPS effect on NF-κB
activation
TIPE2 overexpressed Raw 264.7 cells were treated
with LPS and NF-κB activation was investigated by western blotting.
LPS stimulation enhanced IKKα/β and IκBα phosphorylation in the
cytoplasm (Fig. 4A) and p65, p52,
p50 translocation to the nucleus (Fig.
4B). TIPE2 overexpression inhibites LPS-induced phosphorylation
of IKKα/β and IκBα in the cytoplasm (Fig. 4A) and nuclear translocation of p65,
p50 and p52 (Fig. 4B). These data
indicate that TIPE2 inhibits pro-inflammatory cytokine release by
achieving NF-κB inactivation.
α7 nAchR is involved in nicotine
upregulated TIPE2 expression
Both nicotine and TIPE2 decrease LPS-induced
inflammation by inhibiting NF-κB activation. However, until now,
little is known about the role of TIPE2 in nicotine-mediated
anti-inflammation effect. Nicotine administration increased TIPE2
transcription about 1.5- and 1.51-fold in spleen and thymus in
vivo, respectively (Fig. 5A).
The treatment with nicotine augmented TIPE2 expression in both
macrophages and Raw 264.7 cells in vitro (Fig. 5B and C). To determine the role of
α7 nAChR in nicotine-augmented TIPE2 expression, the cells were
pre-incubated with α-bungarotoxin prior to nicotine treatment. The
treatment with nicotine increased TIPE2 expression, however, the
pretreatment with α-bungarotoxin abrogated the nicotine effect on
TIPE2 upregulation (Fig. 5D).
These results indicate that α7 nAChR is involved in
nicotine-augmented TIPE2 expression.
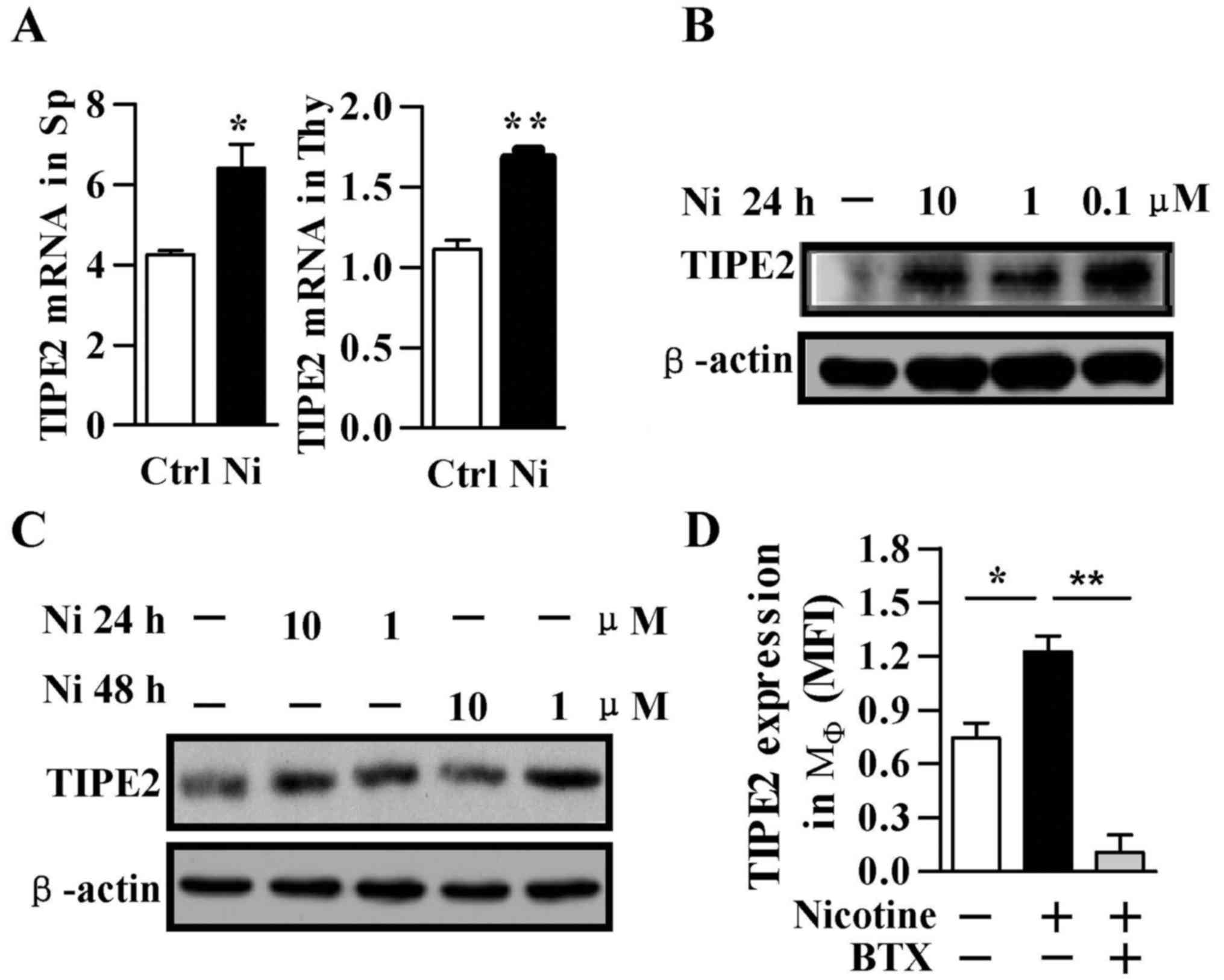 | Figure 5α7 nAchR is involved in
nicotine-increased TIPE2 expression. Balb/c mice (A), murine bone
marrow-derived macrophage (B) and Raw 264.7 cells (C) were treated
with nicotine. TIPE2 expression in spleen/thymus (A), macrophage
(B) and Raw 264.7 cells (C) was determined by real-time PCR and
western blotting, respectively. For in vivo animal test,
mice were subcutaneously injected with nicotine (0.5 µg/10
g, twice per day) for 9 days. Murine macrophage was pretreated with
1 µg/ml α-bungarotoxin prior to 1 µM nicotine
stimulation and the expression of TIPE2 was determined by flow
cytometry (D). Data are given as mean ± SEM, n=3,
*p<0.05, **p<0.01, Student's t-test or
one-way ANOVA with post Newman-Keuls test. A representative out of
three independent experiments is shown. Ni, nicotine; BTX,
α-bungarotoxin. |
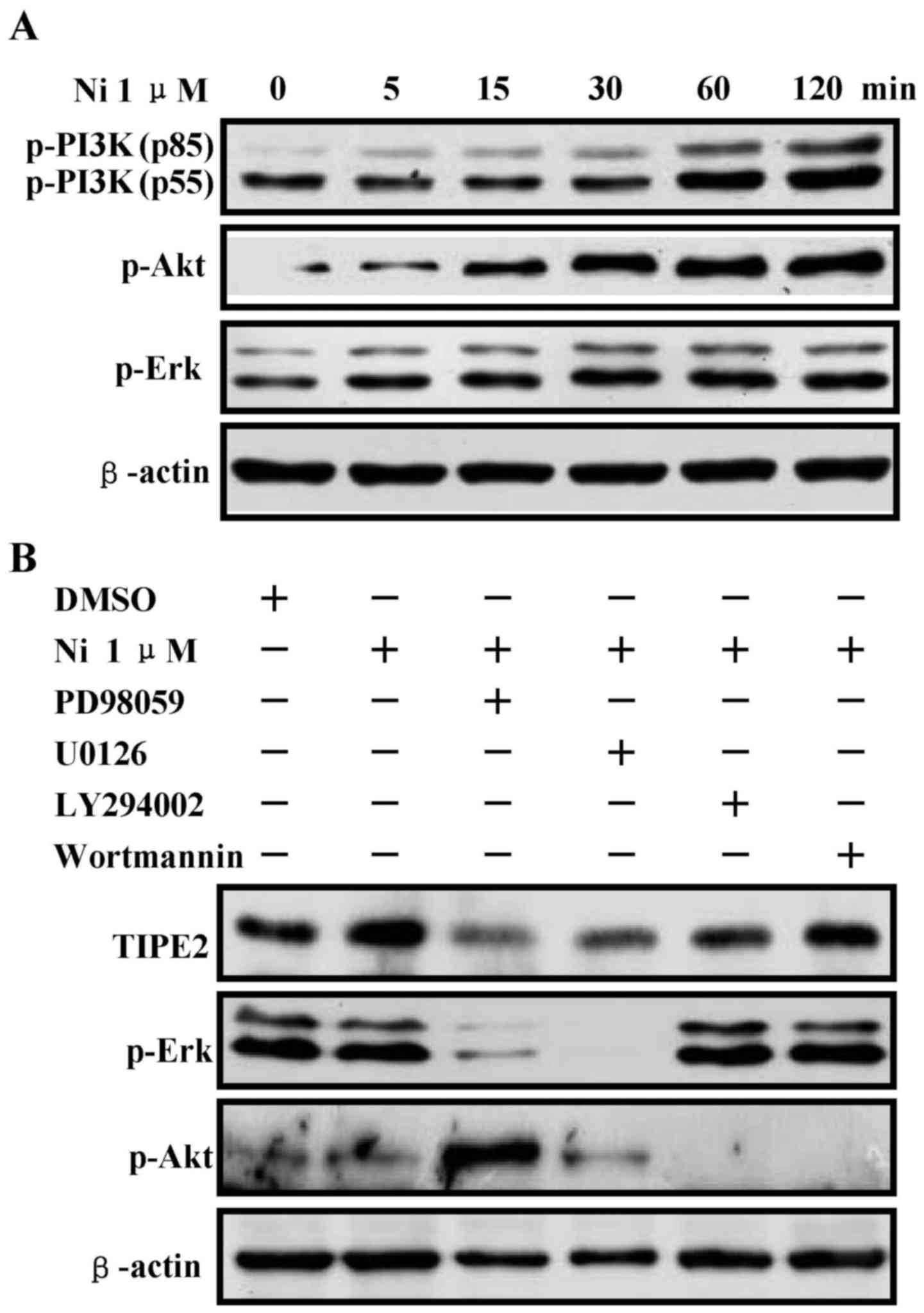
Nicotine upregulates TIPE2 expression via
Erk1/2 and PI3K/Akt pathways
Although α7 nAChR is involved in nicotine-increased
TIPE2 upregulation, the mechanism of nicotine-augmented TIPE2
expression is still unclear. To address this issue, PD98059, U0126,
LY294002 and Wortmannin were used prior to nicotine stimulation and
TIPE2 expression was determined. Consistent with our previous
report (20), nicotine rapidly
increase the phosphorylation of PI3K/Akt and Erk1/2, from 5 to 120
min (Fig. 6A). As these inhibitors
efficiently inhibited the related kinase activities, the
pretreatments of U0126, PD98059, LY294002 and Wortmannin abrogating
the effect of nicotine on TIPE2 upregulation (Fig. 6B), indicate that nicotine-augmented
TIPE2 expression via Erk1/2 and PI3K/Akt pathways.
Nicotine-increased stat3 phosphorylation
contributes to the inhibition of p65 translocation
Nicotine increases stat3 phosphorylation and
suppress inflammatory cytokines production (16,27),
indicating that stat3 might be a negative regulator of
inflammation. To explore the effect of stat3 phosphorylation on p65
translocation, Raw 264.7 cells were pretreated with AG490 prior to
nicotine stimulation. p65 translocation and stat3 phosphorylation
were investigated by western blotting. As upstream kinase of stat3,
Jak2 phosphorylation was increased by the treatment with nicotine
from 5 to 120 min (Fig. 7A). Stat3
phosphorylation was also achieved by the treatment with nicotine
from 60 min to 24 h (Fig. 7A and
B). In contrast to decreased levels of p65 and phosphorylated
stat3 in the cytosol (Fig. 7C),
LPS treatment increased the levels of p65 and phosphorylated stat3
in the nucleus (Fig. 7D),
indicating that LPS enhance nuclear translocation of p65 and
phosphorylated stat3. Compared with LPS-treated cells, the
pretreatment with nicotine increased the levels of p65 and
phosphorylated stat3 in cytosol (Fig.
7C) and decreased the levels of p65 and phosphorylated stat3 in
the nucleus (Fig. 7D).
Importantly, when AG490 was added to inhibit stat3 activities, the
decreased p65 in cytosol and increased p65 in nucleus were also
achieved (Fig. 7C and D). These
data indicate that the phosphorylation of stat3 plays a potential
role in nicotine-mediated NF-κB inactivation.
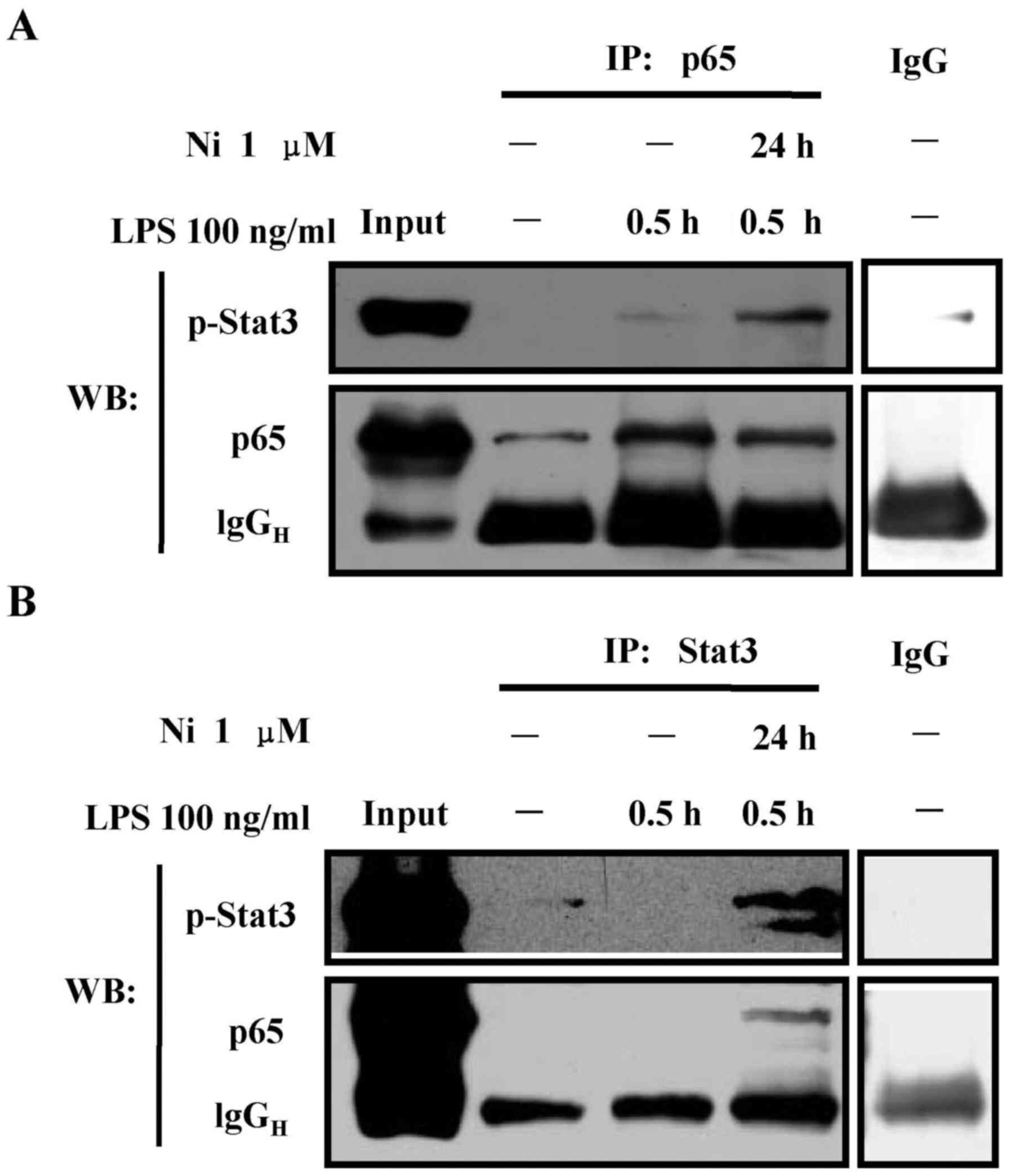
Nicotine augments the interaction of
phosphorylated stat3 and p65
Stat3 inhibited inducible No synthase expression in
mesangial cells by interacting with NF-κB (28). Due to the observation that the
pretreatment with nicotine enhanced stat3 phosphorylation and
inhibited LPS-induced p65 translocation, we speculated that the
inhibitory effect of nicotine on NF-κB activation might be due to
the increased interaction of phosphorylated stat3 and p65. In
Fig. 8A, while LPS treatment had
no effect on the interaction of phosphorylated stat3 and p65,
nicotine pretreatment achieved an obvious band of phosphorylated
stat3 (Fig. 8A). As LPS treatment
increased stat3 phosphorylation from 4 to 6 h in Raw264.7 cells
(29), it is no surprise to find
that phosphorylated stat3 can not be detected in short-term
LPS-stimulated cells (30 min) when total stat3 antibody was applied
to perform co-immunoprecipitation assay (Fig. 8B). Importantly, despite the level
of control heavy chain in nicotine-pretreated cells was slight
higher, the obvious bands of p65 and phosphorylated stat3 revealed
an interaction of phosphorylated stat3 and p65 (Fig. 8B). These observations indicate that
the inhibitory effect of nicotine on NF-κB activation is mediated
by the interaction of phosphorylated stat3 and p65.
Discussion
Inflammation contribute to systemic capillary
leakage syndrome, tissue injury and fatal organ failure (1,2).
Cholinergic agonist was useful for control of depression,
Tourette's syndrome, Parkinson's disease, Crohn's disease and
ulcerative colitis (9–11), indicating that α7 nAChR is crucial
for inflammation regulation (30).
Preserving cytoplasmic levels of inhibitor of NF-κB (IκB) was found
to be essential for nicotine-mediated NF-κB inhibition (5,6).
Whereas, Jak2/stat3 signaling was also documented to facilitate
nicotine's anti-inflammation effect (16). Hence, the exact role of stat3 in
nicotine-mediated cholinergic NF-κB inhibition is still uncertain.
In this study, we demonstrated that nicotine exerts its
anti-inflammatory effect by TIPE2 upregulation and augmented
interaction of phosphorylated stat3 and p65. All these observations
indicate that TIPE2 and stat3 might be potential molecules for
dealing with inflammation-associated diseases.
TIPE2 inhibits TCR-mediated T cell activation and
maintain immune homeostasis (17).
In this study, TIPE2 upregulation via Erk1/2 and PI3K/Akt pathways
was found to contribute to nicotine-mediated NF-κB inhibition.
PI3K/Akt/mToR pathway mediated nicotine-induced tumor growth and
chemoresistance in bladder cancer (31). Other studies also demonstrated that
co-stimulator molecules on dendritic cell can be up-rgulated by
nicotine via Erk1/2 and PI3K/Akt pathway (20,32).
As PI3K and AKT regulated the epigenetic regulator KMT2D and
histone methyltransferase WHSC1 respectively (33,34),
PI3K/AKT-mediated epigenetic regulation might contribute to
nicotine affecting TIPE2 expression. As p38/JNK MAPK and stat3
pathways were also activated by nicotine treatment (Fig. 7) (20). Further studies are needed to
explore epigenetic regulation of these kinases in
nicotine-augmented TIPE2 expression.
Cell viability assay showed that lower doses of
nicotine had no effect on dendritic cell apoptosis, but higher
doses of nicotine actually induced >90% cell programming into
the process of apoptosis, indicating that nicotine is somehow toxic
to cell viability (35).
Short-term exposure to nicotine enhanced lymphocyte c-fos gene
expression, but long-term exposure downregulated nAchR mRNA
expression (36). In our
experiments, nicotine had a maximal effect at 0.1 µM
concentration on TIPE2 upregulation, which can also be found in
other reports (32). This
controversy regarding the effects of nicotine on TIPE2 expression
may be attributed to nicotine concentration used in the
experiments.
NF-κB pathway, which is regulated by IκBα
phosphorylation and degradation (25), is crucial for pro-inflammatory
cytokine expression (24). In this
study, TIPE2 was found to inhibit pro-inflammatory cytokine release
(Fig. 3) and induce NF-κB
inactivation (Fig. 4).
Interestingly, TNF-α induced protein 3 (TNFAIP3, A20), has similar
effect as TIPE2 on NF-κB pathway activation (37). N-terminal of A20 encodes a
deubiquitinating (DUB) domain which mediates the deubiquitination
of K63-polyubiquitinated NF-κB signaling proteins such as TRAF6 and
RIP1, while the C-terminal of A20 encodes seven zinc-finger (ZF)
motifs and confers E3 ubiquitin ligase activity (38). The report that A20 binding to
unanchored K63-linked polyubiquitin chains and NEMO to block IKK's
upstream kinase TAK1 (39)
indicated that a direct non-catalytic mechanism of A20 inhibit
NF-κB activities. As both TIPE2 and A20 have negative regulation
effects on immune function and NF-κB activation, whether TIPE2 had
deubiquitinase enzyme domains and TIPE2 antagonized the
ubiquitination of NEMO-IκB kinase complex (IKK) regulatory subunit
by the interaction with NEMO need further exploration.
Stat3, which is critical for cell differentiation,
growth, apoptosis, innate and adaptive immunity (13), can be activated by nAChR ligation
and regulated pro-inflammatory cytokine release (27). In this study, the phosphorylation
of stat3 was augmented by the treatment with nicotine (Fig. 7). Further investigations revealed
that stat3 phosphorylation prevented nuclear translocation of p65
(Fig. 7). Stat3 inhibited
inducible nitric oxide synthase transcription by interacting with
NF-κB (28). Hence, it is not
surprising to find that the pretreatment of AG490 partially
restored the effect of LPS on p65 translocation. Importantly, the
treatment with nicotine augmented the interaction of p65 and
phosphorylated stat3 (Fig. 8),
indicating that the inhibitory effect of nicotine on NF-κB
activation was mediated by the interaction of phosphorylated stat3
and p65.
Taken together, our studies revealed that the
anti-inflammation effect of nicotine might be due to TIPE2
upregulation and stat3 phosphorylation, providing that TIPE2 and
stat3 might be potential molecules for dealing with
inflammation-associated diseases.
Acknowledgments
We thank Professor Y.H. Chen (University of
Pennsylvania, USA) for kindly providing TIPE2 plasmid and valuable
advice. Also, we thank Jin Hua Su and Fu Chen for excellent animal
care. This study was supported by grants from the State Key
Laboratory of Oncogenes and Related Genes (no. 90-14-05) and by
grants from the National Natural Science Foundation of China (no.
81273203).
References
|
1
|
Matteoli G, Gomez-Pinilla PJ, Nemethova A,
Di Giovangiulio M, Cailotto C, van Bree SH, Michel K, Tracey KJ,
Schemann M, Boesmans W, et al: A distinct vagal anti-inflammatory
pathway modulates intestinal muscularis resident macrophages
independent of the spleen. Gut. 63:938–948. 2014. View Article : Google Scholar
|
|
2
|
Martelli D, McKinley MJ and McAllen RM:
The cholinergic anti-inflammatory pathway: A critical review. Auton
Neurosci. 182:65–69. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bonaz B, Sinniger V and Pellissier S:
Anti-inflammatory properties of the vagus nerve: Potential
therapeutic implications of vagus nerve stimulation. J Physiol.
594:5781–5790. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Han B, Li X and Hao J: The cholinergic
anti-inflammatory pathway: An innovative treatment strategy for
neurological diseases. Neurosci Biobehav Rev. 77:358–368. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guarini S, Altavilla D, Cainazzo MM,
Giuliani D, Bigiani A, Marini H, Squadrito G, Minutoli L, Bertolini
A, Marini R, et al: Efferent vagal fibre stimulation blunts nuclear
factor-kappaB activation and protects against hypovolemic
hemorrhagic shock. Circulation. 107:1189–1194. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Saeed RW, Varma S, Peng-Nemeroff T, Sherry
B, Balakhaneh D, Huston J, Tracey KJ, Al-Abed Y and Metz CN:
Cholinergic stimulation blocks endothelial cell activation and
leukocyte recruitment during inflammation. J Exp Med.
201:1113–1123. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Karin M and Greten FR: NF-kappaB: Linking
inflammation and immunity to cancer development and progression.
Nat Rev Immunol. 5:749–759. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin Y, Bai L, Chen W and Xu S: The
NF-kappaB activation pathways, emerging molecular targets for
cancer prevention and therapy. Expert Opin Ther Targets. 14:45–55.
2010. View Article : Google Scholar
|
|
9
|
Avila J and Díaz-Nido J: Tangling with
hypothermia. Nat Med. 10:460–461. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dong J, Segawa R, Mizuno N, Hiratsuka M
and Hirasawa N: Inhibitory effects of nicotine derived from
cigarette smoke on thymic stromal lymphopoietin production in
epidermal keratinocytes. Cell Immunol. 302:19–25. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Libert C: Inflammation: A nervous
connection. Nature. 421:328–329. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jie Z, Dinwiddie DL, Senft AP and Harrod
KS: Regulation of STAT signaling in mouse bone marrow derived
dendritic cells by respiratory syncytial virus. Virus Res.
156:127–133. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Subramaniam A, Shanmugam MK, Perumal E, Li
F, Nachiyappan A, Dai X, Swamy SN, Ahn KS, Kumar AP, Tan BK, et al:
Potential role of signal transducer and activator of transcription
(STAT)3 signaling pathway in inflammation, survival, proliferation
and invasion of hepatocellular carcinoma. Biochim Biophys Acta.
1835:46–60. 2013.
|
|
14
|
Takeda K, Clausen BE, Kaisho T, Tsujimura
T, Terada N, Förster I and Akira S: Enhanced Th1 activity and
development of chronic enterocolitis in mice devoid of Stat3 in
macrophages and neutrophils. Immunity. 10:39–49. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Welte T, Zhang SS, Wang T, Zhang Z,
Hesslein DG, Yin Z, Kano A, Iwamoto Y, Li E, Craft JE, et al: STAT3
deletion during hematopoiesis causes Crohn's disease-like
pathogenesis and lethality: A critical role of STAT3 in innate
immunity. Proc Natl Acad Sci USA. 100:1879–1884. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
de Jonge WJ, van der Zanden EP, The FO,
Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S,
Akira S, van den Wijngaard RM, et al: Stimulation of the vagus
nerve attenuates macrophage activation by activating the Jak2-STAT3
signaling pathway. Nat Immunol. 6:844–851. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun H, Gong S, Carmody RJ, Hilliard A, Li
L, Sun J, Kong L, Xu L, Hilliard B, Hu S, et al: TIPE2, a negative
regulator of innate and adaptive immunity that maintains immune
homeostasis. Cell. 133:415–426. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Y, Wei X, Liu L, Liu S, Wang Z,
Zhang B, Fan B, Yang F, Huang S, Jiang F, et al: TIPE2, a novel
regulator of immunity, protects against experimental stroke. J Biol
Chem. 287:32546–32555. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gao FG, Wan F and Gu JR: Ex vivo nicotine
stimulation augments the efficacy of therapeutic bone
marrow-derived dendritic cell vaccination. Clin Cancer Res.
13:3706–3712. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jin HJ, Sui HX, Wang YN and Gao FG:
Nicotine up-regulated 4-1BBL expression by activating Mek-PI3K
pathway augments the efficacy of bone marrow-derived dendritic cell
vaccination. J Clin Immunol. 33:246–254. 2013. View Article : Google Scholar
|
|
21
|
Ke SZ, Ni XY, Zhang YH, Wang YN, Wu B and
Gao FG: Camptothecin and cisplatin upregulate ABCG2 and MRP2
expression by activating the ATM/NF-κB pathway in lung cancer
cells. Int J oncol. 42:1289–1296. 2013.PubMed/NCBI
|
|
22
|
Jin HJ, Li HT, Sui HX, Xue MQ, Wang YN,
Wang JX and Gao FG: Nicotine stimulated bone marrow-derived
dendritic cells could augment HBV specific CTL priming by
activating PI3K-Akt pathway. Immunol Lett. 146:40–49. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lu ZG, Liu H, Yamaguchi T, Miki Y and
Yoshida K: Protein kinase Cdelta activates RelA/p65 and nuclear
factor-kappaB signaling in response to tumor necrosis factor-alpha.
Cancer Res. 69:5927–5935. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nyati KK, Masuda K, Zaman MM, Dubey PK,
Millrine D, Chalise JP, Higa M, Li S, Standley DM, Saito K, et al:
TLR4-induced NF-κB and MAPK signaling regulate the IL-6 mRNA
stabilizing protein Arid5a. Nucleic Acids Res. 45:2687–2703. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Buhrmann C, Mobasheri A, Busch F, Aldinger
C, Stahlmann R, Montaseri A and Shakibaei M: Curcumin modulates
nuclear factor kappaB (NF-kappaB)-mediated inflammation in human
tenocytes in vitro: Role of the phosphatidylinositol 3-kinase/Akt
pathway. J Biol Chem. 286:28556–28566. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang H, Liao H, Ochani M, Justiniani M,
Lin X, Yang L, Al-Abed Y, Wang H, Metz C, Miller EJ, et al:
Cholinergic agonists inhibit HMGB1 release and improve survival in
experimental sepsis. Nat Med. 10:1216–1221. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang T, Niu G, Kortylewski M, Burdelya L,
Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola
D, et al: Regulation of the innate and adaptive immune responses by
Stat-3 signaling in tumor cells. Nat Med. 10:48–54. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu Z, Zhang W and Kone BC: Signal
transducers and activators of transcription 3 (STAT3) inhibits
transcription of the inducible nitric oxide synthase gene by
interacting with nuclear factor kappaB. Biochem J. 367:97–105.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lee SB, Lee WS, Shin JS, Jang DS and Lee
KT: Xanthotoxin suppresses LPS-induced expression of iNOS, COX-2,
TNF-α, and IL-6 via AP-1, NF-κB, and JAK-STAT inactivation in RAW
264.7 macrophages. Int Immunopharmacol. 49:21–29. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pavlov VA and Tracey KJ: The vagus nerve
and the inflammatory reflex - linking immunity and metabolism. Nat
Rev Endocrinol. 8:743–754. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yuge K, Kikuchi E, Hagiwara M, Yasumizu Y,
Tanaka N, Kosaka T, Miyajima A and Oya M: Nicotine induces tumor
growth and chemoresistance through activation of the PI3K/Akt/mTOR
pathway in bladder cancer. Mol Cancer Ther. 14:2112–2120. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aicher A, Heeschen C, Mohaupt M, Cooke JP,
Zeiher AM and Dimmeler S: Nicotine strongly activates dendritic
cell-mediated adaptive immunity: Potential role for progression of
atherosclerotic lesions. Circulation. 107:604–611. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Toska E, Osmanbeyoglu HU, Castel P, Chan
C, Hendrickson RC, Elkabets M, Dickler MN, Scaltriti M, Leslie CS,
Armstrong SA, et al: PI3K pathway regulates ER-dependent
transcription in breast cancer through the epigenetic regulator
KMT2D. Science. 355:1324–1330. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li N, Xue W, Yuan H, Dong B, Ding Y, Liu
Y, Jiang M, Kan S, Sun T, Ren J, et al: AKT-mediated stabilization
of histone methyltransferase WHSC1 promotes prostate cancer
metastasis. J Clin Invest. 127:1284–1302. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hu SX, Sui HX, Jin HJ, Ni XY, Liu XX, Xue
MQ, Zhang Y and Gao FG: Lipopolysaccharide and dose of nicotine
determine the effects of nicotine on murine bone marrow-derived
dendritic cells. Mol Med Rep. 5:1005–1010. 2012.PubMed/NCBI
|
|
36
|
Kawashima K and Fujii T: The lymphocytic
cholinergic system and its contribution to the regulation of immune
activity. Life Sci. 74:675–696. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dixit VM, Green S, Sarma V, Holzman LB,
Wolf FW, O'Rourke K, Ward PA, Prochownik EV and Marks RM: Tumor
necrosis factor-alpha induction of novel gene products in human
endothelial cells including a macrophage-specific chemotaxin. J
Biol Chem. 265:2973–2978. 1990.PubMed/NCBI
|
|
38
|
Wertz IE, O'Rourke KM, Zhou H, Eby M,
Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, et al:
De-ubiquitination and ubiquitin ligase domains of A20 downregulate
NF-kappaB signalling. Nature. 430:694–699. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Skaug B, Chen J, Du F, He J, Ma A and Chen
ZJ: Direct, noncatalytic mechanism of IKK inhibition by A20. Mol
Cell. 44:559–571. 2011. View Article : Google Scholar : PubMed/NCBI
|