Introduction
Androgen receptor (AR) is a ligand-dependent
transcription factor and regulates diverse aspects of development,
cell growth and homeostasis (1-3).
The full-length AR (AR-FL) functional domains mainly include a DNA
binding domain (DBD) flanking with the N-terminal domain (NTD) and
C-terminal ligand binding domain (LBD). The constitutively active
AR splicing variants (AR-Vs) originate from aberrant splicing of AR
pre-mRNA via various mechanisms, including the incorporation of
cryptic exons containing a premature stop codon and exon-skipping
events (4-7). The two predominant AR-Vs, AR-V7 and
ARv567es, have lost all or part of the LBD, have become
constitutively active, and enable androgenic signaling during
androgen deprivation therapy (ADT) to drive the development of the
castration-resistant prostate cancers (CRPCs). They function as
AR-FL in binding the androgen response element (ARE) and
interacting with coregulators (8). They can homodimerize and also
heterodimerize with AR-FL in an androgen-independent manner to
drive AR signaling during ADT (9). AR-Vs not only upregulate canonical
AR target genes mainly related to metabolism, secretion and
differentiation, such as PSA and FKBP5, but also AR-Vs-specific
genes UBE2C and CCNA2 associated with cell cycle
progression (10,11). AR-V7 expression is very low in
normal prostate, whereas it is increased in tumors and is further
elevated in CRPC (6,12,13). Nuclear AR-V7 emerges with CRPC in
75% of cases following ADT in comparison with those in <1% prior
to therapy (13,14). AR-V7, particularly in the
circulating tumor cells, is a biomarker for predicting CRPC
response to ADT (8,13,15,16). Nuclear ARv567es is also
significantly elevated in CRPC and metastasis (17). The constitutively active AR-Vs
escape challenges by the current repertoire of agents targeting the
ligand binding domain of AR. Immense efforts have been made to
develop agents targeting AR-Vs in recent years (18-20). However, very few of these have
been approved for clinical trials to date, such as EPI-506 that
binds to the TAU-5 in N-terminal domain (19) and the repurposed anti-helminthic
drug, niclosamide, that promotes the degradation of AR-V7 protein
(20).
Proliferating cell nuclear antigen (PCNA) is a
non-oncogenic protein essential for cell growth and survival. PCNA
encircles DNA and serves as platforms for partner proteins involved
in DNA replication and DNA repair, as well as other cellular
processes (21-23) The partner proteins bind to PCNA
through their PCNA interaction protein (PIP)-box, AlkB homologue 2
PCNA-interacting motif (APIM), and/or other motifs (22,24,25). PCNA is overexpressed in all tumors
(22). PCNA overexpression is
associated with advanced disease and the metastasis of prostate
cancer (26-28). Targeting PCNA as a novel strategy
for cancer therapy was previously explored with small peptides
mimicking the APIM or 'cancer-associated PCNA' (caPCNA) (29,30), and small molecules T2AA targeting
the PIP-box binding cavity of PCNA (31) and AOH1160 targeting caPCNA
(32). Previously, the authors
developed small molecule PCNA inhibitors (PCNA-Is) that block PCNA
relocalization and chromatin association (33-36). PCNA-I1S, identified in the
structure-activity relationship (SAR) analysis of PCNA-Is, is a
more potent PCNA inhibitor than PCNA-I1 (34). These peptides and small molecules
were shown to be well-tolerated in animals and exerted therapeutic
effects against various types of tumors, particularly when combined
with DNA damage drugs (29,30,32,35).
A previous study demonstrated that PCNA interacts
with AR and several proteins involved in DNA synthesis and cell
cycle regulation in replicating tumor cells (37). The aim of the present study was to
investigate the potential role of PCNA in the regulation of AR
activity. A PIP-box consensus sequence was identified at the
N-terminus of AR and it was found that PCNA directly complexes with
AR-FL, AR-V7 and ARv567es, very likely via the PIP-box. PCNA-I1S
inhibits AR-FL transcriptional activity and the expression of the
AR target genes, PSA and p21/WAF, as well as AR-FL and AR-V7
autoregulated by AR signaling. T2AA, a small molecule PIP-box
inhibitor, reduces the PCNA interaction with AR-FL and AR-V7, and
inhibits AR transcriptional activity and the expression of AR
target genes. PCNA-I1 exerts additive inhibitory effects with ADT
on the growth of CRPC cells expressing both AR-FL and AR-V7, but
not on those without AR expression. The present study developed an
AR PIP-box mimicking peptide R9-AR-PIP, which binds to PCNA and
inhibits AR transcriptional activity, the expression of AR target
genes and the growth of AR-expressing cells.
Materials and methods
Reagents
Dihydrotestosterone (DHT, cat. no. D-073), T2AA
(cat. no. SML0794), recombinant AR and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT,
cat. no. M5655), were purchased from Sigma-Aldrich; Merck KGaA.
Antibodies against mouse PCNA (cat. no. 2586) and rabbit PCNA (cat.
no. 13110) and p21/WAF (cat. no. 2946) were purchased from Cell
Signaling Technology, Inc. Antibodies against AR (N20, cat. no.
sc-816 and H-280, cat. no. sc-13062), TMPRSS2 (cat. no. sc-515727)
and GAPDH (cat. no. sc-47724), as well as PCNA siRNA (cat. no.
sc-29440) and control siRNA (cat. no. sc-36869), were purchased
from Santa Cruz Biotechnology, Inc. Enzalutamide (ENZ, cat. no.
S1250) was purchased from Selleckchem. Antibody against
prostate-specific antigen (PSA) (cat. no. A0562) was obtained from
Dako, Agilent Technologies, Inc. The DC Protein assay kit I (cat.
no. 5000111) was purchased from Bio-Rad Laboratories, Inc. The
luciferase assay system (cat. no. E1500) was obtained from Promega
Corporation. Lipofectamine 3000® (cat. no. L3000015) was
obtained from Thermo Fisher Scientific, Inc. IRDye 680LT anti-mouse
fluorescent secondary antibody (cat. no. 926-68022) and IRDye 680LT
anti-rabbit fluorescent secondary antibody (cat. no. 926-68023) for
western blot analysis were from LI-COR Biosciences. The GST-PCNA
expression plasmid was generously provided by Dr Shaochun Wang
(University of Cincinnati, Cincinnati, OH, USA). PCNA-I1 and
PCNA-I1S (33,34,36) were purchased from ChemBridge
Corporation. R9-AR-PIP was synthesized by the custom peptide
service at Biomatik Corporation.
Cells and cell culture
The prostate cancer cell lines, LNCaP (cat. no.
CRL-1740), PC-3 (cat. no. CRL-1435) and 22Rv1 (cat. no. CRL-2505),
were obtained from ATCC. Androgen-independent LNCaP-AI cells were
generated from androgen-dependent LNCaP cells in a previous study
by the authors (38). The cells
were expanded and kept in cryogenic storage for long-term
safekeeping. The cells were authenticated genetically with PCR
identifying the short tandem repeat (STR) and cell-specific
profiling against the ATCC database at the University of Arizona
Genetics Core. The CWR-R1-D567 (R1-D567, cat. no. EMN028-FP) cells
(39) were obtained from
Kerafast. The LNCaP, PC-3, R1-D567 and 22Rv1 cells were cultured in
RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) at
37°C in 5% CO2. The LNCaP-AI cells were cultured in
stripped medium containing RPMI-1640 medium supplemented with 10%
charcoal-dextran treated FBS (sFBS). Cells in exponential growth
phase were harvested by treatment for 2-5 min with a 0.25%
Trypsin-0.02% EDTA solution and resuspended in medium. The
suspensions of single cells with viability >95% (ascertained by
Trypan blue exclusion; data not shown) were used in the present
study.
Knockdown of PCNA expression
Lipofectamine 3000® was used for the
transfection of siRNA according to the protocol provided by the
manufacturer. PC-3 cells (2×105/well) in six-well plates
were transfected with 5 µM siRNA with 10 µl
Lipofectamine 3000 in 100 µl of Opti-MEM medium (Thermo
Fisher Scientific, Inc.) for 3 days. Subsequently, the cell
extracts were prepared and quantitated for western blot
analysis.
Co-immunoprecipitation assay
Co-immunoprecipitation was performed in a modified
radio-immunoprecipitation assay (RIPA) buffer [PBS, 0.1% NP-40,
0.1% sodium deoxycholate, 50 mM NaCl, 1 mM EDTA, 1 mM
dithiothreitol, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 1X
protease inhibitor cocktail]. Mouse PCNA antibody was first
incubated with 1 mg of cell extract at 4°C on a rocker platform for
1-2 h. A total of 50 µl of Protein G plus/protein A agarose
beads (Calbiochem, Thermo Fisher Scientific) was then added, and
the samples were incubated at 4°C on a rocker platform overnight.
The beads were washed with the modified RIPA lysis buffer at 2,500
× g for 5 min each for four times at room temperature and boiled in
SDS loading buffer. The protein samples were subjected to 12%
SDS-PAGE and western blot analysis.
GST-PCNA pull-down assay
GST-PCNA vector was transformed into BL21 bacteria.
The transformed bacteria were cultured in L-broth with the addition
of 100 µM of IPTG to induce GST-fusion protein expression.
The bacteria were then harvested, sonicated and subjected to GST
fusion protein purification by using Glutathione Sepharose 4B
(#17-0756-01, GE Healthcare Bio-sciences AB). For the pull-down
reaction, 5-10 µg of GST-PCNA were incubated with 1 mg of
cell extract or 0.25-0.5 µg of recombinant AR in the
modified RIPA buffer. The samples were incubated at 4°C on a rocker
platform overnight. The beads were washed with the modified RIPA
buffer for four times and boiled in SDS loading buffer. The protein
samples were subjected to 12% SDS-PAGE and western blot
analysis.
Western blot analysis
Western blot analysis was performed as previously
described (40). Briefly,
aliquots of samples with the same amount of protein, determined
using the DC Protein assay kit, were mixed with loading buffer
(62.5 mM Tris-HCl, pH 6.8, 2.3% SDS, 100 mM dithiothreitol and
0.005% bromophenol blue), boiled, fractionated in an 12% SDS-PAGE,
and transferred onto 0.45-µm nitrocellulose membranes
(Bio-Rad Laboratories). The membranes were blocked with 2% fat-free
milk in PBS for 1 h, and probed with primary antibody in PBS
containing 0.01% Tween-20 (PBST) and 1% fat-free milk. The
membranes were incubated overnight at 4°C with primary antibodies
(1:200 dilution for antibodies from Santa Cruz Biotechnology, Inc.
and 1:400 dilution for all other antibodies). The membranes were
then washed four times in PBS and incubated with IRDye 680LT
secondary antibodies with at a 1:1,000 dilution in PBST containing
0.01% Tween 20 (PBST) and 1% fat-free milk for 1 h at room
temperature. After washing four times in PBS, the membranes were
visualized using Odyssey imaging system (Li-COR).
Reporter assay
Cells (105/well) were seeded in 12-well
tissue culture plates. The following day, the cells were
transfected with reporter plasmids (200 ng) without or with AR
expression vector (200 ng) with Lipofectamine 3000®
reagent for 4 h. The cells were then treated with various
concentrations of PCNA-I1S, T2AA, R9-AR-PIP and/or DHT as indicated
in the figures or figure legends for 24 h, respectively.
Subsequently, the cell extracts were prepared and protein
concentration from each sample was determined using the DC Protein
assay kit. Each sample was normalized by protein concentration
relative to the control sample. Luciferase activity was assessed in
a Berthold Detection System (Titertek-Berthold) using the
luciferase assay kit following the manufacturer's instructions. For
each assay, cell extract (20 µl) was used and the reaction
was started by injection of 50 µl of luciferase substrate
(Promega Corporation). Each reaction was measured for 10 sec in a
Sirius luminometer (Berthold Detection Systems). The luciferase
activity was defined as light units/mg protein.
Reverse transcription-quantitative PCR
(RT-qPCR)
RNA from the control and treated 22Rv1 cells was
isolated using the RNeasy Plus Universal Mini kit (Qiagen GmbH).
RNA samples were then treated with the DNase using DNA-free TM kit
(Thermo Fisher Scientific) and subjected to reverse transcription
reactions using High Capacity cDNA Reverse Transcription kits
(Applied Biosystems; Thermo Fisher Scientific). The expression of
AR and PSA was analyzed by RT-PCR using the Fast SYBR-Green Master
Mix (Thermo Fisher Scientific) in a 7300 Real-Time PCR system
(Applied Biosystems, Thermo Fisher Scientific) with primer pairs as
follows: AR, 5′-GGT GAG CAG AGT GCC CTA TC-3′/5′-GGC AGT CTC CAA
ACG CAT GTC-3′ and PSA, 5′-GAC CAC CTG CTA CGC CTC A-3′/5′-AAC TTG
CGC ACA CAC GTC ATT-3′, as previously described (41,42). Following an initial step at 95°C
for 10 min, PCR was performed at 95°C for 3 sec and 60°C for 30 sec
for 40 cycles according to the manufacture instruction. The cycle
threshold values were used to calculate the normalized expression
of target genes against β-actin using Q-Gene software (43).
MTT assay
Cells were plated in 96-well plates and treated as
indicated with enzalutamide (ENZ, 5 µM), R9-AR-PIP (up to 20
µM), and/or PCNA-I1 (150 nM) in normal and/or stripped
medium for 96 h. Viable cells in the control and treated cultures
were stained with MTT as described in a previous study by the
authors (44). Growth inhibition
(%) in the treated cell cultures in comparison with that in the
untreated controls was calculated using the following formula:
(1-A570 of treated wells/A570 of control
wells) ×100, which reflects the effects of the treatments on cell
growth inhibition. The IC50 was defined as the concentration of a
drug that inhibited cell growth by 50% as compared with untreated
control.
PCNA binding assay
The binding and affinity of R9-AR-PIP to PCNA was
measured using microscale thermophoresis (MST) following a
previously described protocol (45). PC-3 cells in 100-mm plates were
transfected with a mammalian expression vector pGFP-PCNA (34,46) or pGFP (Promega Corporation)
encoding GFP-tagged PCNA or GFP (control) using Lipofectamine
3000®, respectively. After two days, the expression
levels of GFP-PCNA and GFP in ~80% cells were confirmed under an
Olympus IX51 inverted fluorescent microscope. The cells were washed
with PBS and scrapped in 10 ml pre-chilled PBS. Following
centrifugation at 250 × g for 5 min at 4°C, the cells were
resuspended in 250 µl of 25 mM Tris-HCl (pH 8.0) containing
1X protease inhibitor mixture and 1 mM PMSF, and sonicated on ice
for 3×10 sec at 30% amplitude using a Sonicator Ultrasonic
Processor (Misonix). The soluble fraction of the cell lysate was
collected following centrifugation at 12,000 × g for 10 min at room
temperature. R9-AR-PIP at increasing concentrations was incubated
with the lysate (1.5 µg in 30 µl) for 1 h at room
temperature in binding buffer (50 mM Tris-HCl, pH7.4, 150 mM NaCl,
10 mM MgCl2, 0.01% Tween-20). The alterations in the
thermophoresis of GFP-PCNA or GFP protein were measured using
Monolith NT.115 (NanoTemper Technologies). The apparent
dissociation constant (Kd) of the binding was calculated
by non-linear regression analysis using MO Affinity Analysis v2.1.3
(NanoTemper Technologies).
Statistical analysis
Data from each assay are expressed as the mean ± SD.
Statistically significant differences between two groups were
determined using a Student's t-test. All statistical analyses were
carried out using Prism 9 software (GraphPad). P<0.05 was
considered to indicate a statistically significant difference.
Results
Interaction between PCNA and AR
The interdomain connector loop (IDCL) on the outer
surface of PCNA trimers interacts with proteins containing the
PIP-box, defined by the consensus sequence Q1-x2-x3-h4-x5-x6-a7-a8,
where 'h' and 'a' represent hydrophobic (ILMV) and aromatic (FYH)
amino acids, respectively (47).
The canonical Q1 docks into a 'Q pocket' and h4, a7 and a8 bind in
a large hydrophobic pocket formed by the IDCL. A previous study
demonstrated that PCNA interacts with AR (37). The present study performed a
sequence analysis of human, mouse and rat AR proteins, and found an
identical consensus sequence of the PIP-box (QLGLGRVY)
located at the amino acid 4 position consisting of Q, L and Y for
Q1, h4 and a8, respectively (Fig.
1).
To determine whether PCNA complexes with AR via the
potential N-terminal PIP-box, the cell extracts from LNCaP
(AR-FL+) and 22Rv1 (AR-FL+/AR-V7+)
cells (48) were subjected to
co-immunoprecipitation assay. It was found that PCNA complexed with
both AR-FL (Fig. 2A-C) and AR-V7
(Fig. 2B and C), and this was
attenuated by the PIP-box inhibitor, T2AA, following treatment in
either the cell culture or by addition to the 22Rv1 cell lysate
during co-IP (Fig. 2C).
Furthermore, GST pull-down assay was performed using the cell
extracts from 22Rv1 cells, as well as R1-D567 cells that only
express ARv567es (39). The cell
extract from AR-negative PC-3 cells was used as a control. It was
found that GST-PCNA complexed with AR-FL, AR-V7 and ARv567es
(Fig. 2D). More importantly,
GST-PCNA also bound to recombinant AR (Fig. 2E). Taken together, the results
from these experiments demonstrated that PCNA bound directly to the
N-terminal domain of AR, very likely via the PIP-box at the
N-terminus of AR.
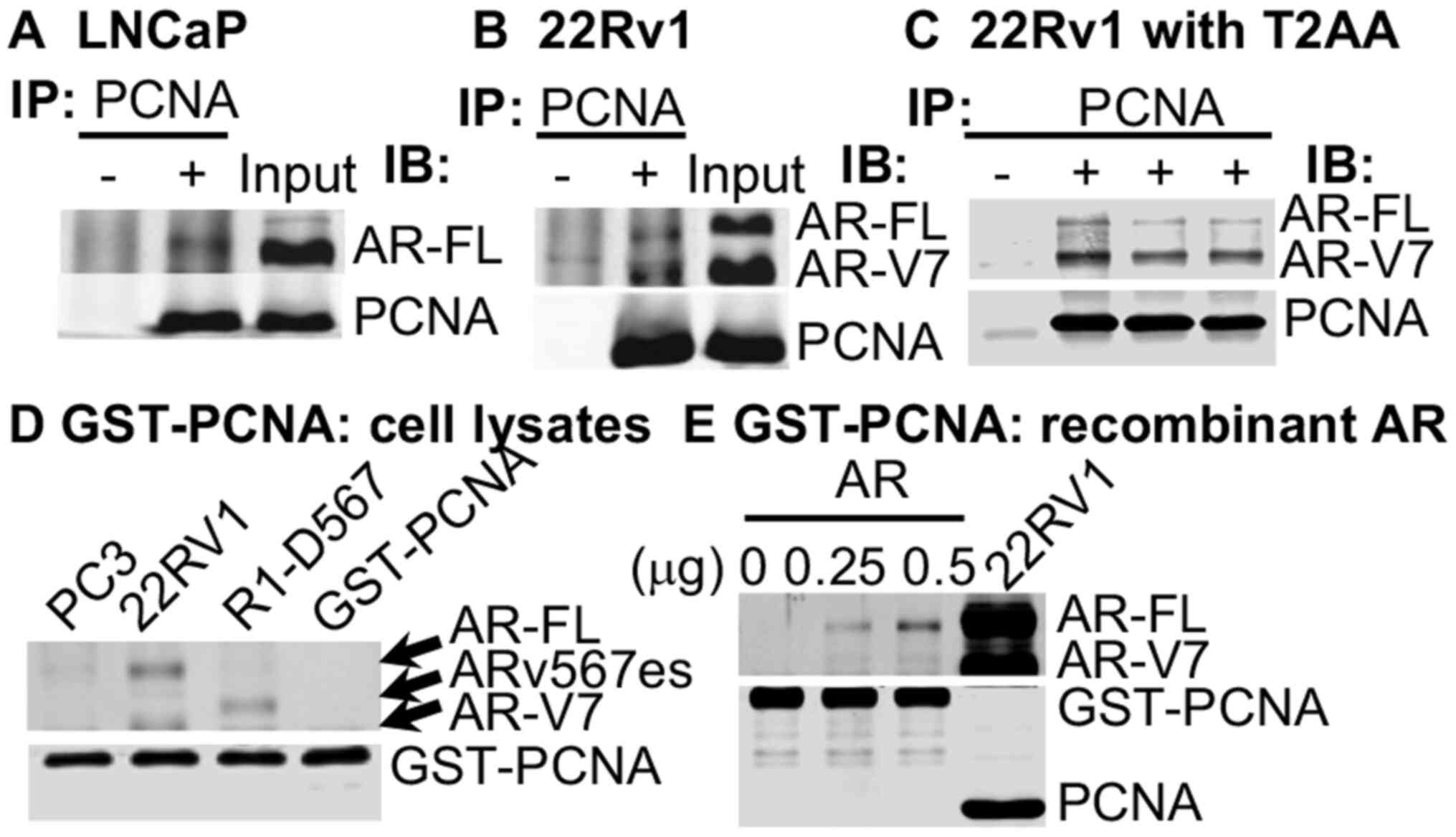 | Figure 2AR is a novel PCNA partner protein.
(A) LNCaP and (B) 22Rv1 cell extracts were used for co-IP with a
mouse antibody to PCNA for IP and with rabbit antibodies to AR (N20
binding to the NTD of AR and PCNA for western blot analysis. (C)
Co-IP was performed with extracts from 22Rv1 cells incubated for 24
h in medium without (lanes 1 and 2) or with 5 µM T2AA (lane
3). T2AA (30 µM) (lane 4) was added to the lysates of
untreated 22Rv1 cells during co-IP. (D) Cell extracts from PC-3,
22Rv1, and R1-D567 cells were subjected to GST pull-down reaction
with GST-PCNA alone as a control. (E) GST-PCNA pull-down reaction
was performed in the presence of 0, 0.25, or 0.5 µg (lanes
1, 2 and 3) of recombinant AR protein, with cell lysate from 22Rv1
cells (lane 4) as a control, followed by western blot analysis
using anti-AR and anti-PCNA antibodies. co-IP,
co-immunoprecipitation; AR, androgen receptor; PCNA, proliferating
cell nuclear antigen; NTD, N-terminal domain. |
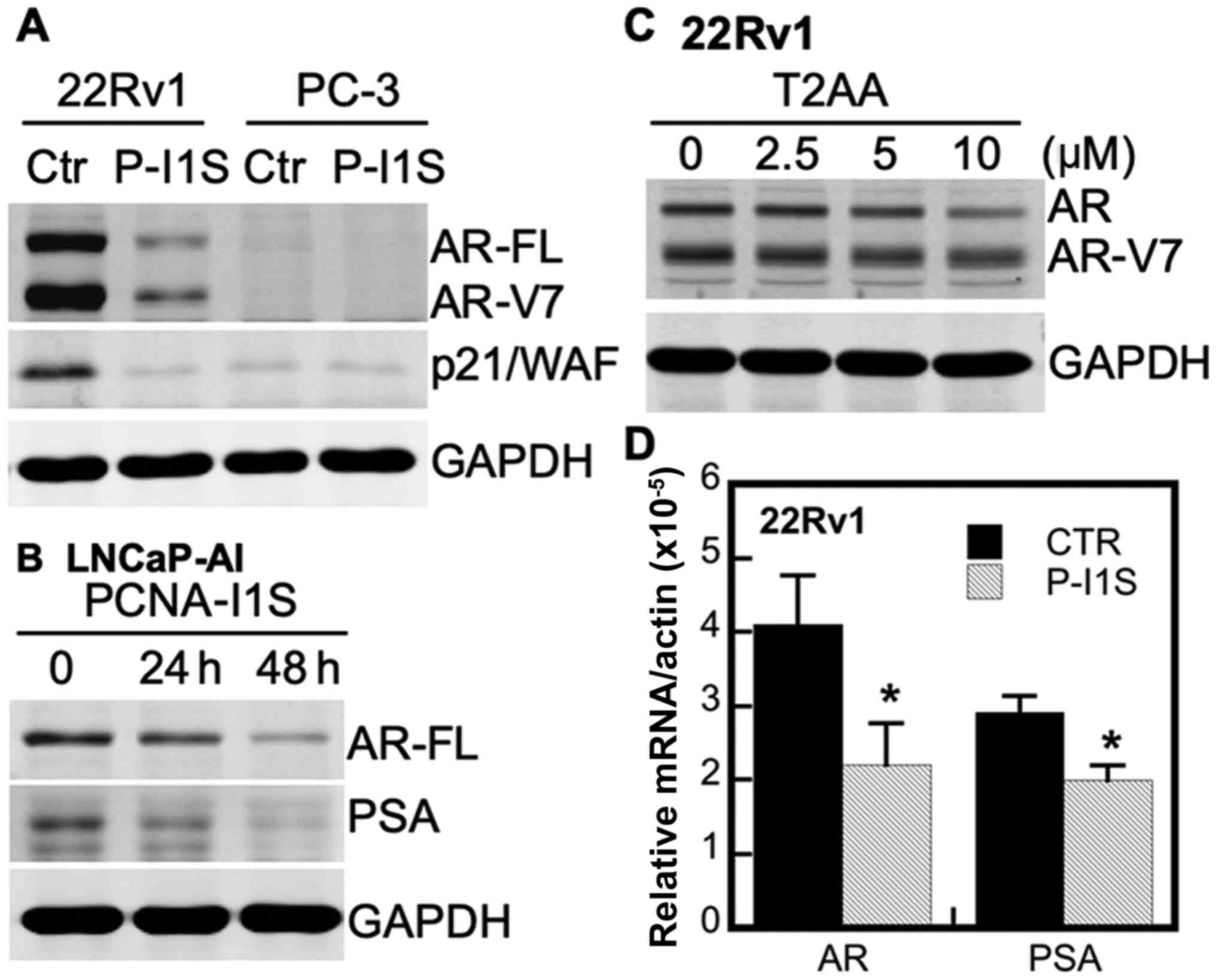
Inhibitory effects of PCNA-I1S and T2AA
on the expression of endogenous canonical AR target genes
The present study further investigated the effects
of PCNA-I1S on the expression of the endogenous canonical AR target
genes, PSA and p21/WAF (49), as
well as AR that is autoregulated by androgens (50), due to the presence of the ARE in
the AR gene (50,51). As shown in Fig. 3A, PCNA-I1S reduced the expression
of p21/WAF in 22Rv1 cells, but not in AR-negative PC-3 cells
(49). PCNA-I1S also
downregulated the expression of both AR-FL and AR-V7 in 22Rv1 cells
(Fig. 3A). Moreover, PCNA-I1S
inhibited the expression of AR-FL and PSA in a time-dependent
manner in the CRPC LNCaP-AI (AR-FL+) cells (38) (Fig.
3B). Similarly, the expression of AR-FL and AR-V7 in the 22Rv1
cells was also attenuated by the PIP-box inhibitor, T2AA, in a
concentration-dependent manner (Fig.
3C). Furthermore, treatment of the CRPC 22Rv1 cells with
PCNA-I1S reduced the mRNA levels of AR and PSA genes by 46 and 31%,
respectively (Fig. 3D).
Inhibitory effects of PCNA-I1S and T2AA
on AR transcriptional activity
The effects of PCNA inhibitors on AR transcriptional
activity were determined in reporter assays using two luciferase
reporters PSA(4.3)-luc driven by PSA promoter containing ARE III
region (-4,086/-3,981 bp) and ARE I region (-231/-143 bp) (52) and p21(-215)-luc driven by p21/WAF
promoter containing an ARE at -200 bp position (49). It was found that PCNA-I1S
inhibited both the basal and DHT-induced promoter activities of PSA
and p21/WAF genes in the LNCaP-AI and 22Rv1 cells (Fig. 4A-C), and also the DHT-induced
promoter activity of the PSA gene in AR-negative PC-3 cells
co-transfected with an AR expression vector (Fig. 4D). The inhibitory effects on PSA
promoter activity were also observed in the 22Rv1 cells treated
with T2AA (Fig. 4E). These AR
functional assays suggest that PCNA promotes AR transcriptional
activity, likely through interaction with AR via the PIP-box.
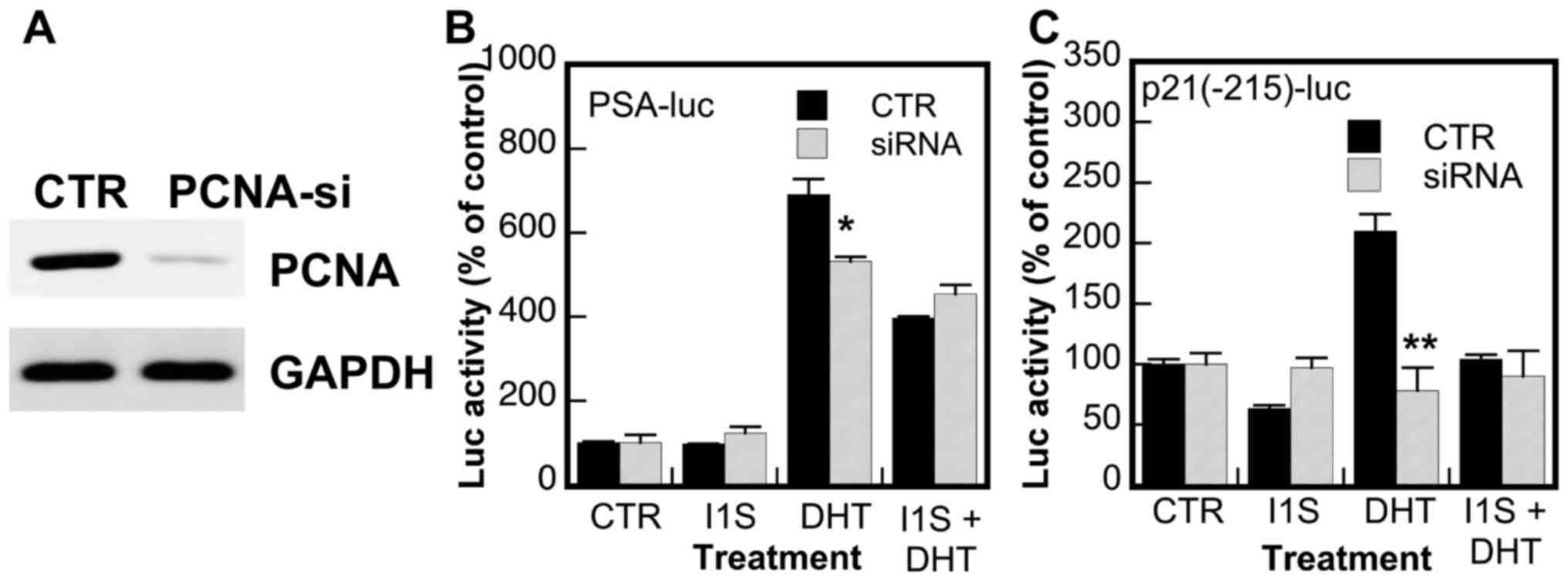
Effects of knockdown of PCNA expression
on AR transcriptional activity and the inhibitory effects of
PCNA-I1S
To validate the role of PCNA in the regulation of AR
transcriptional activity and the target specificity of PCNA-I1S,
the effects of the knockdown of PCNA expression on AR activity and
the inhibitory effects of PCNA-I1S were determined. As shown in
Fig. 5A, the expression of PCNA
was markedly reduced in PC-3 cells transfected with PCNA siRNA. The
knockdown of PCNA expression partially attenuated the DHT-induced
promoter activity of the PSA gene (Fig. 5B) and abolished the DHT-induced
promoter activity of the p21/WAF gene (Fig. 5C), suggesting that PCNA is a
regulator of and promotes AR activity. More importantly, the
inhibitory effects of PCNA-I1S on the DHT-stimulated promoter
activities of the PSA and p21/WAF genes were abolished in the cells
in which the expression of PCNA was knocked down (Fig. 5B and C). On the whole, these
results validate the role of PCNA in the upregulation of AR
transcriptional activity and the target specificity of PCNA-I1S on
PCNA in the suppression of AR activity.
Additive inhibitory effects of ADT and
PCNA-I1 on the growth of AR-positive prostate cancer cells
Given the significant roles of PCNA in the
regulation of AR activity, the present study examined the effects
of the concurrent treatment of PCNA-I1 plus ADT on the growth of
several human prostate cancer cell lines, including
androgen-dependent LNCaP (AR-FL+) cells and CRPC
LNCaP-AI (AR-FL+), 22Rv1
(AR-FL+/AR-V7+) and PC-3 (AR−)
cells. As was expected, ADT (in either cultured in stripped medium)
(Fig. 6A-C) or treatment with 5
µM ENZ (Fig. 6D-F),
significantly inhibited the growth of LNCaP cells (Fig. 6A and D) and, at a lower potency,
also inhibited the growth of CRPC LNCaP-AI and 22Rv1 cells
(Fig. 6B, C and E), but not that
of AR-negative PC-3 cells (Fig.
6F). PCNA-I1 (150 nM) inhibited the growth of all cells and
exerted additive inhibitory effects with ADT in all AR-positive
cells (Fig. 6A-E); however, this
effect was not observed in AR-negative PC-3 cells (Fig. 6F).
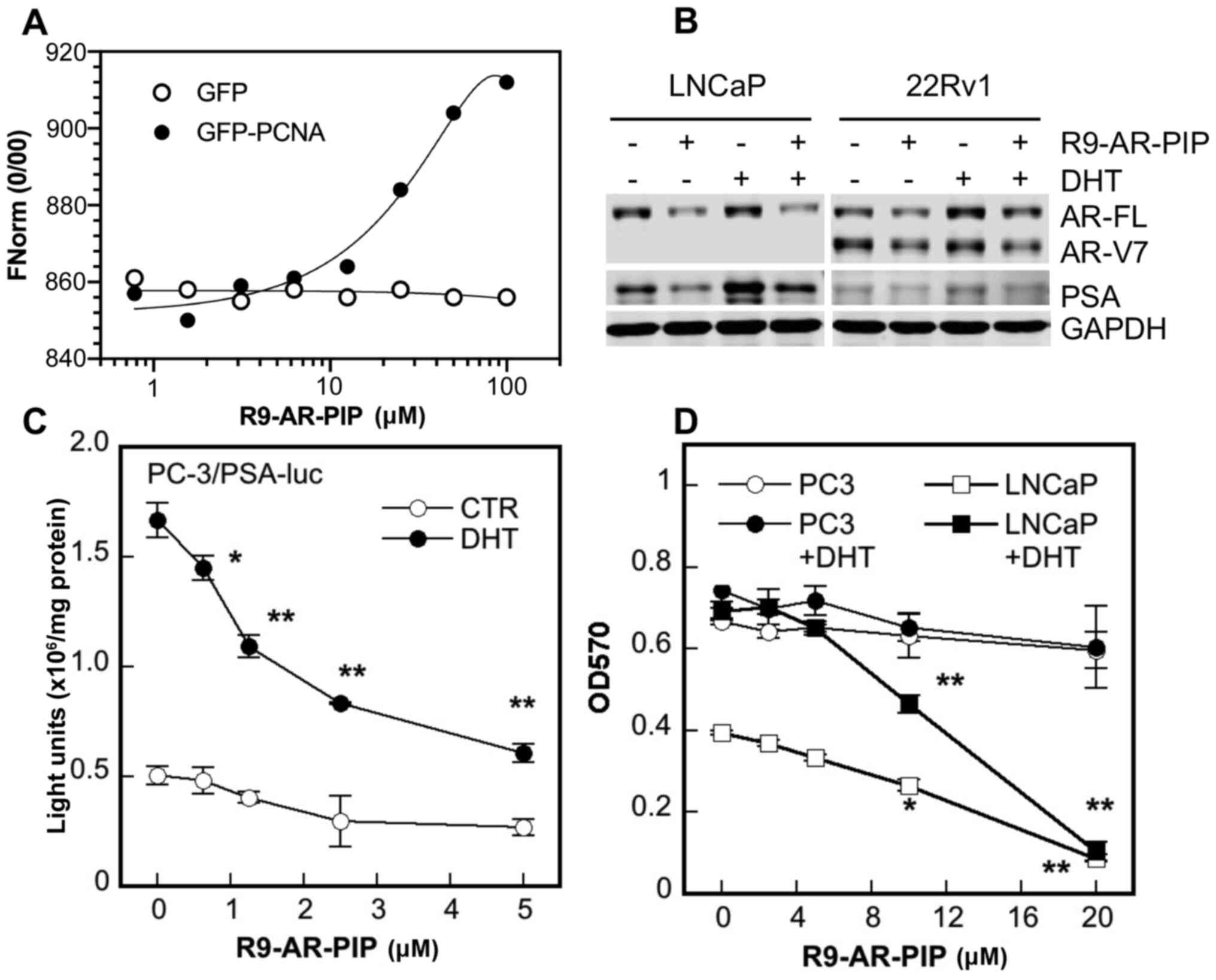
Development of the AR-specific PIP-box
peptide inhibitor, R9-AR-PIP
To further establish the essential role of the AR
PIP-box in AR signaling, the effects of a small AR PIP-box peptide
inhibitor on AR activity were investigated. It has previously been
found that R9-cc-caPeptide, a small peptide with 8 amino acid
residues spanning a portion of the IDCL of caPCNA linked with nine
arginine residues to enhance the penetration of the peptide into
cells and nucleus, inhibits PCNA association to chromatin (30). The peptide also induces
cytotoxicity in culture and attenuates tumor growth in mice
(30). Using the same structural
design, the present study developed the peptide QLGLGRVY-cc-R9
(R9-AR-PIP). It contains eight amino acid residues of the AR
PIP-box sequence (Fig. 1) and
nine arginine residues linked with two cystine residues. The nine
arginine residues are in 'all-D' configuration to minimize
proteolysis.
To measure the binding and affinity of R9-AR-PIP to
PCNA, the lysates of PC-3 cells containing either GFP-PCNA
(34,46) or GFP (control) were incubated with
increasing concentrations of R9-AR-PIP and the apparent
dissociation constant (Kd) was determined by MST.
R9-AR-PIP was found to bind to GFP-PCNA at Kd of
2.73±2.18 µM (n=5) and not to GFP (Fig. 7A). The data presented in Fig. 7B demonstrated that treatment with
R9-AR-PIP reduced both the basal and DHT-induced expression of
AR-FL and PSA proteins in LNCaP cells and AR-FL, AR-V7, and PSA
proteins in 22Rv1 cells. Moreover, R9-AR-PIP inhibited both the
basal and DHT-induced promoter activity of PSA gene in a
concentration-dependent manner in AR-negative PC-3 cells
co-transfected with an AR expression vector (Fig. 7C). Finally, R9-AR-PIP inhibited
the growth of androgen-dependent LNCaP cells both in the absence
and presence of DHT with an IC50 value of 8-10 µM (Fig. 7D). By contrast, the growth
inhibitory effects of the peptide on AR-negative PC-3 cells were
more modest at the same concentrations (Fig. 7D).
Discussion
PCNA is an evolutionally very well conserved
multifunctional protein (53,54). Native PCNA, which is located
mainly in the nucleoplasm as 'free form PCNA', is a ring-shaped
homotrimeric protein joined together through head to tail
interaction (21-23,53,54). To be functional, PCNA must be
linearized or monomerized and relocalized, and serves as platforms
for interaction with its partner proteins containing several
motifs, including the PIP box (22,24,25). The present study identified a
PIP-box consensus sequence at the N-terminus of AR and found that
PCNA complexed with AR-FL, as well as AR-V7 and ARv567es, and this
was attenuated by the PIP-box inhibitor, T2AA. PCNA-I1S and PCNA-I1
are small-molecule PCNA inhibitors that bind to PCNA trimers at the
interfaces of two monomers, stabilize the trimer structure,
interfere with PCNA linearization or monomerization, and attenuate
PCNA relocalization and chromatin association (33-36). The present study found that
PCNA-I1S attenuated AR transcriptional activity and the expression
of the AR target genes, PSA, p21/WAF, AR-FL and AR-V7. The
knockdown of PCNA expression abolished the inhibitory effects of
PCNA-I1S on AR transcriptional activity. Similarly, AR
transcriptional activity and the expression of AR target genes were
also inhibited by T2AA. Moreover, the additive growth inhibitory
effects were demonstrated by targeting PCNA with PCNA-I1 plus ADT
by either the deprivation of androgen or treatment with
antiandrogen enzalutamide only in prostate cancer cells expressing
AR, but not in AR-negative prostate cancer cells. Finally, an AR
PIP-box mimicking peptide inhibitor (R9-AR-PIP) was developed, and
it was found that it binds to PCNA at Kd of
approximately 2.73 µM and inhibited the expression of AR
target genes, AR transcriptional activity and the growth of
AR-expressing cells in a concentration-dependent manner. By
contrast, the growth inhibitory effects of the peptide on
AR-negative PC-3 cells were more modest at the same concentrations.
These data strongly suggest that PCNA interacts with AR through the
PIP-box and enhances AR-mediated signaling.
The exact mechanisms through which PCNA promotes AR
activity in prostate cancer cells remain to be elucidated. One of
the potential mechanisms is that chromatin-associated PCNA serves
as a platform for AR and facilitates AR binding to chromatin. This
notion is supported by several observations: i)
Co-immunoprecipitation assay revealed that PCNA binds to AR and
several other PCNA partner proteins at the S-phase of the cell
cycle, but not at the G1 phase of the cell cycle when PCNA mostly
presents as 'free-form' in the nucleosome (37); ii) PCNA-I1 and PCNA-I1S binds to
PCNA and reduces PCNA chromatin association (34-36), and inhibits AR transcriptional
activity and the expression of AR target genes; iii) the
small-molecule PIP-box specific inhibitor, T2AA, inhibits PCNA
interaction with partner proteins including AR-FL and AR-V7, and
suppresses AR transcriptional activity and the expression of AR
target genes; and iv) targeting the PCNA-AR interaction with AR
PIP-box-specific peptide inhibitor R9-AR-PIP inhibits AR
transcriptional activity, the expression of AR target genes and the
growth of AR-positive cells.
In a previous study by the authors, it was
demonstrated that PCNA-I1 inhibited the growth of tumor cells of
various tissue origins at IC50 concentrations ~9-fold lower than
those in normal cells (36). One
of the important findings of the present study is that PCNA-I1
enhanced the growth inhibitory effects of ADT, either by androgen
ablation or the anti-androgen, ENZ, in both androgen-dependent
prostate cancer cells and CRPC cells. The additive growth
inhibitory effect appears to be AR-dependent, since it was not
observed in AR-negative prostate cancer cells. The potential
underlying mechanisms for this observation are that, in addition to
inhibiting androgen-dependent AR-mediated signaling, PCNA-I1
suppresses PCNA chromatin association (35,36), interrupts the PCNA-AR interaction
and hence attenuates AR activity via the androgen-independent
AR-mediated pathway, such AR phosphorylation by several tyrosine
and serine protein kinases (55),
the overexpression of Vav3 functioning as a co-activator (56) and constitutively active AR-Vs
(5-7), as well as other cellular processes
critical for cell growth (21-23).
In conclusion, the present study found that PCNA
interacted with both AR-FL and AR-Vs very likely through the
PIP-box at the N-terminus of AR and promoted AR transcriptional
activity. In addition, targeting PCNA plus ADT exerted additive
inhibitory effects on the growth of AR-positive prostate cancer
cells. Given that PCNA is preferentially overexpressed in
replicating tumor cells and CRPC overexpresses AR-FL and/or AR-Vs,
targeting the PCNA-AR interaction by R9-AR-PIP to block AR-FL- and
AR-V-mediated signaling may prove to be an innovative and selective
therapeutic strategy against CRPCs, particularly those
overexpressing constitutively active AR-Vs.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
Both authors (SL and ZD) participated in the study
design, conducted the experiments, performed the data analysis, and
wrote or contributed to the writing of the manuscript. Both authors
confirm the authenticity of all the raw data, and have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Dr Shaochun Wang
(University of Cincinnati, Cincinnati, OH, USA) for providing the
GST-PCNA expression plasmid, and Dr Kelsey L. Dillehay (University
of Cincinnati) for providing technical assistance in generating and
purifying GST-PCNA protein used in the study.
Funding
The present study was supported in part by a 1R21CA223049-01
grant from the National Institutes of Health, National Cancer
Institute and Millennium Scholar Funds from the University of
Cincinnati Cancer Center.
Abbreviations:
|
AR
|
androgen receptor
|
|
AR-FL
|
full-length AR
|
|
AR-Vs
|
constitutively active AR splicing
variants
|
|
CRPC
|
castration-resistant prostate
cancer
|
|
DHT
|
dihydrotestosterone
|
|
DBD
|
DNA binding domain
|
|
ENZ
|
enzalutamide
|
|
IDCL
|
interdomain connector loop
|
|
LBD
|
ligand binding domain
|
|
PCNA
|
proliferating cell nuclear antigen
|
|
PIP-box
|
PCNA-interacting protein-box
|
|
PCNA-Is
|
PCNA inhibitors
|
|
PSA
|
prostate-specific antigen
|
References
|
1
|
Tsai MJ and O'Malley BW: Molecular
mechanisms of action of steroid/thyroid receptor superfamily
members. Ann Rev Biochem. 63:451–486. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mangelsdorf DJ and Evans RM: The RXR
heterodimers and orphan receptors. Cell. 83:841–850. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Biron E and Bedard F: Recent progress in
the development of protein-protein interaction inhibitors targeting
androgen receptor-coactivator binding in prostate cancer. J Steroid
Biochem Mol Biol. 161:36–44. 2016. View Article : Google Scholar
|
|
4
|
Dehm SM and Tindall DJ: Alternatively
spliced androgen receptor variants. Endocr Relat Cancer.
18:R183–R196. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lu J, Van der Steen T and Tindall DJ: Are
androgen receptor variants a substitute for the full-length
receptor? Nat Rev Urol. 12:137–144. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu Z, Chen S, Sowalsky AG, Voznesensky OS,
Mostaghel EA, Nelson PS, Cai C and Balk SP: Rapid induction of
androgen receptor splice variants by androgen deprivation in
prostate cancer. Clin Cancer Res. 20:1590–1600. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Antonarakis ES, Lu C, Wang H, Luber B,
Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et
al: AR-V7 and resistance to enzalutamide and abiraterone in
prostate cancer. N Engl J Med. 371:1028–1038. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yin Y, Li R, Xu K, Ding S, Li J, Baek G,
Ramanand SG, Ding S, Liu Z, Gao Y, et al: Androgen receptor
variants mediate DNA repair after prostate cancer irradiation.
Cancer Res. 77:4745–4754. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W,
Gambhir SS, Lee P, Sartor O, Flemington EK, et al: Androgen
receptor splice variants dimerize to transactivate target genes.
Cancer Res. 75:3663–3671. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hu R, Lu C, Mostaghel EA, Yegnasubramanian
S, Gurel M, Tannahill C, Edwards J, Isaacs WB, Nelson PS, Bluemn E,
et al: Distinct transcriptional programs mediated by the
ligand-dependent full-length androgen receptor and its splice
variants in castration-resistant prostate cancer. Cancer Res.
72:3457–3462. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wach S, Taubert H and Cronauer M: Role of
androgen receptor splice variants, their clinical relevance and
treatment options. World J Urol. 38:647–656. 2020. View Article : Google Scholar
|
|
12
|
Guo Z, Yang X, Sun F, Jiang R, Linn DE,
Chen H, Chen H, Kong X, Melamed J, Tepper CG, et al: A novel
androgen receptor splice variant is up-regulated during prostate
cancer progression and promotes androgen depletion-resistant
growth. Cancer Res. 69:2305–2313. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sharp A, Coleman I, Yuan W, Sprenger C,
Dolling D, Rodrigues DN, Russo JW, Figueiredo I, Bertan C, Seed G,
et al: Androgen receptor splice variant-7 expression emerges with
castration resistance in prostate cancer. J Clin Invest.
129:192–208. 2019. View Article : Google Scholar :
|
|
14
|
Honda M, Kimura T, Kamata Y, Tashiro K,
Kimura S, Koike Y, Sato S, Yorozu T, Furusato B, Takahashi H, et
al: Differential expression of androgen receptor variants in
hormone-sensitive prostate cancer xenografts, castration-resistant
sublines, and patient specimens according to the treatment
sequence. Prostate. 79:1043–1052. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Paschalis A, Sharp A, Welti JC, Neeb A,
Raj GV, Luo J, Plymate SR and de Bono JS: Alternative splicing in
prostate cancer. Nature Rev Clin Oncol. 15:663–675. 2018.
View Article : Google Scholar
|
|
16
|
Scher HI, Graf RP, Schreiber NA, Jayaram
A, Winquist E, McLaughlin B, Lu D, Fleisher M, Orr S, Lowes L, et
al: Assessment of the validity of nuclear-localized androgen
receptor splice variant 7 in circulating tumor cells as a
predictive biomarker for castration-resistant prostate cancer. JAMA
Oncol. 4:1179–1186. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun S, Sprenger CC, Vessella RL, Haugk K,
Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, et
al: Castration resistance in human prostate cancer is conferred by
a frequently occurring androgen receptor splice variant. J Clin
Invest. 120:2715–2730. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Van Etten JL, Nyquist M, Li Y, Yang R, Ho
Y, Johnson R, Ondigi O, Voytas DF, Henzler C and Dehm SM: Targeting
a single alternative polyadenylation site coordinately blocks
expression of androgen receptor mRNA splice variants in prostate
cancer. Cancer Res. 77:5228–5235. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang YC, Banuelos CA, Mawji NR, Wang J,
Kato M, Haile S, McEwan IJ, Plymate S and Sadar MD: Targeting
androgen receptor activation Function-1 with EPI to overcome
resistance mechanisms in castration-resistant prostate cancer. Clin
Cancer Res. 22:4466–4477. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz
CT, Evans CP and Gao AC: Niclosamide inhibits androgen receptor
variants expression and overcomes enzalutamide resistance in
castration-resistant prostate cancer. Clin Cancer Res.
20:3198–3210. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Moldovan GL, Pfander B and Jentsch S:
PCNA, the maestro of the replication fork. Cell. 129:665–679. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stoimenov I and Helleday T: PCNA on the
crossroad of cancer. Biochem Soc Transact. 37:605–613. 2009.
View Article : Google Scholar
|
|
23
|
Maga G and Hubscher U: Proliferating cell
nuclear antigen (PCNA): A dancer with many partners. J Cell Sci.
116:3051–3060. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gilljam KM, Feyzi E, Aas PA, Sousa MM,
Muller R, Vagbo CB, Catterall TC, Liabakk NB, Slupphaug G, Drabløs
F, et al: Identification of a novel, widespread, and functionally
important PCNA-binding motif. J Cell Biol. 186:645–654. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gulbis JM, Kelman Z, Hurwitz J, O'Donnell
M and Kuriyan J: Structure of the C-terminal region of
p21(WAF1/CIP1) complexed with human PCNA. Cell. 87:297–306. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lu S, Lee J, Revelo M, Wang X and Dong Z:
Smad3 is overexpressed in advanced human prostate cancer and
necessary for progressive growth of prostate cancer cells in nude
mice. Clin Cancer Res. 13:5692–5702. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Spires SE, Banks ER, Davey DD, Jennings
CD, Wood DP Jr and Cibull ML: Proliferating cell nuclear antigen in
prostatic adenocarcinoma: Correlation with established prognostic
indicators. Urology. 43:660–666. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kallakury BV, Sheehan CE, Rhee SJ, Fisher
HA, Kaufman RP Jr, Rifkin MD and Ross JS: The prognostic
significance of proliferation-associated nucleolar protein p120
expression in prostate adenocarcinoma: A comparison with cyclins A
and B1, Ki-67, proliferating cell nuclear antigen, and p34cdc2.
Cancer. 85:1569–1576. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Muller R, Misund K, Holien T, Bachke S,
Gilljam KM, Vatsveen TK, Ro TB, Bellacchio E, Sundan A and Otterlei
M: Targeting proliferating cell nuclear antigen and its protein
interactions induces apoptosis in multiple myeloma cells. PLoS One.
8:e704302013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smith SJ, Gu L, Phipps EA, Dobrolecki L,
Mabrey KS, Gulley P, Dillehay KL, Dong Z, Fields GB, Chen Y, et al:
A peptide mimicking a region in proliferating cell nuclear antigen
specific to key protein interactions is cytotoxic to breast cancer.
Mol Pharmacol. 87:263–276. 2015. View Article : Google Scholar :
|
|
31
|
Punchihewa C, Inoue A, Hishiki A, Fujikawa
Y, Connelly M, Evison B, Shao Y, Heath R, Kuraoka I, Rodrigues P,
et al: Identification of a small molecule PCNA inhibitor that
disrupts interactions with PIP-Box proteins and inhibits DNA
replication. J Biol Chem. 287:14289–14300. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gu L, Lingeman R, Yakushijin F, Sun E, Cui
Q, Chao J, Hu W, Li H, Hickey RJ, Stark JM, et al: The anticancer
activity of a First-in-class Small-molecule targeting PCNA. Clin
Cancer Res. 24:6053–6065. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lu S and Dong Z: Additive effects of a
small molecular PCNA inhibitor PCNA-I1S and DNA damaging agents on
growth inhibition and DNA damage in prostate and lung cancer cells.
PLoS One. 14:e02238942019. View Article : Google Scholar
|
|
34
|
Dillehay KL, Seibel WL, Zhao D, Lu S and
Dong Z: Target validation and structure-activity analysis of a
series of novel PCNA inhibitors. Pharmacol Res Perspect.
3:e001152015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dillehay KL, Lu S and Dong Z: Antitumor
effects of a novel small molecule targeting PCNA chromatin
association in prostate cancer. Mol Cancer Ther. 13:2817–2826.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tan Z, Wortman M, Dillehay KL, Seibel WL,
Evelyn CR, Smith SJ, Malkas LH, Zheng Y, Lu S and Dong Z:
Small-molecule targeting of proliferating cell nuclear antigen
chromatin association inhibits tumor cell growth. Mol Pharmacol.
81:811–819. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Murthy S, Wu M, Bai VU, Hou Z, Menon M,
Barrack ER, Kim SH and Reddy GP: Role of androgen receptor in
progression of LNCaP prostate cancer cells from G1 to S phase. PLoS
One. 8:e566922013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lu S, Tsai SY and Tsai MJ: Melecular
mechanisms of androgen-independent growth of human prostate cancer
LNCaP-AI cells. Endocrinology. 140:5054–5059. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nyquist MD, Li Y, Hwang TH, Manlove LS,
Vessella RL, Silverstein KA, Voytas DF and Dehm SM:
TALEN-engineered AR gene rearrangements reveal endocrine uncoupling
of androgen receptor in prostate cancer. Proc Natl Acad Sci USA.
110:17492–17497. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Dong Z, Liu Y, Scott KF, Levin L, Gaitonde
K, Bracken RB, Burk B, Zhai Q, Wang J, Oleksowicz L and Lu S:
Secretory phospholipase A2-IIa is involved in prostate cancer
progression and may potentially serve as a biomarker for Prostate
Cancer. Carcinogenesis. 31:1948–1955. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lu S and Dong Z: Overexpression of
secretory phospholipase A2-IIa supports cancer stem cell phenotype
via HER/ERBB-elicited signaling in lung and prostate cancer cells.
Int J Oncol. 50:2113–2122. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lu S, Tan Z, Wortman M, Lu S and Dong Z:
Regulation of heat shock protein 70-1 expression by androgen
receptor and its signaling in human prostate cancer cells. Int J
Oncol. 36:459–467. 2010.PubMed/NCBI
|
|
43
|
Simon P: Q-Gene: Processing quantitative
real-time RT-PCR data. Bioinformatics. 19:1439–1440. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dong ZY, Ward NE, Fan D, Gupta KP and
O'Brian CA: In vitro model for intrinsic drug resistance: Effects
of protein kinase C activators on the chemosensitivity of cultured
human colon cancer cells. Mol Pharmacol. 39:563–569.
1991.PubMed/NCBI
|
|
45
|
Khavrutskii L, Yeh J, Timofeeva O, Tarasov
SG, Pritt S, Stefanisko K and Tarasova N: Protein purification-free
method of binding affinity determination by microscale
thermophoresis. J Vis Exp. 50541:2013.
|
|
46
|
Leonhardt H, Rahn HP, Weinzierl P,
Sporbert A, Cremer T, Zink D and Cardoso MC: Dynamics of DNA
replication factories in living cells. J Cell Biol. 149:271–280.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Slade D: Maneuvers on PCNA Rings during
DNA replication and repair. Genes (Basel). 9:4162018. View Article : Google Scholar
|
|
48
|
Chen Z, Wu D, Thomas-Ahner JM, Lu C, Zhao
P, Zhang Q, Geraghty C, Yan PS, Hankey W, Sunkel B, et al: Diverse
AR-V7 cistromes in castration-resistant prostate cancer are
governed by HoxB13. Proc Natl Acad Sci USA. 115:6810–6815. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lu S, Liu M, Epner DE, Tsai SY and Tsai
MJ: Androgen regulation of the cyclin-dependent kinase inhibitor
p21 gene through an androgen response element in the proximal
promoter. Mol Endocrinol. 13:376–384. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dai JL, Maiorino CA, Gkonos PJ and
Burnstein KL: Androgenic up-regulation of androgen receptor cDNA
expression in androgen-independent prostate cancer cells. Steroids.
61:531–539. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Manin M, Baron S, Goossens K, Beaudoin C,
Jean C, Veyssiere G, Verhoeven G and Morel L: Androgen receptor
expression is regulated by the phosphoinositide 3-kinase/Akt
pathway in normal and tumoral epithelial cells. Biochem J.
366:729–736. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jia L and Coetzee GA: Androgen
receptor-dependent PSA expression in androgen-independent prostate
cancer cells does not involve androgen receptor occupancy of the
PSA locus. Cancer Res. 65:8003–8008. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Krishna TS, Kong XP, Gary S, Burgers PM
and Kuriyan J: Crystal structure of the eukaryotic DNA polymerase
processivity factor PCNA. Cell. 79:1233–1243. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kelman Z and O'Donnell M: Structural and
functional similarities of prokaryotic and eukaryotic DNA
polymerase sliding clamps. Nucleic Acids Res. 23:3613–3620. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Koryakina Y, Ta HQ and Gioeli D: Androgen
receptor phosphorylation: Biological context and functional
consequences. Endocr Relat Cancer. 21:T131–T145. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Dong Z, Liu Y and Lu S, Wang A, Lee K,
Wang LH, Revelo M and Lu S: Vav3 oncogene is overexpressed and
regulates cell growth and androgen receptor activity in human
prostate cancer. Mol Endocrinol. 20:2315–2325. 2006. View Article : Google Scholar : PubMed/NCBI
|