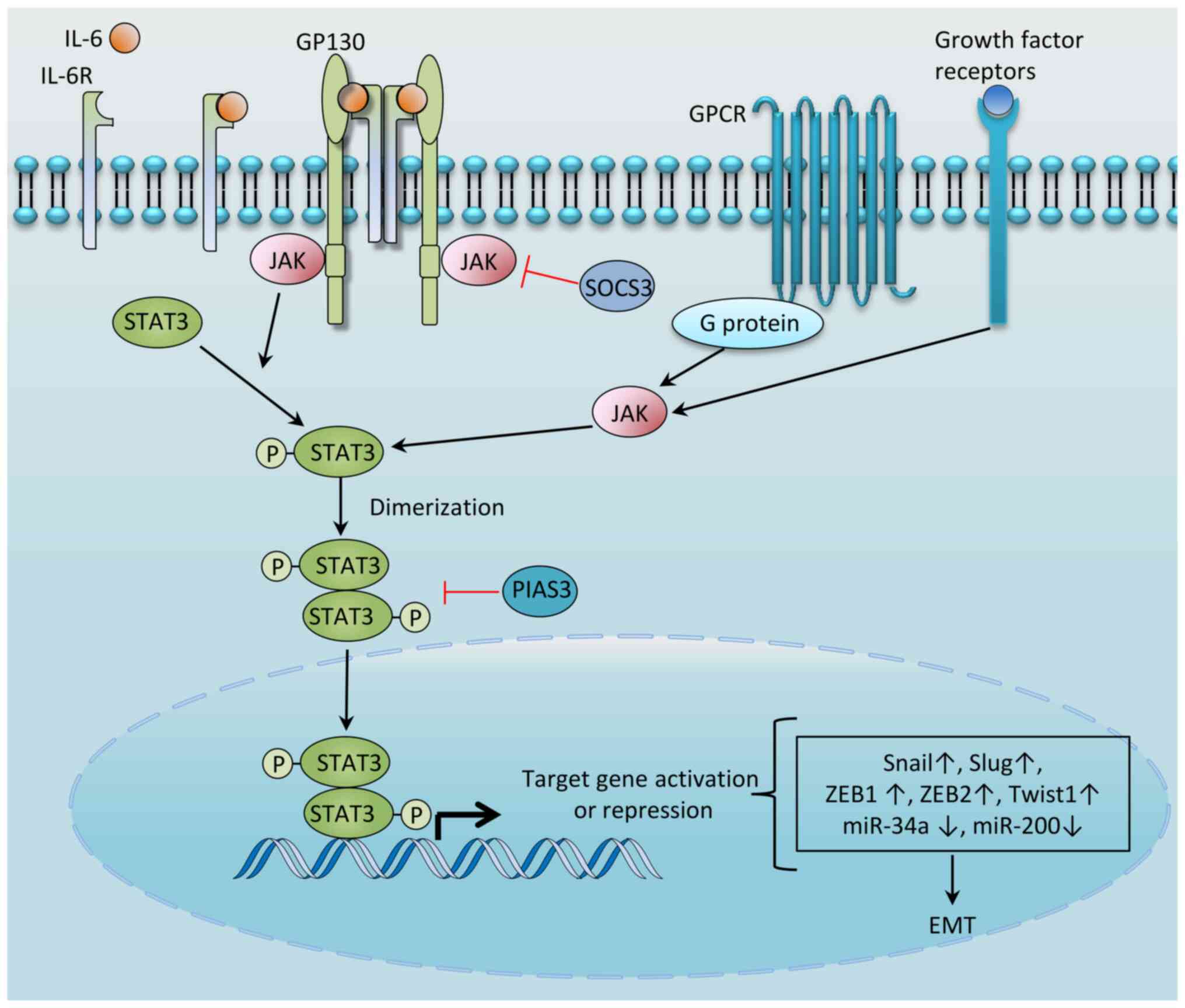
The signal transducer and activator of transcription
(STAT) family of transcription factors (TFs) coordinate cytokine
and growth factor signaling pathways to transcriptionally regulate
a diverse array of cellular processes, such as cellular and
organismal development, proliferation, metabolism, infection,
inflammation and cancer (1).
STAT3, one of seven members of the STAT family (comprising STAT1,
STAT2, STAT3, STAT4, STAT5a, STAT5b and STAT6), has a key role in
the growth and development of various types of human cancer
(2). STAT3 is typically activated
by a wide variety of cytokines [including interleukin (IL)-6,
IL-10, IL-11, IL-31, IL-23, leukaemia inhibitory factor (LIF),
ciliary neurotrophic factor and oncostatin M (OSM)] and growth
factors [including epidermal growth factor receptor (EGFR),
platelet-derived growth factor receptor, fibroblast growth factor
receptor, leptin, granulocyte colony-stimulating factor and peptide
hormones that may be excessively secreted by tumor cells, tumor
stromal cells or immune cells]. These factors bind to their cognate
receptors, inducing conformational changes in the receptors, which
enables the activation of intracellular kinases mainly of the Janus
kinase (JAK) family of non-receptor tyrosine kinases. Once
activated, JAKs transphosphorylate one another and the cytoplasmic
tails of the receptor, forming a docking site for STAT3, which
binds via its SRC homology 2 (SH2) domain. STAT3 is phosphorylated
at Tyr-705 both by JAKs and by non-receptor tyrosine kinases,
including the Src and Abl families of tyrosine kinases (3). Phosphorylated STAT3 undergoes
dimerization via reciprocal interactions between phosphor-Tyr-705
and the SH2 domain, and the homodimer subsequently enters the
nucleus (4) to bind to
palindromic sequences in the genome, thereby initiating the
transcription of hundreds of genes with diverse biological
consequences (3). This pathway is
tightly controlled by negative regulators, including protein
inhibitor of activated STAT3 (PIAS3), protein tyrosine
phosphatases, ubiquitin enzymes and suppressor of cytokine
signaling 3 (SOCS3), which block STAT3 activation either by
directly inhibiting JAK or through inducing its degradation
(5) (Fig. 1). Hyperactivation of the STAT3
signaling pathway is common in diverse types of cancer, and this
typically occurs through several mechanisms, including augmented
cytokine secretion, mutation in upstream kinases or inactivating
mutations in (or epigenetic silencing of) other negative
regulators, such as SOCS (2,3,6,7).
Epithelial-mesenchymal transition (EMT) is a
cellular program that drives plasticity during embryogenesis, wound
healing and cancer progression (9). In various types of cancer, EMT has
been shown to be associated with a large variety of cancer
features, including tumor cell stemness (9), metastasis (9), cancer metabolism (10,11), immune evasion (9,12)
and drug resistance (9), in
addition to adaptation to the microenvironment (9,13-16). EMT is regulated at multiple
levels; physical constraints, hypoxia, inflammation and oncogenic
or metabolic stress act at the first level, whereas the activation
of signaling pathways, including the WNT, hypoxia-inducible factor
(HIF), Notch, transforming growth factor-β (TGF-β), Ras and nuclear
factor-κB (NF-κB) pathways, operates at the second level. These
pathways converge on a set of EMT-activating TFs (EMT-TFs), whose
core set includes SNAI1 (Snail), SNAI2 (Slug), Twist1, zinc finger
E-box binding homeobox 1 (ZEB1) and ZEB2. These TFs are also termed
the 'master' regulators of EMT, and bind to EMT effector genes
(such as E-cadherin, vimentin and N-cadherin), which promote the
loss of epithelial features and the gain of mesenchymal properties
(such as invasion and stem-cell phenotype) (17,18).
E-cadherin acts as the gatekeeper of epithelial
cells and loss of E-cadherin expression is considered a crucial
event in EMT. Snail, Slug and ZEB1 can directly suppress E-cadherin
expression via binding to its promoter (19,20). Twist1 also suppresses E-cadherin
expression but debates regarding its mechanism exist. While some
studies report that Twist1 can directly bind to E-boxes within the
E-cadherin promoter to reduce its expression (21,22), others suggest that Twist1 reduces
expression in an indirect manner such as through PTM (20,23,24). Moreover, loss of E-cadherin
expression is not only a marker of EMT, it also results in the
induction of multiple TFs, including Twist and β-catenin, to
promote EMT (25). Upregulation
of N-cadherin, vimentin and fibronectin is also often observed
during EMT.
In the present review, it is proposed that STAT3
signaling is an integral part of EMT, serving to facilitate the EMT
process via interactions with EMT-TFs, microRNAs (miRNAs), long
non-coding RNAs (lncRNAs) and circular RNAs (circRNAs). The present
review aims to provide both novel insights and a comprehensive
basis for follow-up research.
IL-6 is secreted by multiple cell types within the
tumor microenvironment, including tumor cells, tumor-infiltrating
immune cells and stromal cells (2). There is some evidence to suggest
that the IL-6/STAT3 signaling axis promotes EMT in different types
of cancer. For example, Sullivan et al (26) demonstrated that MCF7, BT474, T47D
and ZR-75-1 cells in a 3D model treated with IL-6 exhibited reduced
expression levels of E-cadherin, a characteristic feature of EMT.
Furthermore, MCF7 cells stably expressing IL-6 (MCF7IL-6
cells), showed characteristics of EMT, including suppression of
E-cadherin expression, induction of vimentin, N-cadherin, Snail and
Twist, and increased invasiveness. Notably, MCF7IL-6
cells also exhibited a reduced expression level of E-cadherin, and
an increased expression of vimentin, in a mice model in
vivo. Similarly, CAL27 cells, a type of head and neck squamous
cell carcinoma (HNSCC) cell line, displayed a decreased level of
E-cadherin expression and enhanced expression of vimentin when
treated with IL-6 for 72 h, which was mitigated by the addition of
a neutralizing anti-IL-6 antibody (27). Additionally, IL-6 overexpression
in HNSCC and immortalized oral epithelial cells was shown to induce
EMT, and these cells also showed higher levels of activation of
STAT3 and Snail compared with the control cells. STAT3 knockdown in
these cells, but not knockdown of AKT or ERK, led to a reversal of
the IL-6-mediated EMT features, suggesting that STAT3 is
responsible for IL-6-mediated EMT (27). In an attempt to understand the
role of IL-6 signaling in prostate tumorigenesis, Rojas et
al (28) treated the P69 and
BPH-1 benign non-tumorigenic prostate epithelial cell lines with
IL-6, which resulted in the induction of EMT, including changes in
the levels of E-cadherin, vimentin, N-cadherin and Snail, and
enhanced motility. Such effects were suppressed by addition of the
JAK2 inhibitor, AG490. IL-6/STAT3-induced EMT has also been
reported in human cervical carcinoma (29). However, there are also reports
indicating IL-6 treatment could not induce EMT in cancer. For
example, treatment of A549, H358 and cancer tissue-originated
spheroid cells with 50 ng/ml IL-6 for 48 h did not lead to EMT
(30). It is possible that this
negative result may be due to an insufficient treatment time with
IL-6. In summary, IL-6 may be effective in inducing EMT in several
cancer models; however, more studies are required to elucidate the
underlying mechanisms, especially with the use of constructed in
vivo models.
Another layer of evidence supporting the role of
STAT3 in EMT is the close association between STAT3 signaling and
the EMT-TFs (Fig. 3).
Snail is the most studied of the EMT-TFs. Numerous
signaling pathways have been found to be associated with the
induction of Snail expression, including the TGF-β, NF-κB, HIF-1α,
Notch and Wnt pathways, reactive oxygen species (ROS) and hypoxia
stress [see the reviews (18,31) for further information]. Snail is
also regulated by the IL-6/STAT3 signaling pathway. The first
reports linking STAT3 and Snail were from studies on zebrafish and
breast cancer. Solute carrier family 39 member 6 (SLC39A6; also
termed LIV-1 or ZIP6), a member of the family of zinc transporter
proteins, was revealed to be upregulated by STAT3 during zebrafish
gastrulation (32) and in EMT in
breast cancer induced by EGF (33). SLC39A6 facilitates the influx of
zinc, which inactivates glycogen synthase kinase-3β (GSK-3β).
Inactivated GSK-3β is unable to phosphorylate and destabilize
Snail, which thereby increases the level of nuclear Snail (33) and promotes EMT. Therefore, STAT3
serves to regulate Snail in an indirect, post-transcriptional
manner.
Treatment with IL-6, or IL-6 overexpression leads to
Snail upregulation at both the mRNA and protein levels in various
types of cancer in vitro, including pancreatic cancer
(34), HNSCC (27), breast cancer (26) and colon cancer (35), and even non-tumorigenic prostate
epithelium cells (28). Such
effects were mediated by STAT3, as suppression of STAT3 led to a
decrease in IL-6-induced Snail upregulation (27,34). In separate studies, TGF-β and
H-Ras were shown to act synergistically to increase Snail
expression (36,37), in which STAT3 also had a role as
STAT3 knockdown ameliorated Snail upregulation by TGF-β and H-Ras
(37). STAT3 was shown to
maintain Snail expression under normal culture conditions, and
STAT3 knockdown or suppression by inhibitors decreased Snail
expression in both breast and prostate cancer (38,39). In a study using hepatocellular
carcinoma (HCC) cells, phosphorylated STAT3 was found to bind to
the Snail promoter; moreover, inhibition of STAT3 by AG490
abrogated hepatitis virus C core-induced expression of Snail
(40).
Snail also regulates STAT3 signaling; for example,
in ARCaP and MCF-7 cells, ectopic overexpression of Snail was shown
to induce further activation of STAT3 (39). Overexpression of Snail also led to
an increase in lactate-induced STAT3 activation in A549 and H1299
cells, whereas Snail knockdown reduced STAT3 activation (44). The underlying mechanism has yet to
be elucidated; however, lactate was demonstrated to induce the
formation of a Snail-enhancer of zeste homolog 2 (EZH2)-STAT3
complex, which enhanced STAT3 activation (44). EZH2 has also been shown to
activate STAT3 via methylation in GBM stem-like cells (45) and in breast cancer cells (46), and therefore, it may be
interesting to investigate whether EZH2 may be required for
Snail-induced STAT3 activation. In hepatitis B virus
(HBV)-associated HCC, the HBV-induced overproduction of ROS was
shown to increase the expression level of Snail, which binds to
E-boxes of the SOCS3 promoter, thereby decreasing SOCS3 expression
via hypermethylation of the SOCS3 promoter, and causing
constitutive activation of STAT3 (47).
Therefore, taken together, the results from several
studies have shown that a positive and mutual regulatory
relationship exists between STAT3 and Snail. STAT3 is able to
increase Snail expression both transcriptionally and
post-transcriptionally, whereas Snail is able to increase the
activation of STAT3 via interacting with STAT3 or suppressing SOCS3
expression.
Slug is another EMT-TF that is important for the EMT
process in cancer. Radiation-resistant A549 cells exhibited
enhanced expression of Slug, which mediated tumor invasion and
resistance. STAT3 small interfering (si)RNA and the STAT3
inhibitor, WP1006, reduced Slug expression and partly restored
tumor cell sensitivity to radiation (48). In HBV-associated HCC,
small-surface antigens promote HCC progression via STAT3-induced
Slug. Treatment with either STAT3 siRNAs or the JAK2 inhibitor,
AG490, was found to reduce the small-surface antigen-induced
upregulation of Slug (49).
STAT3 suppression in brain tumor stem cells (BTSCs)
decreased Slug expression. Furthermore, treatment with EGF, LIF or
OSM led to Slug upregulation, which was reduced by a STAT3
inhibitor, suggesting that these effects were mediated by STAT3. A
ChIP assay revealed that STAT3 bound to the Slug promoter, but not
to the promoters for Snail, Twist, ZEB1 or ZEB2, in BTSCs (50). Lin et al (51) showed that STAT3 binds to the -472
to -463 (TTTTTCAAAA) region of the slug promoter, thereby
increasing Slug expression and enhancing GBM radioresistance.
Plasmacytoma variant translocation 1 (PVT1) is a
well-studied lncRNA that is located at the 8q24.21 region near the
c-Myc oncogene. PVT1 is upregulated by copy number amplification
and is able to promote cancer progression (52,53). Zhao et al (54) showed that PVT1 enhances STAT3
recruitment to the Slug promoter, and transcriptionally enhances
Slug expression in gastric cancer. Treatment with a STAT3 inhibitor
led to a reduction in PVT1-induced Slug upregulation.
Taken together, these results demonstrated that
STAT3 acts as a positive regulator of Slug expression through
binding to its promoter and increasing its transcription.
The Twist protein is a highly conserved TF that
belongs to the family of basic helix-loop-helix proteins. Twist
fulfils a critical role in both embryogenesis and tumorigenesis
(55,56), and its upregulation has been shown
to be associated with numerous types of aggressive tumors,
executing multiple roles in cancer initiation and progression
(56). Several signaling pathways
have been shown to upregulate Twist1 expression in various types of
cancer (56), including NF-κB,
Src, HIF-1α and STAT3. Knockdown of STAT3 protein by RNA
interference in mouse breast cancer cells was shown to block the
expression of Twist and to prevent metastases (57). STAT3 also mediates the IL-6-, EGF-
and Notch-induced upregulation of Twist (58-60). When upregulated, mesoderm-specific
transcript promotes the invasion of breast cancer, and has been
shown to induce Twist-mediated EMT through STAT3 activation
(61). Therefore, diverse
signaling pathways converge on STAT3 to increase Twist expression.
Furthermore, immunohistochemical (IHC) analysis of breast carcinoma
(58,59) and HCC (62) samples revealed that a positive
correlation exists between phosphorylated STAT3 and Twist.
Mechanistically, STAT3 was shown to bind to the promoter of Twist
(58-60), leading to an increase in Twist
expression. Moreover, these studies suggested the same STAT3
binding site in the Twist promoter (-95 to -116).
ZEB1 is not only an EMT-TF, but it is increasingly
being recognized as a crucial regulator of fundamental biological
processes, including stemness vs. differentiation, cell
proliferation vs. senescence and survival vs. apoptosis (63,64). STAT3 is a direct regulator of ZEB1
in various types of cancer. For example, in colon cancer, which
often features STAT3 hyperactivation (65), AG490, an inhibitor of JAK2, was
shown to suppress the expression of ZEB1, but not of ZEB2, Snail,
Slug, Twist1 or Twist2. Similarly, STAT3 knockdown was found to
suppress both ZEB1 expression and the migration of colon cancer
cells (66). STAT3 has also been
shown to bind to the ZEB1 promoter, and mutation of the binding
sites led to a marked reduction both of STAT3 binding and of ZEB1
promoter activity (66). Another
example was provided in a study by Avtanski et al (67), where it was shown that the
constitutively activated form of STAT3 was able to bind to the ZEB1
promoter and to increase the mRNA expression of ZEB1. In addition,
the STAT3 inhibitors, Stattic and honokiol, were shown to reduce
both STAT3 binding and the mRNA expression of ZEB1, and to suppress
EMT in breast cancer (67). The
STAT3/ZEB1 signaling axis was also investigated in
gefitinib-resistant A549 and PC9 cells, wherein increased
activation of STAT3 and the characteristic features of EMT were
displayed, including increased expression levels of ZEB1,
N-cadherin and vimentin, and a decreased expression of E-cadherin,
compared with the parental cells. STAT3 knockdown by siRNA in these
resistant cells led to a reversal of EMT, including ZEB1
downregulation (68). Taken
together, these results support the hypothesis that STAT3 binds
directly to the promoter of ZEB1, enhancing ZEB1 expression to
promote EMT.
Loss of E-cadherin is a hallmark of EMT, and this
phenomenon is associated with increased tumor cell invasion and
spread. In a study by Zhang et al (62), IHC analysis revealed that
activation of STAT3 was conversely correlated with E-cadherin
expression in HCC. In addition, it has been demonstrated that IL-6
treatment leads to E-cadherin downregulation in HCT116 colorectal
carcinoma cells (69). Although
there are two putative STAT3-binding sites in the E-cadherin
promoter region, STAT3 may not directly bind to the E-cadherin
promoter; instead, it may function via regulating the major
EMT-TFs, including Snail, Slug, Twist and ZEB1, to influence
E-cadherin expression (66,69).
The TGF-β superfamily comprises structurally related
growth factors, including TGF-β, activins and bone morphogenetic
proteins. These factors fulfil important roles in morphogenesis
during embryonic development and tissue homeostasis in adults
(70,71). Among them, TGF-β1 is a
well-established potent EMT inducer (72), and adding TGF-β1 to epithelial
cell culture has been shown to be an effective way of inducing EMT.
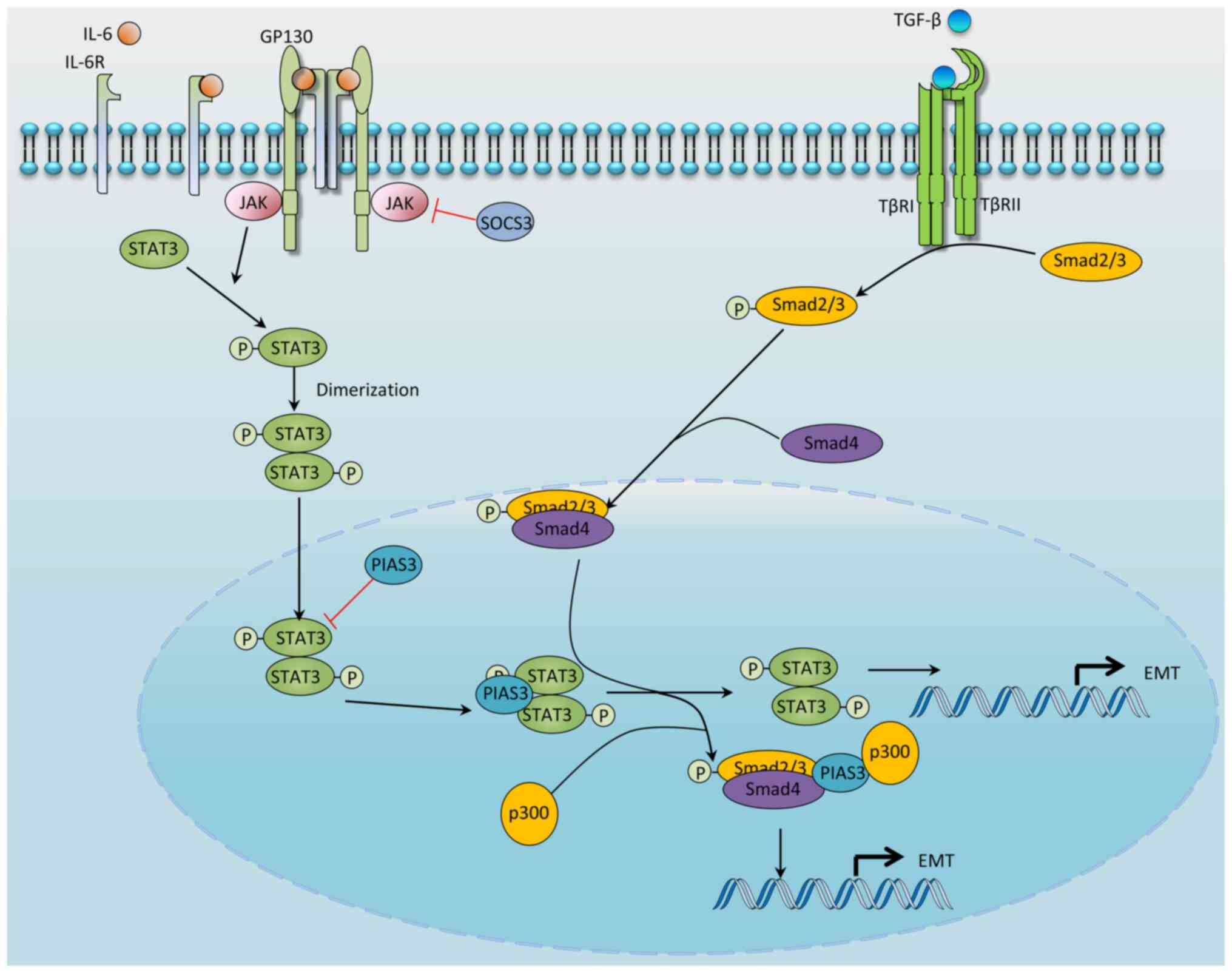
TGF-β1 activates signaling by binding to and promoting the
formation of the single-span transmembrane TGF-β receptor
(TβR)I-TβRII heterocomplex (Fig.
4), which leads to the phosphorylation and activation of the
receptor-regulated Smad (R-Smad) proteins, Smad2 and Smad3. The
activated R-Smad proteins subsequently form a complex with co-Smad
(Smad4), and the complex then translocates to the nucleus, where it
regulates the transcription of a broad range of genes. In addition
to the canonical Smad signaling pathway, TGF-β1 activates other
signaling pathways, including the AKT, ERK, p38/MAPK, GTPase and
STAT3 signaling pathways (71).
These pathways all contribute to the effects elicited by TGF-β1 in
both a context and cell type-dependent manner. In the present
review, the role of STAT3 signaling in TGF-β1-induced EMT is
specifically summarized.
The association between STAT3 and TGF-β1 signaling
is context-dependent in cancer [refer to (71,73) for further information]. During the
early phase of tumorigenesis, STAT3 and TGF-β1 are mutually
antagonistic. Although STAT3 is oncogenic even in the onset of
tumorigenesis (74,75), TGF-β functions both as a tumor
suppressor in pre-malignant cells and as a tumor promoter in
late-stage tumors (76).
TGF-β-induced EMT and Snail expression has been
shown to be enhanced by Ras signaling (77). Through screening a library of
siRNAs, Saitoh et al (37)
identified STAT3 as the mediator molecule that markedly enhances
the Snail promoter activity induced by TGF-β and Ras signaling.
Knockdown or inhibition of STAT3 attenuates TGF-β-induced Snail
upregulation and EMT; moreover, STAT3 mutants that either cannot be
phosphorylated at Tyr-705 or lack transcriptional activity fail to
activate Snail expression. Mechanistically, Smad3 activated by
TGF-β signaling both interacts with and sequesters PIAS3 in the
presence of Ras signaling, thereby causing STAT3 to be released
from its inhibition of PIAS3 and allowing it to positively regulate
Snail expression. Notably, the presence of a PIAS3-Smad3-p300
ternary complex was found to be significantly enhanced in response
to TGF-β, which subsequently increased the activity of Smad3
(78); therefore, PIAS3, upon
dissociation from STAT3, forms the PIAS3-Smad3-p300 ternary
complex, and this represents one of the mechanisms underlying
TGF-β-induced STAT3 activation (Fig.
4).
TGF-β has also been shown to activate STAT3 in colon
cancer, in which TGF-β signaling was inactivated by mutations
(79), suggesting that TGF-β may
activate STAT3 via a mechanism not involving intracellular
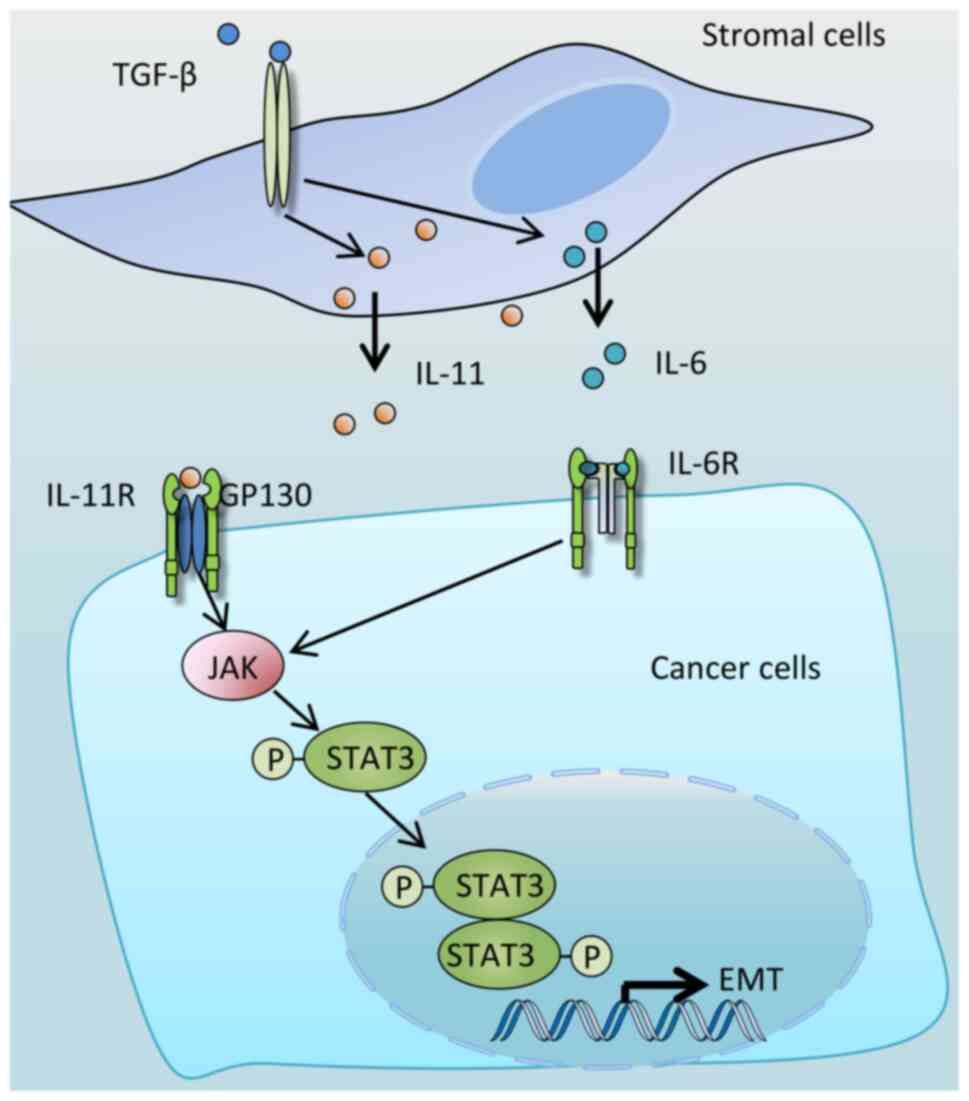
signaling. Indeed, IL-11 was revealed to be the mediator (Fig. 5); TGF-β induces stromal cells to
secrete IL-11, which in turn activates STAT3 in colon cancer cells
through binding to the transmembrane receptor protein, glycoprotein
130 (79). The TGF-β/IL-11/STAT3
signaling axis was also shown to be required for colon cancer
metastasis (79). In addition to
IL-11, in non-small cell lung cancer (NSCLC), HCC and normal human
lung fibroblast cells, TGF-β treatment led to increased secretion
of IL-6. Treatment with either an IL-6 receptor neutralizing
antibody or a JAK/STAT3 inhibitor was found to reduce
TGF-β-mediated STAT3 activation and EMT (80-82). Taken together, these results
suggest that IL-6 is also a mediator of TGF-β-mediated STAT3
activation and EMT (Fig. 5).
Src homology 2-b3 protein (SH2B3; also known as
lymphocyte adapter protein) belongs to the SH2B family of adaptor
proteins and is a negative regulator of JAK/STAT signaling.
Mutations in SH2B3 have been identified in a range of hematological
and inflammatory diseases (83).
Although SH2B3 is considered to act as a negative regulator in
hematological cancer, its role in solid tumors remains
controversial. SH2B3 was reported to act as a tumor promoter in
ovarian (84), breast (85) and anaplastic thyroid cancer
(86) cancer. However, compared
with matched adjacent normal tissues, SH2B3 was found to be
downregulated in colon cancer, and its overexpression led to a
decrease in the invasion rate of colon cancer cells (87). Wang et al (88) also showed that the expression of
SH2B3 is decreased in lung cancer, whereas its overexpression led
to a suppression of malignant phenotypes, including reduced rates
of cell proliferation and invasion. Furthermore, TGF-β was shown to
both reduce SH2B3 expression and activate JAK2/STAT3 and EMT, which
was attenuated by SH2B3 overexpression. Therefore, SH2B3
downregulation may represent an additional mechanism underlying
TGF-β-induced STAT3 activation and EMT.
miRNAs are small (~22-nucleotide) non-protein-coding
RNAs that regulate gene expression by associating with
complementary sequences in the 3′-untranslated region (UTR) of
their target genes, thereby blocking translation. In the field of
cancer, miRNAs can be functionally divided into oncogenic miRNAs
and tumor-suppressor miRNAs (89). Several miRNAs has been shown to be
critical regulators of EMT (18,90,91), including miR-200, miR-34 and
miR-30a, and herein only those miRNAs that are associated with
STAT3-induced EMT are discussed (see Table I).
The miR-34 family members (miR-34a, miR-34b and
miR-34c), and miR-34a in particular, are recognized as master tumor
suppressors (92). Loss of
miR-34a expression occurs in a wide range of tumors, and this miRNA
has been validated as a promising prognostic indicator. To date,
>200 miR-34a targets have been reported, and through these
target genes, miR-34a has been shown to regulate multiple cancer
processes, including the cell cycle, EMT, metastasis, stemness of
cells, apoptosis, senescence and tumor immunity (92,93).
It has been demonstrated that miR-34a suppresses EMT
in various cancer types through targeting a number of key EMT
genes. For instance, miR-34a inhibits EMT through directly
downregulating the expression of the EMT-TFs, Snail (94), ZEB1 (95) and Twist (95), by binding to their 3′-UTRs.
Moreover, Snail and ZEB1 are able to bind to the E-box sequences in
the miR-34a promoter, thereby decreasing miR-34a expression and
forming a double-negative feedback loop maintaining the EMT state
(94). In addition, miR-34a
suppresses several critical EMT signaling pathways, including the
TGF-β [via targeting Smad4 (96)
and TβRII (97)], STAT3 [via
targeting IL-6R (35)], Wnt
(98,99) [via targeting Wnt1 (100,101), transcription factor 7 (TCF7)
(102) and lymphoid enhancer
binding factor 1 (103)], Notch
[via targeting Notch1 (104) and
Notch2 (105)] and AXL [via
targeting AXL (106)] pathways.
All of these signaling molecules and pathways act as enhancers of
EMT (9,17,107,108).
The miR-200 family of miRNAs, including miR-200a,
miR-200b, miR-200c, miR-141 and miR-429, are encoded by two
clusters of hairpin precursors located on human chromosomes 1p36.33
(miR-200b, miR-200a and miR-429 are termed the 'miR-200b/200a/429
cluster') and 12p13.31 (miR-200c and miR-141 are termed the
'miR-200c/141 cluster'). Each of these miRNAs produces a mature-5p
and -3p miRNA (110).
miR-200 is highly expressed in epithelial cancer
cells, and minimally expressed in mesenchymal cancer cells
(110). Overexpression or
knockdown of miR-200 causes changes in the EMT state of cancer
cells by directly targeting ZEB1 and ZEB2 (110-113), which leads to an alteration in
E-cadherin expression, thereby promoting EMT (114-116). Downregulation of miR-200
expression is observed during TGF-β-induced EMT, and overexpression
of miR-200 hinders TGF-β-induced EMT, implying that miR-200 is an
integral component of TGF-β-induced EMT (115,116).
The promoters of both of the aforementioned miR-200
clusters contain ZEB-type E-box elements, and their activities were
shown to be repressed by ZEB1 and ZEB2 (117,118). Therefore, ZEB1/2 and miR-200,
which exert opposite functions on EMT, reciprocally regulate each
other in a double negative feedback loop. There is also evidence to
suggest that STAT3 suppresses miR-200 expression. For instance,
treatment with OSM has been shown to reduce miR-200b and miR-200c
expression in a STAT3-dependent manner to promote EMT (119). Additionally, treatment with the
STAT3 inhibitor, Stattic, leads to a significant upregulation of
miR-200a, miR-200b and miR-429, and a reversal of EMT (120). By contrast, overexpression of
STAT3 leads to a reduction in the expression of these miRNAs, and
an enhancement of EMT (120).
Further study showed that this effect is dependent on EZH2, which
itself is a direct target of STAT3 (121). Therefore, disrupting the
EZH2/miR-200 axis has the effect of attenuating the EMT-promoting
effects of STAT3 (120). Another
study (122) on bladder cancer
also found that EZH2 was able to reduce miR-200 expression and
promote cancer progression, thereby adding a further line of
evidence in support of the existence of a STAT3/EZH2/miR-200
signaling axis in cancer. However, whether STAT3 directly binds to
the promoter of miR-200b/-a/-429 or miR-200c/-141 requires further
study.
miR-30 is a tumor suppressor that inhibits EMT by
directly binding to Snail and downregulating its expression
(123,124). As reported in AML12 murine
hepatocytes (124) and HNSCC
(125), TGF-β1 treatment led to
the induction of EMT concomitant with the downregulation of miR-30.
The ectopic expression of miR-30 mimics inhibited both the EMT
phenotype (125) and
TGF-β1-induced EMT (124,125).
miR-30 was also shown to negatively regulate the expression of
Snail though direct targeting of its 3′-UTR sites (124). STAT3 activated by TGF-β1 binds
to the promoter of metastasis-associated lung adenocarcinoma
transcript 1 (MALAT1), thereby increasing its expression.
Upregulated MALAT1 sponges miR-30a, leading to a decrease in
miR-30a expression (125).
Therefore, it has been shown that TGF-β1 is also able to promote
EMT through the STAT3/MALAT1/miR-30 signaling axis.
miR-218, acting as a tumor suppressor, was shown to
be downregulated in various cancer types compared with the normal
surrounding cells (134).
miR-218 suppress EMT in several cancer models, including lung
cancer [via targeting of roundabout guidance receptor 1,
EGFR-coamplified and overexpressed protein (135) and Slug/ZEB2 (136)], cervical cancer (via targeting
of Scm-like with four MBT domains 1 and defective in cullin
neddylation 1, domain containing 1) (137), HCC (via targeting of serpin
mRNA-binding protein 1) (138),
glioma cells (via targeting of lipoma HMGIC fusion partner-like 3)
(139), colorectal cancer (CRC;
via targeting of connective tissue growth factor) (140) and gastric cancer (via targeting
of WASP family member 3) (141).
STAT3 directly interacts with a locus downstream of the miR-218
gene, inhibiting its expression by recruiting the transcriptional
repressor, BCL2-associated transcription factor 1 (142). Therefore, it seems plausible
that STAT3 enhances EMT by directly inhibiting miR-218 expression;
however, to date, this has not been confirmed experimentally.
lncRNAs comprise a large class of regulatory RNA
molecules, are generally >200 nucleotides in length and are
considered to lack evident protein-coding potential (143-145). lncRNAs fulfil crucial roles in
diverse biological processes, including EMT (144,145), and perform their functions
through modifying gene expression at either the transcriptional or
the post-transcriptional level, or by interacting with DNA, RNA (by
complementary base-pairing) or proteins (by adapting specific
secondary structures) (143).
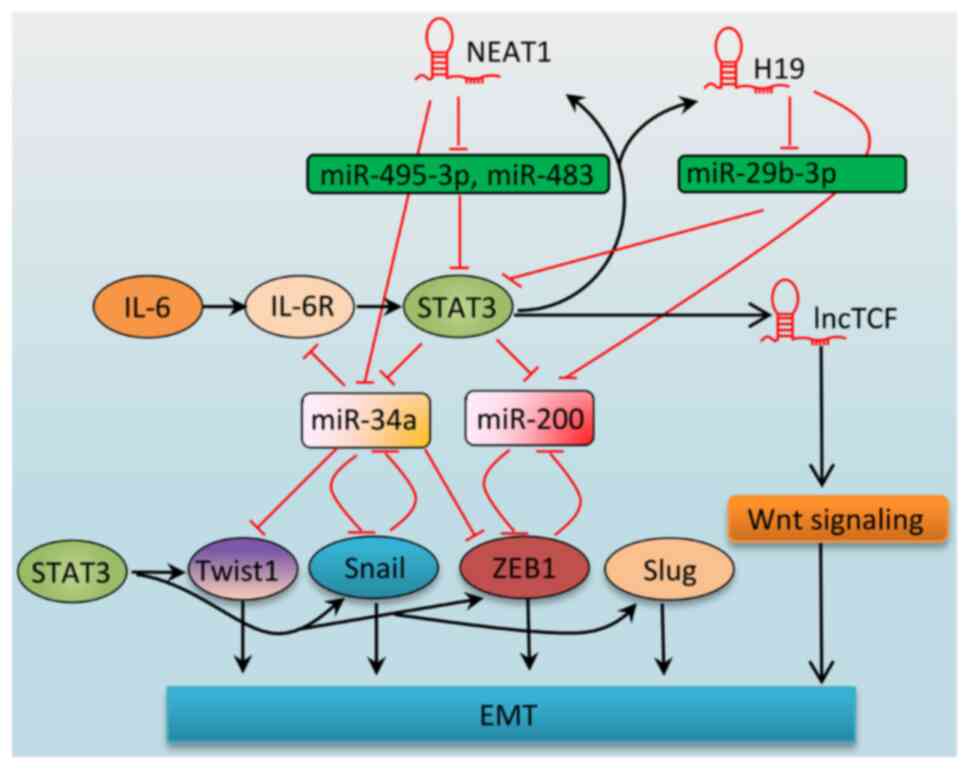
A growing body of evidence has shown that STAT3
signaling is regulated by, and also regulates an increasing number
of, lncRNAs (146-148). A dual relationship exists
between lncRNAs and STAT3 signaling as they influence each other to
promote cancer progression. STAT3 regulates the expression of
lncRNAs to enhance EMT; however, lncRNAs also modulate STAT3
expression or activity to coordinate EMT (Fig. 3 and Table II). For instance, nuclear
paraspeckle assembly transcript 1 (NEAT1), the most extensively
studied lncRNA, which is abnormally expressed in numerous types of
cancer, has been shown to drive tumor initiation, progression and
drug resistance (149), and is
also an enhancer of EMT in different types of cancer (150-152). STAT3 enhances NEAT1 expression
by binding to its promoter (153). In osteosarcoma cells, NEAT1 was
found to increase STAT3 expression by sponging miR-483 to promote
EMT (154). Additionally, NEAT1
has been shown to sponge miR-361 (155) and miR-495-3p (156), leading to the upregulation of
STAT3. Therefore, a positive loop exists between NEAT1 and STAT3,
as they mutually enhance each other's oncogenic function.
H19 imprinted maternally expressed transcript (H19)
is another widely studied potent EMT enhancer (157-159). Several mechanisms have been
suggested to explain the effects of H19 (158,159). For instance, H19 sponges miR-200
to upregulate ZEB1 and it sponges miR-138 to increase the level of
SRY-box transcription factor 4 to enhance EMT (160,161). Additionally, H19 has been shown
to associate with EZH2 to both enhance β-catenin expression and
decrease E-cadherin expression (162). Moreover, studies have revealed
that STAT3 is an important downstream mediator of the EMT-promoting
function of H19. miR-29b-3p targets STAT3, leading to a decrease in
its expression (163,164), and H19 has been shown to promote
EMT by targeting miR-29b-3p to increase STAT3 expression (165). In prostate cancer cells,
miR-675-3p, a non-coding RNA transcribed from the first exon of
H19, was reported to mediate the EMT function of H19 by
downregulating the STAT3 inhibitor, SOCS5 (166). Considering that STAT3 also
upregulates H19 transcriptionally (167), STAT3/H19 may constitute a
positive loop to induce EMT.
IL-6 has been shown to increase the level of
lncTCF7 expression via STAT3 binding to the lncTCF promoter, and
knockdown of lncTCF7 expression impaired EMT induced by IL-6 in HCC
(168), suggesting the
involvement of lncTCF7 in IL-6-induced EMT. KIAA0087 is a recently
identified tumor suppressor lncRNA, the expression of which is
reduced in endometrial carcinoma (169) and is associated with overall
survival in NSCLC (170). Gong
et al (171) demonstrated
that KIAA0087 was also downregulated in osteosarcoma compared with
normal tissues, and its downregulation was found to promote cell
growth, metastasis and EMT through releasing the sponging effect of
miR-411-3p, which mediates reductions in the level of SOCS1 and
activation of the JAK2/STAT3 pathway.
The lncRNA cancer susceptibility 11 (CASC11; also
known as CARLo-7, LINC00990 and MYMLR) was also found to be
upregulated in various types of cancer (172), and functions as a oncogene to
promote cancer progression, including EMT. CASC11 has also been
shown to be associated with poor prognosis (172,173) and to enhance bladder cancer cell
proliferation, invasion and EMT through activating the
Wnt/β-catenin and STAT3 signaling pathways (173). Additionally, CASC11 knockdown
was shown to reduce EMT in HCC (174). Notably, four STAT3 binding sites
exist in the CASC11 promoter, and deletion of the first site
significantly decreases CASC11 promoter activity. In addition,
manipulation of STAT3 expression changes CASC11 expression
accordingly (174). Therefore,
these studies have collectively shown that STAT3 acts as a TF,
promoting CASC11 expression to enhance cancer EMT. Additionally,
STAT3 signaling appears to operate downstream of CASC11, mediating
CASC11-induced EMT. However, the detailed underlying mechanisms of
this requires further investigation.
The expression of lncRNA AB073614 was found to be
significantly higher in the tumor tissues of various cancer types
compared with that in the surrounding normal tissues, including
ovarian cancer (175,176), cervical cancer (177), glioma (178,179) and CRC (180,181), and has been shown to facilitate
invasion, proliferation and EMT (179,181). AB073614 knockdown in colon
cancer cells reversed EMT, along with decreased STAT3 activation.
Furthermore, a JAK2 inhibitor, AT9283, blocked the effects of
AB073614, suggesting that STAT3 may be involved in the EMT-inducing
role of AB073614 (181).
DLGAP1-AS1, an oncogenic lncRNA, that has been identified in
several types of cancer, and it was shown to be upregulated in
tumor tissues, where it enhanced tumor progression, EMT and drug
resistance (182). Lin et
al (183) showed that
DLGAP1-AS1, through sponging miR-26a/b-5p which directly targets
IL-6, promoted STAT3 signaling. STAT3 reciprocally increased the
expression of DLGAP1-AS1, thereby forming a positive feedback loop
that facilitates EMT in HCC. DLGAP1-AS1 knockdown inhibits EMT in
HCC, and treatment with IL-6 is able to partially restore EMT
suppressed by knockdown of DLGAP1-AS1.
The lncRNA, PVT1, has been shown to facilitate EMT
by physically interacting with activated STAT3, which then enhances
STAT3 binding to the Slug promoter, increasing Slug expression to
facilitate EMT (54). Indeed,
several studies have revealed that PVT1 is an EMT inducer (52,53,184-187). Furthermore, STAT3 was also shown
to upregulate PVT1 expression through binding to its promoter
(14) and therefore, PVT1 and
STAT3 form a positive regulatory loop to enhance cancer
progression. Taken together, PVT1 has been demonstrated to
participate in the regulation of the STAT3-EMT signaling axis.
FEZ family zinc finger antisense 1 (FEZF1-AS1) is a
novel oncogenic lncRNA that is upregulated in various types of
human cancer, and is associated with various aspects of
carcinogenesis, including cell proliferation, invasion, metastasis
and EMT (188-191). It was reported that FEZF1-AS1
could activate STAT3 in ovarian cancer and CRC (192,193). Conversely, Knockdown of
FEZF1-AS1 was found to reduce cell proliferation and EMT, and to
enhance apoptosis, concomitant with a decreased activation of STAT3
(194). Furthermore, JAK2
overexpression notably restored the attenuated EMT following
FEZF1-AS1 knockdown, suggesting that the JAK2/STAT3 signaling axis
participates in mediating the effect of FEZF1-AS1 on EMT (194).
circRNAs are a class of RNAs that are
single-stranded and circular, lacking 5′-caps and 3′-tails.
circRNAs are stable, difficult to cleave and resistant to RNA
exonuclease or RNase degradation (195-197), and function through modulating
transcription and splicing, regulating the stability and
translation of cytoplasmic mRNAs, interfering with signaling
pathways and serving as templates for translation (198). With the rapid development of
sequencing technology, novel circRNAs have been discovered, and
their characteristics and functions are being revealed (198). Dissecting the roles and
mechanisms of circRNAs is a cancer research 'hotspot', and are also
promising targets for cancer therapy (199-201).
An increasing number of studies have reported that
circRNAs regulate EMT by targeting EMT-TFs or EMT-associated
signaling pathways (195,202,203).
Unfortunately, at present and to the best of our knowledge, no
study has surveyed the role of circRNAs in the STAT3-EMT signaling
axis or the role of STAT3 in the circRNA-EMT axis in any great
detail. Previously published studies (204-207) have shown that certain circRNAs
are able to induce or reduce EMT, concomitant with enhanced or
reduced activation of STAT3. However, whether or not STAT3 is
required for these circRNA-induced EMT changes has not yet been
studied; therefore, at present, the STAT3-circRNA-EMT axis requires
further investigation.
Due to the critical tumor-promoting role, the STAT3
pathway has been intensely pursued as a therapeutic target. The
inhibitors of the STAT3 pathway can be divided into direct STAT3
inhibitors, JAK inhibitors and IL-6/IL-6R inhibitors.
STAT3 itself is a TF that lacks enzymatic activity,
and therefore the development of inhibitors has been difficult.
Generally, direct inhibitors of STAT3 can be classified into three
categories: peptides, small molecules and oligonucleotides
(208,209).
STAT3 activation requires an interaction between
the SH2 domain and phosphorylated Tyr-705; therefore, it is
plausible that a peptide mimicking the sequence containing
phosphorylated Tyr-705 would be able to bind to the SH2 domain of
STAT3 and inhibit its activation and activity (209). Indeed, a 6-amino acid
Tyr-phosphorylated peptide (PY*LKTK) can bind to the STAT3 SH2
domain, thus blocking STAT3 dimerization, DNA binding and gene
regulation (210). Mimics or
modification of PY*LKTK such as peptidomimetic ISS-610 (211) and PM-73G (212), also suppress STAT3 activity.
However, these agents are challenged by potency, cellular
permeability, stability and potential immunogenicity, which hinder
their clinical development (2,209,213).
Another group of STAT3 inhibitors are small
molecules mainly targeting the SH2 domain (209). The number of inhibitors reported
is large; however, only a few have entered into early phase
clinical trials. For instance, C188-9 (also termed TTI-101), which
targets the STAT3 SH2 domain, inhibits STAT3 activation in
vitro (214) and alleviates
inflammation and the severity of colitis in a T-cell transfer
colitis model in vivo (215). C188-9 also suppressed HNSCC
growth in a nude mice xenograft model (216). The clinical trial of this
inhibitor in humans (NCT03195699) is still ongoing. Another two
STAT3 SH2 domain inhibitors, OPB-31121 and OPB-51602, highly
suppress STAT3 activation and display potent cancer suppression
in vitro and in mouse models (217-220). However, early phase clinical
trials of OPB-31121 and OPB-51602 showed very limited clinical
activity (2), and the reasons for
this failure are not currently known. A lack of specificity due to
a high similarity of the SH2 domain of STAT3 and other STAT family
members may be involved.
ASOs are short oligonucleotides that can base-pair
with complementary RNA and trigger post hybridization mechanisms to
modulate gene expression (221).
One example is AZD9150 (danvatirsen), which targets the 3′-UTR
region of the STAT3 gene (222).
Clinical studies have shown that it is well tolerated (223-225), decreased the tumor-initiating
potential of neuroblastoma cells (222) and suppressed leukemic cell
growth (226). The
tumor-suppressive effect of danvatirsen may be related to tumor
stromal cells, which preferentially uptake danvatirsen and suppress
tumor growth (227). Clinical
trials for HCC (NCT01839604), HNSCC (NCT05814666), CRC
(NCT02983578) and NSCLC (NCT02983578) are ongoing to evaluate the
safety and activity of danvatirsen.
TF decoy oligonucleotides are short double-stranded
DNA molecules that bind to TFs, thus blocking the interaction
between TFs and DNA. Leong et al (228) designed a STAT3 decoy composed of
a 15-bp double-stranded oligonucleotide representing the STAT3
responsive element within the c-Fos promoter. The decoy inhibited
STAT3 transcriptional activity by competitively interfering with
phosphorylated STAT3 dimers binding to the promoter region of STAT3
target genes, thereby inhibiting STAT3-mediated gene regulation.
Further studies showed that the decoy suppressed growth of HNSCC
(229) and lung cancer (230) cells in xenograft models via
daily intratumoral injection. Additionally, a phase 0 clinical
trial (NCT00696176) demonstrated that this STAT3 decoy abrogated
target gene expression in HNSCC tumors. Although encouraging
effects were observed, the decoy was unstable in serum and
short-lived, which restricted its usage (231). To overcome this barrier, Sen
et al (231) designed a
cyclic STAT3 decoy by linking the oligonucleotide strands using
hexaethylene glycol spacers. This modified decoy had a long
half-life in serum (~12 vs. ~1.5 h, compared with the parental
decoy), making it suitable for intravenous (IV) administration.
Indeed, in HNSCC (231) and
NSCLC (232) xenograft mice,
daily IV injections of the modified decoy significantly prevented
tumor growth, concomitant with decreased expression of STAT3 target
genes. Other modifying strategies have also been applied. For
instance, Zhang et al (233) linked the same STAT3 decoy to the
Toll-like receptor 9 (TLR9) ligand. This STAT3 decoy conjugate also
had a long half-life and targeted TLR9+ immune cells
(dendritic cells and B cells) and the majority of acute myeloid
leukemia cells from patients, including leukemia stem/progenitor
cells preferentially. In preclinical studies, daily IV injections
of the STAT3 decoy conjugate markedly reduced myeloid leukemia
progression in a mouse model (233).
Although oligodeoxynucleotides inhibitors of STAT3
provide great specificity and potency, their poor cell membrane
penetration, rapid degradation and the lack of effective targeted
delivery carriers remain the major obstacles that impede their use
against solid tumors clinically.
PROTAC technology has emerged as a promising
strategy for developing novel drugs, and acts by inducing targeted
protein degradation through ubiquitination-mediated proteasomal
degradation (234,235). A STAT3-targeting PROTAC molecule
can bind to STAT3 specifically on one side and to an E3 ligase on
the other side, thus inducing specific degradation of STAT3.
Since 2015, the field of PROTAC technology has
grown rapidly and currently at least 20 PROTACs have entered
clinical trials, including KT-333, which targets STAT3 (235,239). PROTAC technology provide routes
to target proteins once considered 'undruggable', and some of these
PROTACs exhibit superior potency and efficacy against cancer. For
instance it was reported that SD-36 induced complete and long-term
tumor regression at doses of either 100 mg/kg weekly or 50 mg/kg
twice weekly for 4 weeks in animal models (236). However, there are several
challenges to overcome, especially the adverse effects caused by
protein degradation in healthy tissues when PROTACs are
administered orally or intravenously (239).
Indirect inhibitors of STAT3 target the upstream or
downstream components of the STAT3 signaling pathway, for which
hundreds of compounds have been identified, mainly JAK (2,6)
and IL-6/IL-6R (2,6,240) inhibitors.
The JAK family consists of four non-receptor
tyrosine protein kinases (JAK1, JAK2, JAK3 and TYK2). JAKs
incorporate signals from various cytokines and growth factor
receptors and principally activate STATs. Targeting JAKs to
interfere with the signaling of the JAK/STAT pathway has been
successful, which is best illustrated by the fact that eight
pan-JAKs or selective JAK inhibitors have been approved to treat
rheumatoid arthritis (RA), atopic dermatitis and myeloproliferative
neoplasm (MPN) (241). These
inhibitors are tofacitinib, baricitinib, delgocitinib, peficitinib,
ruxolitinib, upadacitinib, filgotinib and abrocitinib. Several JAK
inhibitors, including the aforementioned eight inhibitors, are in
clinical trials to evaluate their efficacy and safety in leukemia
(242) and solid tumors.
However, no JAK inhibitors are currently approved to treat these
diseases. A clinical investigation showed an inadequate clinical
response and serious adverse events following the treatment of
solid tumors with the JAK inhibitor, AZD1480 (243).
Another strategy to suppress STAT3 signaling is
targeting IL-6 and its receptor, IL-6R. Indeed, there have been
several such antibody drugs used in the clinic including
siltuximab, tocilizumab and sarilumab. Siltuximab, a chimeric
antibody against IL-6, is currently used in the clinic to treat
multicentric Castleman disease, which was approved in 2014
(244). Tocilizumab, a humanized
anti-IL-6R inhibitor, has already successfully entered the clinic
to treat RA. Sarilumab, an anti-IL-6R antibody, was also approved
in 2017 for the treatment of RA. In addition, these inhibitors were
widely evaluated in clinical trials for solid and hematological
malignancies. However, anti-IL6 or anti-IL-6R antibodies do not
demonstrated clinical efficacy in various types of cancer (245). For instance siltuximab
monotherapy has not shown significant activity in pretreated
castration-resistant prostate cancer (CRPC) (246), NSCLC (247), HNSCC (247), CRC (247) or multiple myeloma (245). Additionally, siltuximab plus
mitoxantrone/prednisone (M/P) treatment did not show a more
superior effect than M/P treatment alone in patients with
metastatic CRPC (248). A number
of clinical trials using tocilizumab to treat patients with cancer
are ongoing, most of which are combination therapies; however, no
results have been published. Sarilumab is also currently in the
preclinical stages.
There are several possible explanations for this
lack of efficacy of IL-6/IL-6R inhibitors. First, the large number
of tumor-promoting cytokines in the tumor microenvironment may
limit efficacy of therapeutically targeting a single one. Second,
cancer plasticity and heterogeneity could enable tumor cell
resistance to IL-6 and IL-6R therapies.
In conclusion, the STAT3 pathway is a central
signaling node that regulates a plethora of cancer hallmarks. The
hyperactivation of STAT3 facilitates cancer progression, drug
resistance, metastasis and EMT. Various newly identified mechanisms
and regulatory proteins, miRNAs, lncRNAs and circRNAs have been
shown to be integral members of the STAT3/EMT axis. A great effort
has already been made to develop inhibitors that suppress the
IL-6/STAT3 axis via targeting IL-6, IL-6R, JAKs or STAT3 itself
(2,3), some of which have been approved for
the treatment of inflammatory diseases or MPN. There have also been
several preclinical studies that demonstrated that some compounds
suppress EMT through the STAT3 pathway (66,67). However, no inhibitors have yet
been approved for solid tumors. In contrast to monotherapy,
combination therapies involving STAT3 pathway inhibitors with
chemotherapy, radiotherapy and immune checkpoint inhibitors could
be considered to enhance efficacy and reduce side effects.
Furthermore, if we consider that tens of thousands of non-coding
RNAs have been identified by high-throughput RNA sequencing, but
only a small percentage of these have been functionally
characterized (159), we may
anticipate that the number of known non-coding RNAs involved in the
STAT3-EMT axis will increase in the future. This rapidly expanding
area will provide increasing therapeutic targets for STAT3
signaling suppression. For instance, miR34, a molecule downstream
of STAT3 that also acts as a regulator of STAT3 signaling, has also
been evaluated for its potential as a cancer therapeutic agent in a
clinical Phase I study (NCT01829971) (249). In addition, biomarkers to
predict therapy responders are urgently needed. Technological
advances such as single cell profiling, may increase the
understanding of the response of cancer to STAT3 inhibitors at the
single cell level and provide opportunities to stratify
patients.
Not applicable.
GZ, SH and SL performed the literature review and
wrote the manuscript. YW and WC revised the manuscript. All authors
have read and approved the final version of the manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
Not applicable.
The present study was supported by the Shandong Provincial
Natural Science Foundation (grant no. ZR2020MH212).
|
1
|
Philips RL, Wang Y, Cheon H, Kanno Y,
Gadina M, Sartorelli V, Horvath CM, Darnell JE Jr, Stark GR and
O'Shea JJ: The JAK-STAT pathway at 30: Much learned, much more to
do. Cell. 185:3857–3876. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Johnson DE, O'Keefe RA and Grandis JR:
Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev
Clin Oncol. 15:234–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huynh J, Chand A, Gough D and Ernst M:
Therapeutically exploiting STAT3 activity in cancer-using tissue
repair as a road map. Nat Rev Cancer. 19:82–96. 2019. View Article : Google Scholar
|
|
4
|
Cimica V, Chen HC, Iyer JK and Reich NC:
Dynamics of the STAT3 transcription factor: Nuclear import
dependent on Ran and importin-β1. PLoS One. 6:e201882011.
View Article : Google Scholar
|
|
5
|
Garbers C, Aparicio-Siegmund S and
Rose-John S: The IL-6/gp130/STAT3 signaling axis: Recent advances
towards specific inhibition. Curr Opin Immunol. 34:75–82. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Buchert M, Burns CJ and Ernst M: Targeting
JAK kinase in solid tumors: Emerging opportunities and challenges.
Oncogene. 35:939–951. 2016. View Article : Google Scholar
|
|
7
|
Yu H, Lee H, Herrmann A, Buettner R and
Jove R: Revisiting STAT3 signalling in cancer: New and unexpected
biological functions. Nat Rev Cancer. 14:736–746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Diallo M and Herrera F: The role of
understudied post-translational modifications for the behavior and
function of signal transducer and activator of transcription 3.
FEBS J. 289:6235–6255. 2022. View Article : Google Scholar
|
|
9
|
Brabletz S, Schuhwerk H, Brabletz T and
Stemmler MP: Dynamic EMT: A multi-tool for tumor progression. EMBO
J. 40:e1086472021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hua W, Ten Dijke P, Kostidis S, Giera M
and Hornsveld M: TGFβ-induced metabolic reprogramming during
epithelial-to-mesenchymal transition in cancer. Cell Mol Life Sci.
77:2103–2123. 2020. View Article : Google Scholar
|
|
11
|
Lai X, Li Q, Wu F, Lin J, Chen J, Zheng H
and Guo L: Epithelial-mesenchymal transition and metabolic
switching in cancer: Lessons from somatic cell reprogramming. Front
Cell Dev Biol. 8:7602020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Terry S, Savagner P, Ortiz-Cuaran S,
Mahjoubi L, Saintigny P, Thiery JP and Chouaib S: New insights into
the role of EMT in tumor immune escape. Mol Oncol. 11:824–846.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stemmler MP, Eccles RL, Brabletz S and
Brabletz T: Non-redundant functions of EMT transcription factors.
Nat Cell Biol. 21:102–112. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guo H, Zhuang K, Ding N, Hua R, Tang H, Wu
Y, Yuan Z, Li T and He S: High-fat diet induced cyclophilin B
enhances STAT3/lncRNA-PVT1 feedforward loop and promotes growth and
metastasis in colorectal cancer. Cell Death Dis. 13:8832022.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Akhmetkaliyev A, Alibrahim N, Shafiee D
and Tulchinsky E: EMT/MET plasticity in cancer and Go-or-Grow
decisions in quiescence: The two sides of the same coin? Mol
Cancer. 22:902023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lambert AW and Weinberg RA: Linking EMT
programmes to normal and neoplastic epithelial stem cells. Nat Rev
Cancer. 21:325–338. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nieto MA, Huang RYJ, Jackson RA and Thiery
JP: EMT: 2016. Cell. 166:21–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zheng H and Kang Y: Multilayer control of
the EMT master regulators. Oncogene. 33:1755–1763. 2014. View Article : Google Scholar
|
|
19
|
Puisieux A, Brabletz T and Caramel J:
Oncogenic roles of EMT-inducing transcription factors. Nat Cell
Biol. 16:488–494. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Thiery JP, Acloque H, Huang RYJ and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Vesuna F, van Diest P, Chen JH and Raman
V: Twist is a transcriptional repressor of E-cadherin gene
expression in breast cancer. Biochem Biophys Res Commun.
367:235–241. 2008. View Article : Google Scholar
|
|
22
|
Yang MH, Hsu DS, Wang HW, Wang HJ, Lan HY,
Yang WH, Huang CH, Kao SY, Tzeng CH, Tai SK, et al: Bmi1 is
essential in Twist1-induced epithelial-mesenchymal transition. Nat
Cell Biol. 12:982–992. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: At the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shamir ER, Pappalardo E, Jorgens DM,
Coutinho K, Tsai WT, Aziz K, Auer M, Tran PT, Bader JS and Ewald
AJ: Twist1-induced dissemination preserves epithelial identity and
requires E-cadherin. J Cell Biol. 204:839–856. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Onder TT, Gupta PB, Mani SA, Yang J,
Lander ES and Weinberg RA: Loss of E-cadherin promotes metastasis
via multiple downstream transcriptional pathways. Cancer Res.
68:3645–3654. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sullivan NJ, Sasser AK, Axel AE, Vesuna F,
Raman V, Ramirez N, Oberyszyn TM and Hall BM: Interleukin-6 induces
an epithelial-mesenchymal transition phenotype in human breast
cancer cells. Oncogene. 28:2940–2947. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yadav A, Kumar B, Datta J, Teknos TN and
Kumar P: IL-6 promotes head and neck tumor metastasis by inducing
epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling
pathway. Mol Cancer Res. 9:1658–1667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rojas A, Liu G, Coleman I, Nelson PS,
Zhang M, Dash R, Fisher PB, Plymate SR and Wu JD: IL-6 promotes
prostate tumorigenesis and progression through autocrine
cross-activation of IGF-IR. Oncogene. 30:2345–2355. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Miao JW, Liu LJ and Huang J:
Interleukin-6-induced epithelial-mesenchymal transition through
signal transducer and activator of transcription 3 in human
cervical carcinoma. Int J Oncol. 45:165–176. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shintani Y, Fujiwara A, Kimura T, Kawamura
T, Funaki S, Minami M and Okumura M: IL-6 secreted from
cancer-associated fibroblasts mediates chemoresistance in NSCLC by
increasing epithelial-mesenchymal transition signaling. J Thorac
Oncol. 11:1482–1492. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Baulida J, Diaz VM and Herreros AG:
Snail1: A transcriptional factor controlled at multiple levels. J
Clin Med. 8:7572019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yamashita S, Miyagi C, Fukada T, Kagara N,
Che YS and Hirano T: Zinc transporter LIVI controls
epithelial-mesenchymal transition in zebrafish gastrula organizer.
Nature. 429:298–302. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hogstrand C, Kille P, Ackland ML, Hiscox S
and Taylor KM: A mechanism for epithelial-mesenchymal transition
and anoikis resistance in breast cancer triggered by zinc channel
ZIP6 and STAT3 (signal transducer and activator of transcription
3). Biochem J. 455:229–237. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang C, Yang G, Jiang T, Zhu G, Li H and
Qiu Z: The effects and mechanisms of blockage of STAT3 signaling
pathway on IL-6 inducing EMT in human pancreatic cancer cells in
vitro. Neoplasma. 58:396–405. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rokavec M, Öner MG, Li H, Jackstadt R,
Jiang L, Lodygin D, Kaller M, Horst D, Ziegler PK, Schwitalla S, et
al: IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated
colorectal cancer invasion and metastasis. J Clin Invest.
124:1853–1867. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Peinado H, Quintanilla M and Cano A:
Transforming growth factor beta-1 induces snail transcription
factor in epithelial cell lines: Mechanisms for epithelial
mesenchymal transitions. J Biol Chem. 278:21113–21123. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Saitoh M, Endo K, Furuya S, Minami M,
Fukasawa A, Imamura T and Miyazawa K: STAT3 integrates cooperative
Ras and TGF-β signals that induce Snail expression. Oncogene.
35:1049–1057. 2016. View Article : Google Scholar
|
|
38
|
Kim M and Lim J, Yang Y, Lee M and Lim J:
N-myc downstream-regulated gene 2 (NDRG2) suppresses the
epithelial-mesenchymal transition (EMT) in breast cancer cells via
STAT3/Snail signaling. Cancer Lett. 354:33–42. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Burton LJ, Smith BA, Smith BN, Loyd Q,
Nagappan P, McKeithen D, Wilder CL, Platt MO, Hudson T and
Odero-Marah VA: Muscadine grape skin extract can antagonize
Snail-cathepsin L-mediated invasion, migration and
osteoclastogenesis in prostate and breast cancer cells.
Carcinogenesis. 36:1019–1027. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou JJ, Meng Z, He XY, Cheng D, Ye HL,
Deng XG and Chen RF: Hepatitis C virus core protein increases Snail
expression and induces epithelial-mesenchymal transition through
the signal transducer and activator of transcription 3 pathway in
hepatoma cells. Hepatol Res. 47:574–583. 2017. View Article : Google Scholar
|
|
41
|
Liu WH, Chen MT, Wang ML, Lee YY, Chiou
GY, Chien CS, Huang PI, Chen YW, Huang MC, Chiou SH, et al:
Cisplatin-selected resistance is associated with increased motility
and stem-like properties via activation of STAT3/Snail axis in
atypical teratoid/rhabdoid tumor cells. Oncotarget. 6:1750–1768.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liang H, Chen G, Li J and Yang F: Snail
expression contributes to temozolomide resistance in glioblastoma.
Am J Transl Res. 11:4277–4289. 2019.PubMed/NCBI
|
|
43
|
Dai X, Ahn KS, Wang LZ, Kim C,
Deivasigamni A, Arfuso F, Um JY, Kumar AP, Chang YC, Kumar D, et
al: Ascochlorin enhances the sensitivity of doxorubicin leading to
the reversal of epithelial-to-mesenchymal transition in
hepatocellular carcinoma. Mol Cancer Ther. 15:2966–2976. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xie Q, Zhu Z, He Y, Zhang Z, Zhang Y, Wang
Y, Luo J, Peng T, Cheng F, Gao J, et al: A lactate-induced
Snail/STAT3 pathway drives GPR81 expression in lung cancer cells.
Biochim Biophys Acta Mol Basis Dis. 1866:1655762020. View Article : Google Scholar
|
|
45
|
Kim E, Kim M, Woo DH, Shin Y, Shin J,
Chang N, Oh YT, Kim H, Rheey J, Nakano I, et al: Phosphorylation of
EZH2 activates STAT3 signaling via STAT3 methylation and promotes
tumorigenicity of glioblastoma stem-like cells. Cancer Cell.
23:839–852. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhao Y, Hu Z, Li J and Hu T: EZH2
exacerbates breast cancer by methylating and activating STAT3
directly. J Cancer. 12:5220–5230. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yuan K, Lei Y, Chen HN, Chen Y, Zhang T,
Li K, Xie N, Wang K, Feng X, Pu Q, et al: HBV-induced ROS
accumulation promotes hepatocarcinogenesis through Snail-mediated
epigenetic silencing of SOCS3. Cell Death Differ. 23:616–627. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim JY, Kim HJ, Jung CW, Lee TS, Kim EH
and Park MJ: CXCR4 uses STAT3-mediated slug expression to maintain
radioresistance of non-small cell lung cancer cells: Emerges as a
potential prognostic biomarker for lung cancer. Cell Death Dis.
12:482021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wu S, Ye S and Lin X, Chen Y, Zhang Y,
Jing Z, Liu W, Chen W and Lin X and Lin X: Small hepatitis B virus
surface antigen promotes malignant progression of hepatocellular
carcinoma via endoplasmic reticulum stress-induced FGF19/JAK2/STAT3
signaling. Cancer Lett. 499:175–187. 2021. View Article : Google Scholar
|
|
50
|
Chesnelong C, Hao X, Cseh O, Wang AY,
Luchman HA and Weiss S: SLUG directs the precursor state of human
brain tumor stem cells. Cancers (Basel). 11:16352019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lin JC, Tsai JT, Chao TY, Ma HI and Liu
WH: The STAT3/Slug axis enhances radiation-induced tumor invasion
and cancer stem-like properties in radioresistant glioblastoma.
Cancers (Basel). 10:5122018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhou DD, Liu XF, Lu CW, Pant OP and Liu
XD: Long non-coding RNA PVT1: Emerging biomarker in digestive
system cancer. Cell Prolif. 50:e123982017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Dong L, Wang H, Gao Y, Wang S and Wang W:
Long non-coding RNA PVT1 promotes the proliferation, migration and
EMT process of ovarian cancer cells by regulating CTGF. Oncol Lett.
25:712023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhao J, Wu J, Qin Y, Zhang W, Huang G and
Qin L: LncRNA PVT1 induces aggressive vasculogenic mimicry
formation through activating the STAT3/Slug axis and
epithelial-to-mesenchymal transition in gastric cancer. Cell Oncol
(Dordr). 43:863–876. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Barnes RM and Firulli AB: A twist of
insight-the role of Twist-family bHLH factors in development. Int J
Dev Biol. 53:909–924. 2009. View Article : Google Scholar
|
|
56
|
Qin Q, Xu Y, He T, Qin C and Xu J: Normal
and disease-related biological functions of Twist1 and underlying
molecular mechanisms. Cell Res. 22:90–106. 2012. View Article : Google Scholar :
|
|
57
|
Ling X and Arlinghaus RB: Knockdown of
STAT3 expression by RNA interference inhibits the induction of
breast tumors in immunocompetent mice. Cancer Res. 65:2532–2536.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola
D, Mansour M, Xu LM, Costanzo C, Cheng JQ and Wang LH: Twist is
transcriptionally induced by activation of STAT3 and mediates STAT3
oncogenic function. J Biol Chem. 283:14665–14673. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei
Y, Abbruzzese JL, Hortobagyi GN and Hung MC: Epidermal growth
factor receptor cooperates with signal transducer and activator of
transcription 3 to induce epithelial-mesenchymal transition in
cancer cells via up-regulation of TWIST gene expression. Cancer
Res. 67:9066–9076. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hsu KW, Hsieh RH, Huang KH, Fen-Yau Li A,
Chi CW, Wang TY, Tseng MJ, Wu KJ and Yeh TS: Activation of the
Notch1/STAT3/Twist signaling axis promotes gastric cancer
progression. Carcinogenesis. 33:1459–1467. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kim MS, Lee HS, Kim YJ, Lee DY, Kang SG
and Jin W: MEST induces Twist-1-mediated EMT through STAT3
activation in breast cancers. Cell Death Differ. 26:2594–2606.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhang CH, Xu GL, Jia WD, Li JS, Ma JL, Ren
WH, Ge YS, Yu JH, Liu WB and Wang W: Activation of STAT3 signal
pathway correlates with twist and E-cadherin expression in
hepatocellular carcinoma and their clinical significance. J Surg
Res. 174:120–129. 2012. View Article : Google Scholar
|
|
63
|
Cheng L, Zhou MY, Gu YJ, Chen L and Wang
Y: ZEB1: New advances in fibrosis and cancer. Mol Cell Biochem.
476:1643–1650. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Caramel J, Ligier M and Puisieux A:
Pleiotropic roles for ZEB1 in cancer. Cancer Res. 78:30–35. 2018.
View Article : Google Scholar
|
|
65
|
Lu R, Zhang YG and Sun J: STAT3 activation
in infection and infection-associated cancer. Mol Cell Endocrinol.
451:80–87. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Xiong H, Hong J, Du W, Lin YW, Ren LL,
Wang YC, Su WY, Wang JL, Cui Y, Wang ZH and Fang JY: Roles of STAT3
and ZEB1 proteins in E-cadherin down-regulation and human
colorectal cancer epithelial-mesenchymal transition. J Biol Chem.
287:5819–5832. 2012. View Article : Google Scholar :
|
|
67
|
Avtanski DB, Nagalingam A, Bonner MY,
Arbiser JL, Saxena NK and Sharma D: Honokiol inhibits
epithelial-mesenchymal transition in breast cancer cells by
targeting signal transducer and activator of transcription
3/Zeb1/E-cadherin axis. Mol Oncol. 8:565–580. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Liu Z, Ma L, Sun Y, Yu W and Wang X:
Targeting STAT3 signaling overcomes gefitinib resistance in
non-small cell lung cancer. Cell Death Dis. 12:5612021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Huang YH, Chen HK, Hsu YF, Chen HC, Chuang
CH, Huang SW and Hsu MJ: Src-FAK signaling mediates interleukin
6-induced HCT116 colorectal cancer epithelial-mesenchymal
transition. Int J Mol Sci. 24:66502023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Shi Q and Chen YG: Interplay between
TGF-beta signaling and receptor tyrosine kinases in tumor
development. Sci China Life Sci. 60:1133–1141. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Derynck R and Budi EH: Specificity,
versatility, and control of TGF-β family signaling. Sci Signal.
12:eaav51832019. View Article : Google Scholar
|
|
72
|
Katsuno Y and Derynck R: Epithelial
plasticity, epithelial-mesenchymal transition, and the TGF-β
family. Dev Cell. 56:726–746. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Itoh Y, Saitoh M and Miyazawa K:
Smad3-STAT3 crosstalk in pathophysiological contexts. Acta Biochim
Biophys Sin (Shanghai). 50:82–90. 2018. View Article : Google Scholar
|
|
74
|
Sun CY, Nie J, Huang JP, Zheng GJ and Feng
B: Targeting STAT3 inhibition to reverse cisplatin resistance.
Biomed Pharmacother. 117:1091352019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Bowman T, Broome MA, Sinibaldi D, Wharton
W, Pledger WJ, Sedivy JM, Irby R, Yeatman T, Courtneidge SA and
Jove R: Stat3-mediated Myc expression is required for Src
transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci
USA. 98:7319–7324. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Yu Y and Feng XH: TGF-β signaling in cell
fate control and cancer. Curr Opin Cell Biol. 61:56–63. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Horiguchi K, Shirakihara T, Nakano A,
Imamura T, Miyazono K and Saitoh M: Role of Ras signaling in the
induction of snail by transforming growth factor-beta. J Biol Chem.
284:245–253. 2009. View Article : Google Scholar
|
|
78
|
Long J, Wang G, Matsuura I, He D and Liu
F: Activation of Smad transcriptional activity by protein inhibitor
of activated STAT3 (PIAS3). Proc Natl Acad Sci USA. 101:99–104.
2004. View Article : Google Scholar :
|
|
79
|
Calon A, Espinet E, Palomo-Ponce S,
Tauriello DV, Iglesias M, Céspedes MV, Sevillano M, Nadal C, Jung
P, Zhang XH, et al: Dependency of colorectal cancer on a
TGF-β-driven program in stromal cells for metastasis initiation.
Cancer Cell. 22:571–584. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Abulaiti A, Shintani Y, Funaki S, Nakagiri
T, Inoue M, Sawabata N, Minami M and Okumura M: Interaction between
non-small-cell lung cancer cells and fibroblasts via enhancement of
TGF-β signaling by IL-6. Lung Cancer. 82:204–213. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Liu RY, Zeng Y, Lei Z, Wang L, Yang H, Liu
Z, Zhao J and Zhang HT: JAK/STAT3 signaling is required for
TGF-β-induced epithelial-mesenchymal transition in lung cancer
cells. Int J Oncol. 44:1643–1651. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wang B, Liu T, Wu JC, Luo SZ, Chen R, Lu
LG and Xu MY: STAT3 aggravates TGF-β1-induced hepatic
epithelial-to-mesenchymal transition and migration. Biomed
Pharmacother. 98:214–221. 2018. View Article : Google Scholar
|
|
83
|
Morris R, Butler L, Perkins A, Kershaw NJ
and Babon JJ: The Role of LNK (SH2B3) in the regulation of JAK-STAT
signalling in haematopoiesis. Pharmaceuticals (Basel). 15:242021.
View Article : Google Scholar
|
|
84
|
Ding LW, Sun QY, Lin DC, Chien W, Hattori
N, Dong XM, Gery S, Garg M, Doan NB, Said JW, et al: LNK (SH2B3):
Paradoxical effects in ovarian cancer. Oncogene. 34:1463–1474.
2015. View Article : Google Scholar
|
|
85
|
Lv J, Yu W, Zhang Y, Cao X, Han L, Hu H
and Wang C: LNK promotes the growth and metastasis of triple
negative breast cancer via activating JAK/STAT3 and ERK1/2 pathway.
Cancer Cell Int. 20:1242020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zhong ZM, Chen X, Qi X, Wang XM, Li CY,
Qin RJ, Wang SQ, Liang J, Zeng MS and Sun CZ: Adaptor protein LNK
promotes anaplastic thyroid carcinoma cell growth via 14-3-3 ε/γ
binding. Cancer Cell Int. 20:112020. View Article : Google Scholar
|
|
87
|
Pan J, Peng R, Cheng N, Chen F and Gao B:
LNK protein: Low expression in human colorectal carcinoma and
relationship with tumor invasion. Biomed Pharmacother.
121:1094672020. View Article : Google Scholar
|
|
88
|
Wang LN, Zhang ZT, Wang L, Wei HX, Zhang
T, Zhang LM, Lin H, Zhang H and Wang SQ: TGF-β1/SH2B3 axis
regulates anoikis resistance and EMT of lung cancer cells by
modulating JAK2/STAT3 and SHP2/Grb2 signaling pathways. Cell Death
Dis. 13:4722022. View Article : Google Scholar
|
|
89
|
Dragomir MP, Knutsen E and Calin GA:
Classical and noncanonical functions of miRNAs in cancers. Trends
Genet. 38:379–394. 2022. View Article : Google Scholar
|
|
90
|
Gregory PA, Bracken CP, Bert AG and
Goodall GJ: MicroRNAs as regulators of epithelial-mesenchymal
transition. Cell Cycle. 7:3112–3118. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Hao J, Zhang Y, Deng M, Ye R, Zhao S, Wang
Y, Li J and Zhao Z: MicroRNA control of epithelial-mesenchymal
transition in cancer stem cells. Int J Cancer. 135:1019–1027. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zhang L, Liao Y and Tang L: MicroRNA-34
family: A potential tumor suppressor and therapeutic candidate in
cancer. J Exp Clin Cancer Res. 38:532019. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Li WJ, Wang Y, Liu R, Kasinski AL, Shen H,
Slack FJ and Tang DG: MicroRNA-34a: Potent tumor suppressor, cancer
stem cell inhibitor, and potential anticancer therapeutic. Front
Cell Dev Biol. 9:6405872021. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Siemens H, Jackstadt R, Hünten S, Kaller
M, Menssen A, Götz U and Hermeking H: miR-34 and SNAIL form a
double-negative feedback loop to regulate epithelial-mesenchymal
transitions. Cell Cycle. 10:4256–4271. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Imani S, Wei C, Cheng J, Khan MA, Fu S,
Yang L, Tania M, Zhang X, Xiao X, Zhang X and Fu J: MicroRNA-34a
targets epithelial to mesenchymal transition-inducing transcription
factors (EMT-TFs) and inhibits breast cancer cell migration and
invasion. Oncotarget. 8:21362–21379. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Huang G, Du MY, Zhu H, Zhang N, Lu ZW,
Qian LX, Zhang W, Tian X, He X and Yin L: MiRNA-34a reversed
TGF-β-induced epithelial-mesenchymal transition via suppression of
SMAD4 in NPC cells. Biomed Pharmacother. 106:217–224. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ma ZL, Hou PP, Li YL, Wang DT, Yuan TW,
Wei JL, Zhao BT, Lou JT, Zhao XT, Jin Y and Jin YX: MicroRNA-34a
inhibits the proliferation and promotes the apoptosis of non-small
cell lung cancer H1299 cell line by targeting TGFβR2. Tumour Biol.
36:2481–2490. 2015. View Article : Google Scholar
|
|
98
|
Jiang L and Hermeking H: miR-34a and
miR-34b/c suppress intestinal tumorigenesis. Cancer Res.
77:2746–2758. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Li B, Guo X, Li N, Chen Q, Shen J, Huang
X, Huang G and Wang F: WNT1, a target of miR-34a, promotes cervical
squamous cell carcinoma proliferation and invasion by induction of
an E-P cadherin switch via the WNT/β-catenin pathway. Cell Oncol
(Dordr). 43:489–503. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Hashimi ST, Fulcher JA, Chang MH, Gov L,
Wang S and Lee B: MicroRNA profiling identifies miR-34a and miR-21
and their target genes JAG1 and WNT1 in the coordinate regulation
of dendritic cell differentiation. Blood. 114:404–414. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Si W, Li Y, Shao H, Hu R, Wang W, Zhang K
and Yang Q: MiR-34a inhibits breast cancer proliferation and
progression by targeting Wnt1 in Wnt/β-catenin signaling pathway.
Am J Med Sci. 352:191–199. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Chen WY, Liu SY, Chang YS, Yin JJ, Yeh HL,
Mouhieddine TH, Hadadeh O, Abou-Kheir W and Liu YN: MicroRNA-34a
regulates WNT/TCF7 signaling and inhibits bone metastasis in
Ras-activated prostate cancer. Oncotarget. 6:441–457. 2015.
View Article : Google Scholar :
|
|
103
|
Wang X, Zhao Y, Lu Q, Fei X, Lu C, Li C
and Chen H: MiR-34a-5p inhibits proliferation, migration, invasion
and epithelial-mesenchymal transition in esophageal squamous cell
carcinoma by targeting LEF1 and inactivation of the Hippo-YAP1/TAZ
signaling pathway. J Cancer. 11:3072–3081. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Kang L, Mao J, Tao Y, Song B, Ma W, Lu Y,
Zhao L, Li J, Yang B and Li L: MicroRNA-34a suppresses the breast
cancer stem cell-like characteristics by downregulating Notch1
pathway. Cancer Sci. 106:700–708. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Fan F, Zhuang J, Zhou P, Liu X and Luo Y:
MicroRNA-34a promotes mitochondrial dysfunction-induced apoptosis
in human lens epithelial cells by targeting Notch2. Oncotarget.
8:110209–110220. 2017. View Article : Google Scholar
|
|
106
|
Mudduluru G, Ceppi P, Kumarswamy R,
Scagliotti GV, Papotti M and Allgayer H: Regulation of Axl receptor
tyrosine kinase expression by miR-34a and miR-199a/b in solid
cancer. Oncogene. 30:2888–2899. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Marcucci F, Stassi G and De Maria R:
Epithelial-mesenchymal transition: A new target in anticancer drug
discovery. Nat Rev Drug Discov. 15:311–325. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Zhang G, Wang M, Zhao H and Cui W:
Function of Axl receptor tyrosine kinase in non-small cell lung
cancer. Oncol Lett. 15:2726–2734. 2018.PubMed/NCBI
|
|
109
|
Avtanski DB, Nagalingam A, Kuppusamy P,
Bonner MY, Arbiser JL, Saxena NK and Sharma D: Honokiol abrogates
leptin-induced tumor progression by inhibiting Wnt1-MTA1-β-catenin
signaling axis in a microRNA-34a dependent manner. Oncotarget.
6:16396–16410. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Cavallari I, Ciccarese F, Sharova E, Urso
L, Raimondi V, Silic-Benussi M, D'Agostino DM and Ciminale V: The
miR-200 family of microRNAs: Fine tuners of epithelial-mesenchymal
transition and circulating cancer biomarkers. Cancers (Basel).
13:58742021. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Peter ME: Let-7 and miR-200 microRNAs:
Guardians against pluripotency and cancer progression. Cell Cycle.
8:843–852. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Feng X, Wang Z, Fillmore R and Xi Y:
MiR-200, a new star miRNA in human cancer. Cancer Lett.
344:166–173. 2014. View Article : Google Scholar :
|
|
113
|
Georgakopoulos-Soares I, Chartoumpekis DV,
Kyriazopoulou V and Zaravinos A: EMT factors and metabolic pathways
in cancer. Front Oncol. 10:4992020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Park SM, Gaur AB, Lengyel E and Peter ME:
The miR-200 family determines the epithelial phenotype of cancer
cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes
Dev. 22:894–907. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Korpal M, Lee ES, Hu G and Kang Y: The
miR-200 family inhibits epithelial-mesenchymal transition and
cancer cell migration by direct targeting of E-cadherin
transcriptional repressors ZEB1 and ZEB2. J Biol Chem.
283:14910–14914. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Gregory PA, Bert AG, Paterson EL, Barry
SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y and Goodall GJ:
The miR-200 family and miR-205 regulate epithelial to mesenchymal
transition by targeting ZEB1 and SIP1. Nat Cell Biol. 10:593–601.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Bracken CP, Gregory PA, Kolesnikoff N,
Bert AG, Wang J, Shannon MF and Goodall GJ: A double-negative
feedback loop between ZEB1-SIP1 and the microRNA-200 family
regulates epithelial-mesenchymal transition. Cancer Res.
68:7846–7854. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Burk U, Schubert J, Wellner U, Schmalhofer
O, Vincan E, Spaderna S and Brabletz T: A reciprocal repression
between ZEB1 and members of the miR-200 family promotes EMT and
invasion in cancer cells. EMBO Rep. 9:582–589. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Guo L, Chen C, Shi M, Wang F, Chen X, Diao
D, Hu M, Yu M, Qian L and Guo N: Stat3-coordinated
Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain
oncostatin M-driven epithelial-mesenchymal transition. Oncogene.
32:5272–5282. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Wang Y, Guo W, Li Z, Wu Y, Jing C, Ren Y,
Zhao M, Kong L, Zhang C, Dong J, et al: Role of the EZH2/miR-200
axis in STAT3-mediated OSCC invasion. Int J Oncol. 52:1149–1164.
2018.PubMed/NCBI
|
|
121
|
Pan YM, Wang CG, Zhu M, Xing R, Cui JT, Li
WM, Yu DD, Wang SB, Zhu W, Ye YJ, et al: STAT3 signaling drives
EZH2 transcriptional activation and mediates poor prognosis in
gastric cancer. Mol Cancer. 15:792016. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Martínez-Fernández M, Dueñas M, Feber A,
Segovia C, García-Escudero R, Rubio C, López-Calderón FF,
Díaz-García C, Villacampa F, Duarte J, et al: A Polycomb-mir200
loop regulates clinical outcome in bladder cancer. Oncotarget.
6:42258–42275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Yang X, Chen Y and Chen L: The versatile
role of microRNA-30a in human cancer. Cell Physiol Biochem.
41:1616–1632. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Zhang J, Zhang H, Liu J, Tu X, Zang Y, Zhu
J, Chen J, Dong L and Zhang J: miR-30 inhibits TGF-β1-induced
epithelial-to-mesenchymal transition in hepatocyte by targeting
Snail1. Biochem Biophys Res Commun. 417:1100–1105. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Wang Y, Wu C, Zhang C, Li Z, Zhu T, Chen
J, Ren Y, Wang X, Zhang L and Zhou X: TGF-β-induced STAT3
overexpression promotes human head and neck squamous cell carcinoma
invasion and metastasis through malat1/miR-30a interactions. Cancer
Lett. 436:52–62. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Pfeffer SR, Yang CH and Pfeffer LM: The
role of miR-21 in cancer. Drug Dev Res. 76:270–277. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Singh A, Singh AK, Giri R, Kumar D, Sharma
R, Valis M, Kuca K and Garg N: The role of microRNA-21 in the onset
and progression of cancer. Future Med Chem. 13:1885–1906. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Yan LX, Liu YH, Xiang JW, Wu QN, Xu LB,
Luo XL, Zhu XL, Liu C, Xu FP, Luo DL, et al: PIK3R1 targeting by
miR-21 suppresses tumor cell migration and invasion by reducing
PI3K/AKT signaling and reversing EMT, and predicts clinical outcome
of breast cancer. Int J Oncol. 48:471–484. 2016. View Article : Google Scholar :
|
|
129
|
Tang Y, Zhao Y, Ran J and Wang Y:
MicroRNA-21 promotes cell metastasis in cervical cancer through
modulating epithelial-mesenchymal transition. Oncol Lett.
19:3289–3295. 2020.PubMed/NCBI
|
|
130
|
Su C, Cheng X, Li Y, Han Y, Song X, Yu D,
Cao X and Liu Z: MiR-21 improves invasion and migration of
drug-resistant lung adenocarcinoma cancer cell and transformation
of EMT through targeting HBP1. Cancer Med. 7:2485–2503. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Bian Z, Ji W, Xu B, Huo Z, Huang H, Huang
J, Jiao J, Shao J and Zhang X: Noncoding RNAs involved in the STAT3
pathway in glioma. Cancer Cell Int. 21:4452021. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Tse J, Pierce T, Carli ALE, Alorro MG,
Thiem S, Marcusson EG, Ernst M and Buchert M: Onco-miR-21 promotes
Stat3-dependent gastric cancer progression. Cancers (Basel).
14:2642022. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Yue X, Zhao Y, Zhang C, Li J, Liu Z, Liu J
and Hu W: Leukemia inhibitory factor promotes EMT through
STAT3-dependent miR-21 induction. Oncotarget. 7:3777–3790. 2016.
View Article : Google Scholar :
|
|
134
|
Lu YF, Zhang L, Waye MM, Fu WM and Zhang
JF: MiR-218 mediates tumorigenesis and metastasis: Perspectives and
implications. Exp Cell Res. 334:173–182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Li YJ, Zhang W, Xia H, Zhang BS, Chen P,
Zhao YL and Li J: miR-218 suppresses epithelial-to-mesenchymal
transition by targeting Robo1 and Ecop in lung adenocarcinoma
cells. Future Oncol. 13:2571–2582. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Shi ZM, Wang L, Shen H, Jiang CF, Ge X, Li
DM, Wen YY, Sun HR, Pan MH, Li W, et al: Downregulation of miR-218
contributes to epithelial-mesenchymal transition and tumor
metastasis in lung cancer by targeting Slug/ZEB2 signaling.
Oncogene. 36:2577–2588. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Jiang Z, Song Q, Zeng R, Li J, Li J, Lin
X, Chen X, Zhang J and Zheng Y: MicroRNA-218 inhibits EMT,
migration and invasion by targeting SFMBT1 and DCUN1D1 in cervical
cancer. Oncotarget. 7:45622–45636. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Zhang L, Li J, Wang Q, Meng G, Lv X, Zhou
H, Li W and Zhang J: The relationship between microRNAs and the
STAT3-related signaling pathway in cancer. Tumour Biol.
39:10104283177198692017. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Li Z, Qian R, Zhang J and Shi X:
MiR-218-5p targets LHFPL3 to regulate proliferation, migration, and
epithelial-mesenchymal transitions of human glioma cells. Biosci
Rep. 39:BSR201808792019. View Article : Google Scholar
|
|
140
|
Lun W, Wu X, Deng Q and Zhi F: MiR-218
regulates epithelial-mesenchymal transition and angiogenesis in
colorectal cancer via targeting CTGF. Cancer Cell Int. 18:832018.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Wang G, Fu Y, Liu G, Ye Y and Zhang X:
miR-218 inhibits proliferation, migration, and EMT of gastric
cancer cells by targeting WASF3. Oncol Res. 25:355–364. 2017.
View Article : Google Scholar
|
|
142
|
Mathew LK, Huangyang P, Mucaj V, Lee SS,
Skuli N, Eisinger-Mathason TS, Biju K, Li B, Venneti S, Lal P, et
al: Feedback circuitry between miR-218 repression and RTK
activation in glioblastoma. Sci Signal. 8:ra422015. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Mattick JS, Amaral PP, Carninci P,
Carpenter S, Chang HY, Chen LL, Chen R, Dean C, Dinger ME,
Fitzgerald KA, et al: Long non-coding RNAs: Definitions, functions,
challenges and recommendations. Nat Rev Mol Cell Biol. 24:430–447.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Shen D, Peng H, Xia C, Deng Z, Tong X,
Wang G and Qian K: The role of long non-coding RNAs in
epithelial-mesenchymal transition-related signaling pathways in
prostate cancer. Front Mol Biosci. 9:9390702022. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Mercer TR, Dinger ME and Mattick JS: Long
non-coding RNAs: Insights into functions. Nat Rev Genet.
10:155–159. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Rahbar Farzam O, Najafi S, Amini M, Rahimi
Z, Dabbaghipour R, Zohdi O, Asemani Shahgoli G, Baradaran B and
Akbari B: Interplay of miRNAs and lncRNAs in STAT3 signaling
pathway in colorectal cancer progression. Cancer Cell Int.
24:162024. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Liu S, Li W, Liang L, Zhou Y and Li Y: The
regulatory relationship between transcription factor STAT3 and
noncoding RNA. Cell Mol Biol Lett. 29:42024. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Ashrafizadeh M, Zarrabi A, Orouei S,
Zarrin V, Rahmani Moghadam E, Zabolian A, Mohammadi S, Hushmandi K,
Gharehaghajlou Y, Makvandi P, et al: STAT3 pathway in gastric
cancer: Signaling, therapeutic targeting and future prospects.
Biology (Basel). 9:1262020.PubMed/NCBI
|
|
149
|
Knutsen E, Harris AL and Perander M:
Expression and functions of long non-coding RNA NEAT1 and isoforms
in breast cancer. Br J Cancer. 126:551–561. 2022. View Article : Google Scholar :
|
|
150
|
Dong P, Xiong Y, Yue J, Hanley SJB,
Kobayashi N, Todo Y and Watari H: Long Non-coding RNA NEAT1: A
novel target for diagnosis and therapy in human tumors. Front
Genet. 9:4712018. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Yang X, Qu S, Wang L, Zhang H, Yang Z,
Wang J, Dai B, Tao K, Shang R, Liu Z, et al: PTBP3 splicing factor
promotes hepatocellular carcinoma by destroying the splicing
balance of NEAT1 and pre-miR-612. Oncogene. 37:6399–6413. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Wang H and Zheng G: LncRNA NEAT1 promotes
proliferation, migration, invasion and epithelial-mesenchymal
transition process in TGF-β2-stimulated lens epithelial cells
through regulating the miR-486-5p/SMAD4 axis. Cancer Cell Int.
20:5292020. View Article : Google Scholar
|
|
153
|
Chen Q, Cai J, Wang Q, Wang Y, Liu M, Yang
J, Zhou J, Kang C, Li M and Jiang C: Long noncoding RNA NEAT1,
regulated by the EGFR pathway, contributes to glioblastoma
progression through the WNT/β-catenin pathway by scaffolding EZH2.
Clin Cancer Res. 24:684–695. 2018. View Article : Google Scholar
|
|
154
|
Chen Y, Li J, Xiao JK, Xiao L, Xu BW and
Li C: The lncRNA NEAT1 promotes the epithelial-mesenchymal
transition and metastasis of osteosarcoma cells by sponging miR-483
to upregulate STAT3 expression. Cancer Cell Int. 21:902021.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Dong P, Xiong Y, Yue J, Xu D, Ihira K,
Konno Y, Kobayashi N, Todo Y and Watari H: Long noncoding RNA NEAT1
drives aggressive endometrial cancer progression via
miR-361-regulated networks involving STAT3 and tumor
microenvironment-related genes. J Exp Clin Cancer Res. 38:2952019.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Xia D, Yao R, Zhou P, Wang C, Xia Y and Xu
S: LncRNA NEAT1 reversed the hindering effects of miR-495-3p/STAT3
axis and miR-211/PI3K/AKT axis on sepsis-relevant inflammation. Mol
Immunol. 117:168–179. 2020. View Article : Google Scholar
|
|
157
|
Ghafouri-Fard S, Esmaeili M and Taheri M:
H19 lncRNA: Roles in tumorigenesis. Biomed Pharmacother.
123:1097742020. View Article : Google Scholar
|
|
158
|
Hashemi M, Moosavi MS, Abed HM, Dehghani
M, Aalipour M, Heydari EA, Behroozaghdam M, Entezari M,
Salimimoghadam S, Gunduz ES, et al: Long non-coding RNA (lncRNA)
H19 in human cancer: From proliferation and metastasis to therapy.
Pharmacol Res. 184:1064182022. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Liu SJ, Dang HX, Lim DA, Feng FY and Maher
CA: Long noncoding RNAs in cancer metastasis. Nat Rev Cancer.
21:446–460. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Chen X, Li Y, Zuo C, Zhang K, Lei X, Wang
J, Yang Y, Zhang J, Ma K, Wang S, et al: Long non-coding RNA H19
regulates glioma cell growth and metastasis via miR-200a-mediated
CDK6 and ZEB1 expression. Front Oncol. 11:7576502021. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Zhao Y, Feng C, Li Y, Ma Y and Cai R:
LncRNA H19 promotes lung cancer proliferation and metastasis by
inhibiting miR-200a function. Mol Cell Biochem. 460:1–8. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Luo M, Li Z, Wang W, Zeng Y, Liu Z and Qiu
J: Long non-coding RNA H19 increases bladder cancer metastasis by
associating with EZH2 and inhibiting E-cadherin expression. Cancer
Lett. 333:213–221. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Xu JX, Yang Y, Zhang X and Luan XP:
MicroRNA-29b promotes cell sensitivity to temozolomide by targeting
STAT3 in glioma. Eur Rev Med Pharmacol Sci. 24:1922–1931.
2020.PubMed/NCBI
|
|
164
|
Fang JH, Zheng ZY, Liu JY, Xie C, Zhang ZJ
and Zhuang SM: Regulatory role of the MicroRNA-29b-IL-6 signaling
in the formation of vascular mimicry. Mol Ther Nucleic Acids.
8:90–100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Liu L and Lu S: lncRNA H19 promotes
viability and epithelial-mesenchymal transition of lung
adenocarcinoma cells by targeting miR-29b-3p and modifying STAT3.
Int J Oncol. 54:929–941. 2019.PubMed/NCBI
|
|
166
|
Wang F, Rong L, Zhang Z, Li M, Ma L, Ma Y,
Xie X, Tian X and Yang Y: LncRNA H19-derived miR-675-3p promotes
epithelial-mesenchymal transition and stemness in human pancreatic
cancer cells by targeting the STAT3 pathway. J Cancer.
11:4771–4782. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Sasaki N, Hirano K, Shichi Y, Gomi F,
Yoshimura H, Matsushita A, Toyoda M and Ishiwata T: Gp130-mediated
STAT3 activation contributes to the aggressiveness of pancreatic
cancer through H19 long non-coding RNA expression. Cancers (Basel).
14:20552022. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Wu J, Zhang J, Shen B, Yin K, Xu J, Gao W
and Zhang L: Long noncoding RNA lncTCF7, induced by IL-6/STAT3
transactivation, promotes hepatocellular carcinoma aggressiveness
through epithelial-mesenchymal transition. J Exp Clin Cancer Res.
34:1162015. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Li J, Jiang ZZ, Li YY, Tang WT, Yin J and
Long XP: LncRNA CHRF promotes TGF-β1 induced EMT in alveolar
epithelial cells by inhibiting miR-146a up-regulating L1CAM
expression. Exp Lung Res. 47:198–209. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Tan WX, Sun G, Shangguan MY, Gui Z, Bao Y,
Li YF and Jia ZH: Novel role of lncRNA CHRF in cisplatin resistance
of ovarian cancer is mediated by miR-10b induced EMT and STAT3
signaling. Sci Rep. 10:147682020. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Gong H, Tao Y, Xiao S, Li X, Fang K, Wen
J, He P and Zeng M: LncRNA KIAA0087 suppresses the progression of
osteosarcoma by mediating the SOCS1/JAK2/STAT3 signaling pathway.
Exp Mol Med. 55:831–843. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Ghafouri-Fard S, Harsij A, Hussen BM,
Taheri M and Sharifi G: A review on the role of CASC11 in cancers.
Front Cell Dev Biol. 11:11311992023. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Huang H, Fan X, Zhang X, Xie Y and Ji Z:
LncRNA CARLo-7 facilitates proliferation, migration, invasion, and
EMT of bladder cancer cells by regulating Wnt/β-catenin and
JAK2/STAT3 signaling pathways. Transl Androl Urol. 9:2251–2261.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Han Y, Chen M, Wang A and Fan X:
STAT3-induced upregulation of lncRNA CASC11 promotes the cell
migration, invasion and epithelial-mesenchymal transition in
hepatocellular carcinoma by epigenetically silencing PTEN and
activating PI3K/AKT signaling pathway. Biochem Biophys Res Commun.
508:472–479. 2019. View Article : Google Scholar
|
|
175
|
Cheng Z, Guo J, Chen L, Luo N, Yang W and
Qu X: A long noncoding RNA AB073614 promotes tumorigenesis and
predicts poor prognosis in ovarian cancer. Oncotarget.
6:25381–25389. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Zeng S, Liu S, Feng J, Gao J and Xue F:
Upregulation of lncRNA AB073614 functions as a predictor of
epithelial ovarian cancer prognosis and promotes tumor growth in
vitro and in vivo. Cancer Biomark. 24:421–428. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Guo LY, Qin CF, Zou HX, Song MY, Gong ML
and Chen C: LncRNA AB073614 promotes the proliferation and inhibits
apoptosis of cervical cancer cells by repressing RBM5. Eur Rev Med
Pharmacol Sci. 23:2374–2379. 2019.PubMed/NCBI
|
|
178
|
Hu L, Lv QL, Chen SH, Sun B, Qu Q, Cheng
L, Guo Y, Zhou HH and Fan L: Up-regulation of long non-coding RNA
AB073614 predicts a poor prognosis in patients with glioma. Int J
Environ Res Public Health. 13:4332016. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Li J, Wang YM and Song YL: Knockdown of
long noncoding RNA AB073614 inhibits glioma cell proliferation and
migration via affecting epithelial-mesenchymal transition. Eur Rev
Med Pharmacol Sci. 20:3997–4002. 2016.PubMed/NCBI
|
|
180
|
Wang Y, Kuang H, Xue J, Liao L, Yin F and
Zhou X: LncRNA AB073614 regulates proliferation and metastasis of
colorectal cancer cells via the PI3K/AKT signaling pathway. Biomed
Pharmacother. 93:1230–1237. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Xue J, Liao L, Yin F, Kuang H, Zhou X and
Wang Y: LncRNA AB073614 induces epithelial-mesenchymal transition
of colorectal cancer cells via regulating the JAK/STAT3 pathway.
Cancer Biomark. 21:849–858. 2018. View Article : Google Scholar
|
|
182
|
Zhao F, Tan F, Tang L, Du Z, Chen X, Yang
Y, Zhou G and Yuan C: Long non-coding RNA DLGAP1-AS1 and
DLGAP1-AS2: Two novel oncogenes in multiple cancers. Curr Med Chem.
30:2822–2834. 2023. View Article : Google Scholar
|
|
183
|
Lin Y, Jian Z, Jin H, Wei X, Zou X, Guan R
and Huang J: Long non-coding RNA DLGAP1-AS1 facilitates
tumorigenesis and epithelial-mesenchymal transition in
hepatocellular carcinoma via the feedback loop of
miR-26a/b-5p/IL-6/JAK2/STAT3 and Wnt/β-catenin pathway. Cell Death
Dis. 11:342020. View Article : Google Scholar
|
|
184
|
Zheng X, Hu H and Li S: High expression of
lncRNA PVT1 promotes invasion by inducing epithelial-to-mesenchymal
transition in esophageal cancer. Oncol Lett. 12:2357–2362. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Zhang X, Feng W, Zhang J, Ge L, Zhang Y,
Jiang X, Peng W, Wang D, Gong A and Xu M: Long non-coding RNA PVT1
promotes epithelial-mesenchymal transition via the TGF-β/Smad
pathway in pancreatic cancer cells. Oncol Rep. 40:1093–1102.
2018.PubMed/NCBI
|
|
186
|
Wang L, Xiao B, Yu T, Gong L, Wang Y,
Zhang X, Zou Q and Zuo Q: lncRNA PVT1 promotes the migration of
gastric cancer by functioning as ceRNA of miR-30a and regulating
Snail. J Cell Physiol. 236:536–548. 2021. View Article : Google Scholar
|
|
187
|
Chang Z, Cui J and Song Y: Long noncoding
RNA PVT1 promotes EMT via mediating microRNA-186 targeting of
Twist1 in prostate cancer. Gene. 654:36–42. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Zhou Y, Xu S, Xia H, Gao Z, Huang R, Tang
E and Jiang X: Long noncoding RNA FEZF1-AS1 in human cancers. Clin
Chim Acta. 497:20–26. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Wang H, Wu Y, Wang Z, Chen Y, Mo J, Guan
W, Zhang Y and Yao H: The LncRNA FEZF1-AS1 promotes tumor
proliferation in colon cancer by regulating the mitochondrial
protein PCK2. Oncol Res. 29:201–215. 2022. View Article : Google Scholar
|
|
190
|
Zhang G, Yang W, Li D, Li X, Huang J,
Huang R and Luo J: lncRNA FEZF1-AS1 promotes migration, invasion
and epithelial-mesenchymal transition of retinoblastoma cells by
targeting miR-1236-3p. Mol Med Rep. 22:3635–3644. 2020.PubMed/NCBI
|
|
191
|
He R, Zhang FH and Shen N: LncRNA
FEZF1-AS1 enhances epithelial-mesenchymal transition (EMT) through
suppressing E-cadherin and regulating WNT pathway in non-small cell
lung cancer (NSCLC). Biomed Pharmacother. 95:331–338. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Zhao X, Cheng Z and Wang J: Long noncoding
RNA FEZF1-AS1 promotes proliferation and inhibits apoptosis in
ovarian cancer by activation of JAK-STAT3 pathway. Med Sci Monit.
24:8088–8095. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Bian Z, Zhang J, Li M, Feng Y, Wang X,
Zhang J, Yao S, Jin G, Du J, Han W, et al: LncRNA-FEZF1-AS1
promotes tumor proliferation and metastasis in colorectal cancer by
regulating PKM2 signaling. Clin Cancer Res. 24:4808–4819. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Wang YD, Sun XJ, Yin JJ, Yin M, Wang W,
Nie ZQ and Xu J: Long non-coding RNA FEZF1-AS1 promotes cell
invasion and epithelial-mesenchymal transition through JAK2/STAT3
signaling pathway in human hepatocellular carcinoma. Biomed
Pharmacother. 106:134–141. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Shang BQ, Li ML, Quan HY, Hou PF, Li ZW,
Chu SF, Zheng JN and Bai J: Functional roles of circular RNAs
during epithelial-to-mesenchymal transition. Mol Cancer.
18:1382019. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Wei X, Shi Y, Dai Z, Wang P, Meng X and
Yin B: Underlying metastasis mechanism and clinical application of
exosomal circular RNA in tumors (review). Int J Oncol. 58:289–297.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Sharma AR, Banerjee S, Bhattacharya M,
Saha A, Lee SS and Chakraborty C: Recent progress of circular RNAs
in different types of human cancer: Technological landscape,
clinical opportunities and challenges (review). Int J Oncol.
60:562022. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Liu CX and Chen LL: Circular RNAs:
Characterization, cellular roles, and applications. Cell.
185:2016–2034. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Xue C, Li G, Zheng Q, Gu X, Bao Z, Lu J
and Li L: The functional roles of the circRNA/Wnt axis in cancer.
Mol Cancer. 21:1082022. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Long F, Lin Z, Li L, Ma M, Lu Z, Jing L,
Li X and Lin C: Comprehensive landscape and future perspectives of
circular RNAs in colorectal cancer. Mol Cancer. 20:262021.
View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Kristensen LS, Jakobsen T, Hager H and
Kjems J: The emerging roles of circRNAs in cancer and oncology. Nat
Rev Clin Oncol. 19:188–206. 2022. View Article : Google Scholar
|
|
202
|
Tang X, Ren H, Guo M, Qian J, Yang Y and
Gu C: Review on circular RNAs and new insights into their roles in
cancer. Comput Struct Biotechnol J. 19:910–928. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Chen S, Chen C, Hu Y, Song G and Shen X:
The diverse roles of circular RNAs in pancreatic cancer. Pharmacol
Ther. 226:1078692021. View Article : Google Scholar : PubMed/NCBI
|
|
204
|
Song J, Liu Q, Han L, Song T, Huang S,
Zhang X, He Q, Liang C, Zhu S and Xiong B:
Hsa_circ_0009092/miR-665/NLK signaling axis suppresses colorectal
cancer progression via recruiting TAMs in the tumor
microenvironment. J Exp Clin Cancer Res. 42:3192023. View Article : Google Scholar : PubMed/NCBI
|
|
205
|
Wu M, Sun T and Xing L: Circ_0004913
inhibits cell growth, metastasis, and glycolysis by absorbing
miR-184 to regulate HAMP in hepatocellular carcinoma. Cancer
Biother Radiopharm. 38:708–719. 2023.
|
|
206
|
Li G, Kong J, Dong S, Niu H, Wu S and Sun
W: Circular BANP knockdown inhibits the malignant progression of
residual hepatocellular carcinoma after insufficient radiofrequency
ablation. Chin Med J (Engl). 135:1578–1587. 2022.Epub ahead of
print. View Article : Google Scholar : PubMed/NCBI
|
|
207
|
He SL, Zhao X and Yi SJ: CircAHNAK
upregulates EIF2B5 expression to inhibit the progression of ovarian
cancer by modulating the JAK2/STAT3 signaling pathway.
Carcinogenesis. 43:941–955. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
208
|
Zou S, Tong Q, Liu B, Huang W, Tian Y and
Fu X: Targeting STAT3 in cancer immunotherapy. Mol Cancer.
19:1452020. View Article : Google Scholar : PubMed/NCBI
|
|
209
|
Beebe JD, Liu JY and Zhang JT: Two decades
of research in discovery of anticancer drugs targeting STAT3, how
close are we? Pharmacol Ther. 191:74–91. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
210
|
Turkson J, Ryan D, Kim JS, Zhang Y, Chen
Z, Haura E, Laudano A, Sebti S, Hamilton AD and Jove R:
Phosphotyrosyl peptides block Stat3-mediated DNA binding activity,
gene regulation, and cell transformation. J Biol Chem.
276:45443–45455. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
211
|
Turkson J, Kim JS, Zhang S, Yuan J, Huang
M, Glenn M, Haura E, Sebti S, Hamilton AD and Jove R: Novel
peptidomimetic inhibitors of signal transducer and activator of
transcription 3 dimerization and biological activity. Mol Cancer
Ther. 3:261–269. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
212
|
Auzenne EJ, Klostergaard J, Mandal PK,
Liao WS, Lu Z, Gao F, Bast RC Jr, Robertson FM and McMurray JS: A
phosphopeptide mimetic prodrug targeting the SH2 domain of Stat3
inhibits tumor growth and angiogenesis. J Exp Ther Oncol.
10:155–162. 2012.
|
|
213
|
Wong ALA, Hirpara JL, Pervaiz S, Eu JQ,
Sethi G and Goh BC: Do STAT3 inhibitors have potential in the
future for cancer therapy? Expert Opin Investig Drugs. 26:883–887.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
214
|
Redell MS, Ruiz MJ, Alonzo TA, Gerbing RB
and Tweardy DJ: Stat3 signaling in acute myeloid leukemia:
Ligand-dependent and -independent activation and induction of
apoptosis by a novel small-molecule Stat3 inhibitor. Blood.
117:5701–5709. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
215
|
Peng HY, Wang L, Das JK, Kumar A, Ballard
DJ, Ren Y, Xiong X, de Figueiredo P, Yang JM and Song J: Control of
CD4+ T cells to restrain inflammatory diseases via
eukaryotic elongation factor 2 kinase. Signal Transduct Target
Ther. 8:4152023. View Article : Google Scholar
|
|
216
|
Bharadwaj U, Eckols TK, Xu X, Kasembeli
MM, Chen Y, Adachi M, Song Y, Mo Q, Lai SY and Tweardy DJ:
Small-molecule inhibition of STAT3 in radioresistant head and neck
squamous cell carcinoma. Oncotarget. 7:26307–26330. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
217
|
Genini D, Brambilla L, Laurini E, Merulla
J, Civenni G, Pandit S, D'Antuono R, Perez L, Levy DE, Pricl S, et
al: Mitochondrial dysfunction induced by a SH2 domain-targeting
STAT3 inhibitor leads to metabolic synthetic lethality in cancer
cells. Proc Natl Acad Sci USA. 114:E4924–E4933. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
218
|
Kim MJ, Nam HJ, Kim HP, Han SW, Im SA, Kim
TY, Oh DY and Bang YJ: OPB-31121, a novel small molecular
inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an
antitumor activity in gastric cancer cells. Cancer Lett.
335:145–152. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
219
|
Hayakawa F, Sugimoto K, Harada Y,
Hashimoto N, Ohi N, Kurahashi S and Naoe T: A novel STAT inhibitor,
OPB-31121, has a significant antitumor effect on leukemia with
STAT-addictive oncokinases. Blood Cancer J. 3:e1662013. View Article : Google Scholar : PubMed/NCBI
|
|
220
|
Brambilla L, Genini D, Laurini E, Merulla
J, Perez L, Fermeglia M, Carbone GM, Pricl S and Catapano CV:
Hitting the right spot: Mechanism of action of OPB-31121, a novel
and potent inhibitor of the signal transducer and activator of
transcription 3 (STAT3). Mol Oncol. 9:1194–1206. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
221
|
Crooke ST, Baker BF, Crooke RM and Liang
XH: Antisense technology: An overview and prospectus. Nat Rev Drug
Discov. 20:427–453. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
222
|
Odate S, Veschi V, Yan S, Lam N, Woessner
R and Thiele CJ: Inhibition of STAT3 with the generation 2.5
antisense oligonucleotide, azd9150, decreases neuroblastoma
tumorigenicity and increases Chemosensitivity. Clin Cancer Res.
23:1771–1784. 2017. View Article : Google Scholar :
|
|
223
|
Reilley MJ, McCoon P, Cook C, Lyne P,
Kurzrock R, Kim Y, Woessner R, Younes A, Nemunaitis J, Fowler N, et
al: STAT3 antisense oligonucleotide AZD9150 in a subset of patients
with heavily pretreated lymphoma: Results of a phase 1b trial. J
Immunother Cancer. 6:1192018. View Article : Google Scholar : PubMed/NCBI
|
|
224
|
Roschewski M, Patel MR, Reagan PM, Saba
NS, Collins GP, Arkenau HT, de Vos S, Nuttall B, Acar M, Burke K,
et al: Phase I study of acalabrutinib plus danvatirsen (AZD9150) in
relapsed/refractory diffuse large B-cell lymphoma including
circulating tumor DNA biomarker assessment. Clin Cancer Res.
29:3301–3312. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
225
|
Cascone T, Kar G, Spicer JD,
García-Campelo R, Weder W, Daniel DB, Spigel DR, Hussein M,
Mazieres J, Oliveira J, et al: Neoadjuvant durvalumab alone or
combined with novel immuno-oncology agents in resectable lung
cancer: The phase II NeoCOAST platform trial. Cancer Discov.
13:2394–2411. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
226
|
Shastri A, Choudhary G, Teixeira M,
Gordon-Mitchell S, Ramachandra N, Bernard L, Bhattacharyya S, Lopez
R, Pradhan K, Giricz O, et al: Antisense STAT3 inhibitor decreases
viability of myelodysplastic and leukemic stem cells. J Clin
Invest. 128:5479–5488. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
227
|
Proia TA, Singh M, Woessner R, Carnevalli
L, Bommakanti G, Magiera L, Srinivasan S, Grosskurth S, Collins M,
Womack C, et al: STAT3 antisense oligonucleotide remodels the
suppressive tumor microenvironment to enhance immune activation in
combination with anti-PD-L1. Clin Cancer Res. 26:6335–6349. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
228
|
Leong PL, Andrews GA, Johnson DE, Dyer KF,
Xi S, Mai JC, Robbins PD, Gadiparthi S, Burke NA, Watkins SF and
Grandis JR: Targeted inhibition of Stat3 with a decoy
oligonucleotide abrogates head and neck cancer cell growth. Proc
Natl Acad Sci USA. 100:4138–4143. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
229
|
Xi S, Gooding WE and Grandis JR: In vivo
antitumor efficacy of STAT3 blockade using a transcription factor
decoy approach: Implications for cancer therapy. Oncogene.
24:970–979. 2005. View Article : Google Scholar
|
|
230
|
Zhang X, Zhang J, Wang L, Wei H and Tian
Z: Therapeutic effects of STAT3 decoy oligodeoxynucleotide on human
lung cancer in xenograft mice. BMC Cancer. 7:1492007. View Article : Google Scholar : PubMed/NCBI
|
|
231
|
Sen M, Thomas SM, Kim S, Yeh JI, Ferris
RL, Johnson JT, Duvvuri U, Lee J, Sahu N, Joyce S, et al:
First-in-human trial of a STAT3 decoy oligonucleotide in head and
neck tumors: Implications for cancer therapy. Cancer Discov.
2:694–705. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
232
|
Njatcha C, Farooqui M, Kornberg A, Johnson
DE, Grandis JR and Siegfried JM: STAT3 cyclic decoy demonstrates
robust antitumor effects in non-small cell lung cancer. Mol Cancer
Ther. 17:1917–1926. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
233
|
Zhang Q, Hossain DMS, Duttagupta P,
Moreira D, Zhao X, Won H, Buettner R, Nechaev S, Majka M, Zhang B,
et al: Serum-resistant CpG-STAT3 decoy for targeting survival and
immune checkpoint signaling in acute myeloid leukemia. Blood.
127:1687–1700. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
234
|
Pettersson M and Crews CM: PROteolysis
targeting chimeras (PROTACs)-past, present and future. Drug Discov
Today Technol. 31:15–27. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
235
|
Békés M, Langley DR and Crews CM: PROTAC
targeted protein degraders: The past is prologue. Nat Rev Drug
Discov. 21:181–200. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
236
|
Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy
K, McEachern D, Chen J, Yang CY, Liu Z, Wang M, et al: A potent and
selective small-molecule degrader of STAT3 achieves complete tumor
regression in vivo. Cancer Cell. 36:498–511.e17. 2019. View Article : Google Scholar :
|
|
237
|
Jin J, Wu Y, Zhao Z, Wu Y, Zhou YD, Liu S,
Sun Q, Yang G, Lin J, Nagle DG, et al: Small-molecule PROTAC
mediates targeted protein degradation to treat STAT3-dependent
epithelial cancer. JCI Insight. 7:e1606062022. View Article : Google Scholar : PubMed/NCBI
|
|
238
|
Shih PC, Naganuma M, Tsuji G, Demizu Y and
Naito M: Development of decoy oligonucleotide-warheaded chimeric
molecules targeting STAT3. Bioorg Med Chem. 95:1175072023.
View Article : Google Scholar : PubMed/NCBI
|
|
239
|
He X, Weng Z and Zou Y: Progress in the
controllability technology of PROTAC. Eur J Med Chem.
265:1160962024. View Article : Google Scholar : PubMed/NCBI
|
|
240
|
Kang S, Tanaka T, Narazaki M and Kishimoto
T: Targeting interleukin-6 signaling in clinic. Immunity.
50:1007–1023. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
241
|
Shen P, Wang Y, Jia X, Xu P, Qin L, Feng
X, Li Z and Qiu Z: Dual-target Janus kinase (JAK) inhibitors:
Comprehensive review on the JAK-based strategies for treating solid
or hematological malignancies and immune-related diseases. Eur J
Med Chem. 239:1145512022. View Article : Google Scholar : PubMed/NCBI
|
|
242
|
Liang D, Wang Q, Zhang W, Tang H, Song C,
Yan Z, Liang Y and Wang H: JAK/STAT in leukemia: A clinical update.
Mol Cancer. 23:252024. View Article : Google Scholar : PubMed/NCBI
|
|
243
|
Plimack ER, Lorusso PM, McCoon P, Tang W,
Krebs AD, Curt G and Eckhardt SG: AZD1480: A phase I study of a
novel JAK2 inhibitor in solid tumors. Oncologist. 18:819–820. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
244
|
Deisseroth A, Ko CW, Nie L, Zirkelbach JF,
Zhao L, Bullock J, Mehrotra N, Del Valle P, Saber H, Sheth C, et
al: FDA approval: Siltuximab for the treatment of patients with
multicentric Castleman disease. Clin Cancer Res. 21:950–954. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
245
|
Rossi JF, Lu ZY, Jourdan M and Klein B:
Interleukin-6 as a therapeutic target. Clin Cancer Res.
21:1248–1257. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
246
|
Dorff TB, Goldman B, Pinski JK, Mack PC,
Lara PN Jr, Van Veldhuizen PJ Jr, Quinn DI, Vogelzang NJ, Thompson
IM Jr and Hussain MH; Clinical and correlative results of SWOG
S0354: A phase II trial of CNTO328 (siltuximab), a monoclonal
antibody against interleukin-6, in chemotherapy-pretreated patients
with castration-resistant prostate cancer. Clin Cancer Res.
16:3028–3034. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
247
|
Angevin E, Tabernero J, Elez E, Cohen SJ,
Bahleda R, van Laethem JL, Ottensmeier C, Lopez-Martin JA, Clive S,
Joly F, et al: A phase I/II, multiple-dose, dose-escalation study
of siltuximab, an anti-interleukin-6 monoclonal antibody, in
patients with advanced solid tumors. Clin Cancer Res. 20:2192–2204.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
248
|
Fizazi K, De Bono JS, Flechon A,
Heidenreich A, Voog E, Davis NB, Qi M, Bandekar R, Vermeulen JT,
Cornfeld M and Hudes GR: Randomised phase II study of siltuximab
(CNTO 328), an anti-IL-6 monoclonal antibody, in combination with
mitoxantrone/prednisone versus mitoxantrone/prednisone alone in
metastatic castration-resistant prostate cancer. Eur J Cancer.
48:85–93. 2012. View Article : Google Scholar
|
|
249
|
Beg MS, Brenner AJ, Sachdev J, Borad M,
Kang YK, Stoudemire J, Smith S, Bader AG, Kim S and Hong DS: Phase
I study of MRX34, a liposomal miR-34a mimic, administered twice
weekly in patients with advanced solid tumors. Invest New Drugs.
35:180–188. 2017. View Article : Google Scholar
|
|
250
|
Liu S, Wang L, Li Y, Cui Y, Wang Y and Liu
C: Long non-coding RNA CHRF promotes proliferation and mesenchymal
transition (EMT) in prostate cancer cell line PC3 requiring
up-regulating microRNA-10b. Biol Chem. 400:1035–1045. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
251
|
Li Y and Xu X: The long noncoding RNA
cardiac hypertrophy-related factor plays oncogenic roles in
hepatocellular carcinoma by downregulating microRNA-211. J Cell
Biochem. 120:13361–13371. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
252
|
Tao Y, Han T, Zhang T, Ma C and Sun C:
LncRNA CHRF-induced miR-489 loss promotes metastasis of colorectal
cancer via TWIST1/EMT signaling pathway. Oncotarget. 8:36410–36422.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
253
|
Gong J, Wang Y and Shu C: LncRNA CHRF
promotes cell invasion and migration via EMT in gastric cancer. Eur
Rev Med Pharmacol Sci. 24:1168–1176. 2020.PubMed/NCBI
|