Introduction
Ataxia-telangiectasia (A-T) is an infrequent
autosomal recessive disorder (OMIM 208900) caused by mutations in
the ATM gene encoding the ATM protein, and it was first described
in 1957 by Boder et al (1).
The ATM gene is located on human chromosome 11q22-q23 and is made
up of 66 exons (4 non-coding and 62 coding) spanning 150 kb of
genomic DNA. The ATM gene codes a Ser/Thr kinase (ATM protein)
involved in DNA repair that phosphorylates almost two dozen
distinct substrates that function in cell signaling to control the
cell cycle, repair double-strand DNA breaks, respond to oxidative
stress and regulate transcription. The molecular pathogenesis of
A-T is abnormal signal transduction of DNA repair and DNA damage
(2). The worldwide prevalence of A-T
is estimated to be between 1 in 40,000 and 1 in 100,000 live
births, and there is no sex predilection (3). More than 1,000 mutations have been
reported to date (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=ATM)
throughout the gene. Genomic instability affects immunoglobulin
coding-related sites and the development of malignant neoplasms.
This interference increases the risk of infection and tumorigenesis
(4). According to statistics,
approximately one-third of A-T patients develop a malignant
neoplasm during their lifetime, with a greatly increased incidence
of malignant neoplasms in homozygous affected individuals.
Heterozygous carriers of the ATM gene are common among the close
relatives of patients with A-T. The risk of dying from a malignant
neoplasm in A-T heterozygotes was estimated to be over 5 times the
respective risk in the general population (5). ATM gene mutations can be identified
efficiently and reliably by exon sequencing, which is widely used
in the diagnosis of immune deficiency, heredopathia and cancer,
providing clinicians with quick guidance on subsequent healthcare
management strategies (6,7). In order to enhance the understanding of
A-T among clinicians and to promote the early identification and
treatment of diseases, we herein report a case of A-T with a
hematological malignancy and analyze the clinical data and the gene
sequence of the whole exome in the patient's pedigree.
Case report
A 7-year-old male patient was hospitalized at
Xiangya Hospital of Central South University (Changsha, China) in
September 2017, with complaints of recurrent fever and
osteoarticular pain for 20 days that did not improve with
antibiotic therapy. The findings on physical examination included
superficial lymphadenectasis with movable and non-tender nodes
palpated in the neck, axilla and groin (maximum size, ~1.5×1 cm),
without hepatosplenomegaly. The neurological evaluation revealed
instability of gait, abnormal finger-nose test and positive
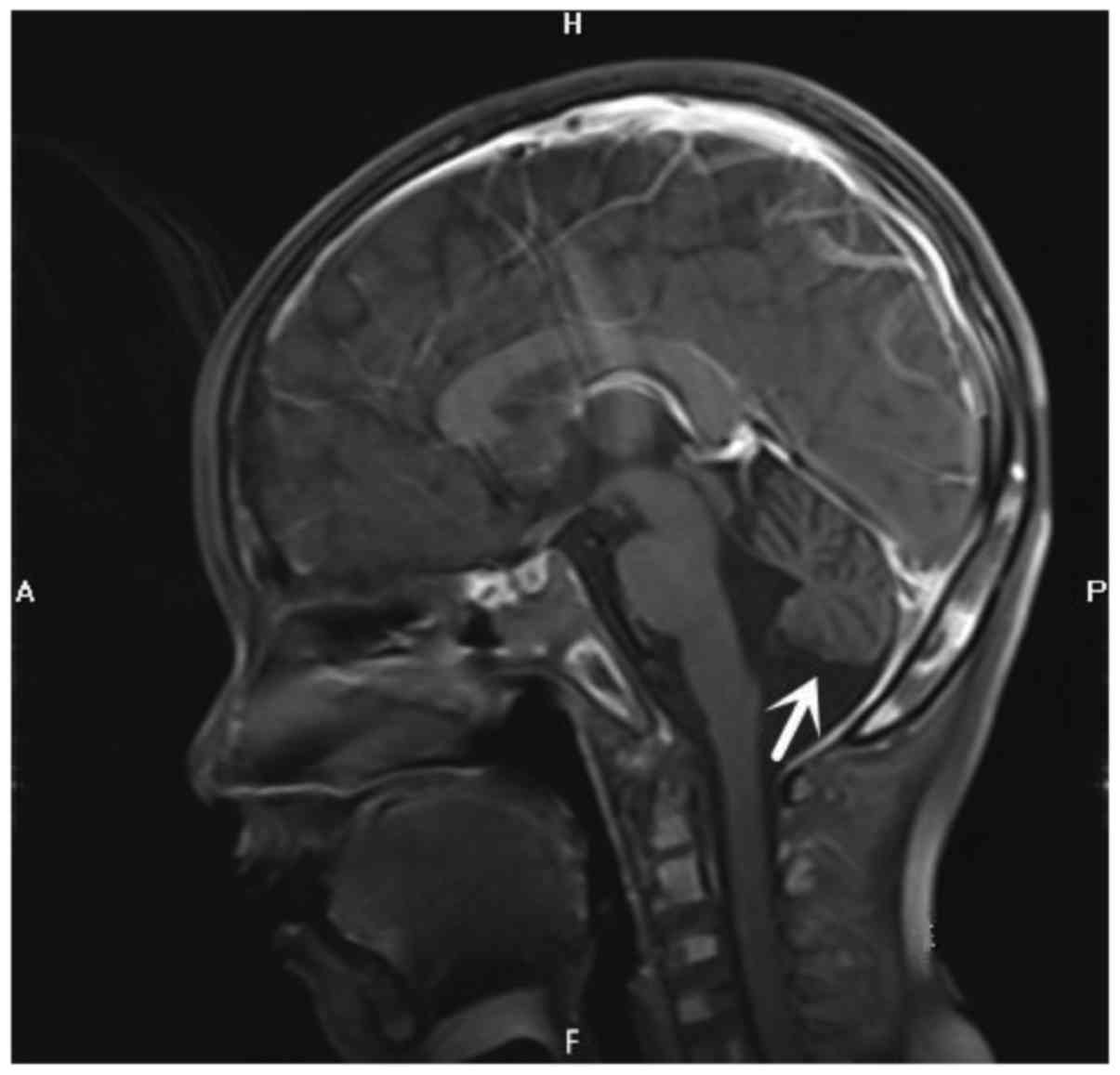
Romberg's sign. Craniocerebral magnetic resonance imaging
examination revealed cerebellar atrophy (Fig. 1). The patient's complete blood count
(Table I) revealed a differential
count of neutrophils 9.0×109/l (91.1%), lymphocytes
0.5×109/l (5.4%) and monocytes 0.3×109/l
(3.4%). The patient had microcytic hypochromic anemia (hemoglobin
110 g/l, mean corpuscular volume 80.6 fl, mean corpuscular
hemoglobin 26.6 pg and mean corpuscular hemoglobin concentration
330 g/l). A computed tomography examination of the chest, abdomen
and pelvis revealed multiple nodules in the liver and kidney. Bone
scans revealed multiple bone metabolic abnormalities throughout the
body, including the ribs, vertebrae, tibia and fibula, raising the
suspicion of tumor invasion. Immunological testing (Table I) indicated IgA deficiency. Bone
marrow cell morphology examination revealed medullary proliferative
activity, with primitive and immature lymphocytes accounting for
~78.5%. Flow cytometry analysis of the bone marrow revealed a group
of abnormal cells accounting for 22% of karyocytes, exhibiting
weaker expression of CD45 compared with monocytes and slightly more
prominent side scatter compared with lymphocytes; these cells were
positive for HLA-DR, CD10, CD19, CD20, CD22, CD38, FMC-7 and
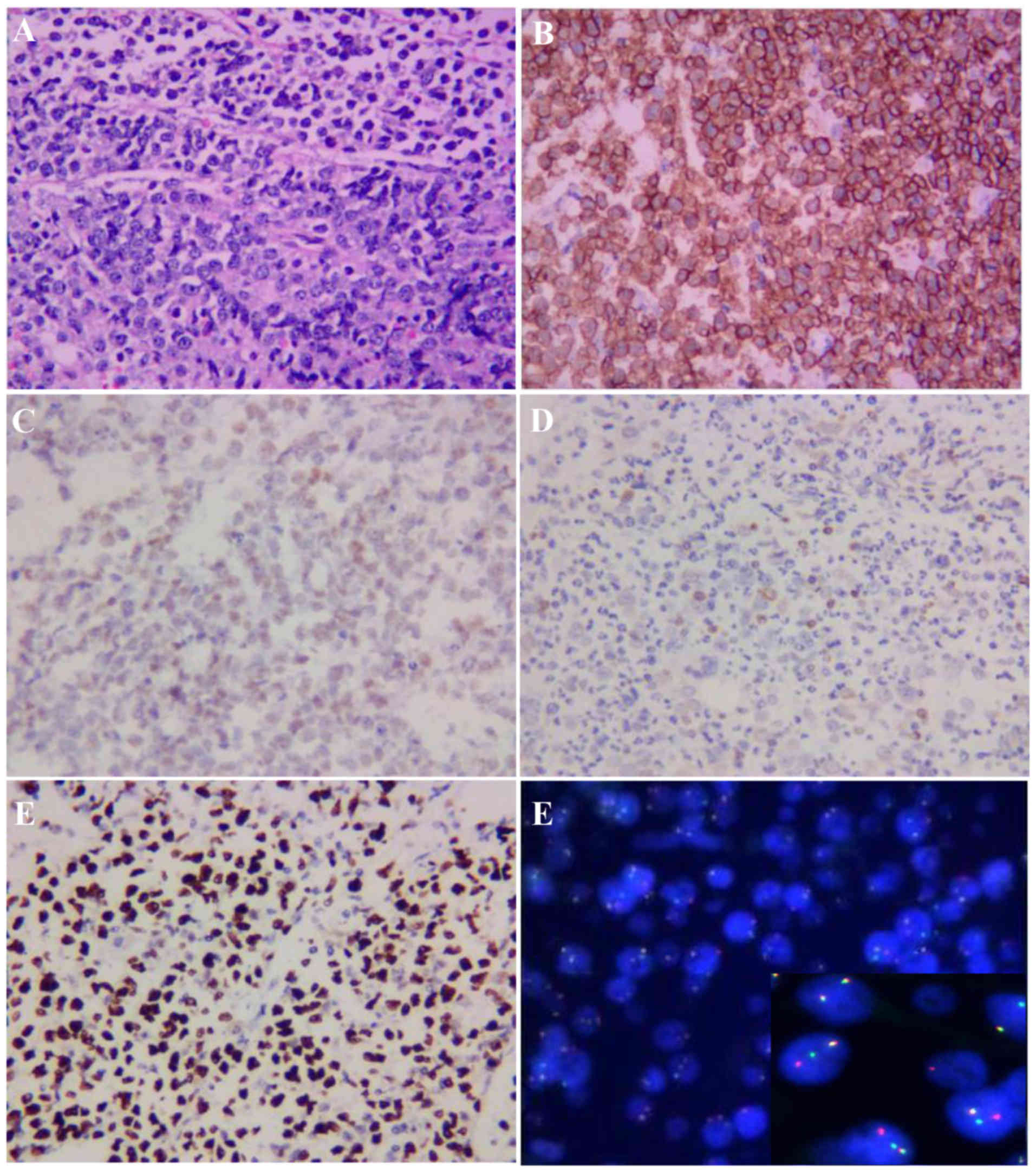
cCD79a. Immunohistochemical staining revealed the following
phenotype: Terminal deoxynucleotidyl transferase−,
CD10+, C-myc+ (60%), CD20+, B-cell
lymphoma (Bcl)-2− and Bcl-6+. The MIB1
proliferation index was ~100%. The results of fluorescence in
situ hybridization were positive for C-MYC gene rearrangement
(Fig. 2), suggesting a diagnosis of
Burkitt leukemia.
 | Table I.Complete blood count and immunological
test in the present case. |
Table I.
Complete blood count and immunological
test in the present case.
| Tests | Value | Normal range |
|---|
| White blood cell
count (×109/l) | 9.9 | 3.5–9.5 |
| Red blood cell count
(×1012/l) | 4.13 | 4.3–5.8 |
| Platelet count
(×109/l) | 581 | 125–350 |
| Hemoglobin (g/l) | 110 | 130–175 |
| Neutrophils
(×109/l) | 9.0 | 1.8–6.3 |
| Lymphocytes
(×109/l) | 0.5 | 1.1–3.2 |
| Mean corpuscular
volume (fl) | 80.6 | 82.0–100.0 |
| Mean corpuscular
hemoglobin (pg) | 26.6 | 27.0–34.0 |
| Mean corpuscular
hemoglobin concentration (g/l) | 330.0 | 316–354 |
| Immunoglobulin G
(g/l) | 13.6 | 7.23–16.85 |
| Immunoglobulin A
(mg/l) | <66.7 | 690-3,820 |
| Immunoglobulin M
(mg/l) | 2,410 | 630-2,770 |
The patient's previous medical history included at
least seven upper respiratory infections annually. He was the
second child of the family and the perinatal history was
unremarkable. The patient's grandfather had been diagnosed with
nasopharyngeal carcinoma, but there was no family history of
immunodeficiency or neurological disease. The patient started
walking at the age of 18 months, with occasional falls. The
symptoms of gait instability gradually increased over time,
eventually developing into hypotonia and gait dysfunction. The
patient was diagnosed with cerebellar ataxia and received
rehabilitation training for 6 months; however, his balance did not
improve over time.
The patient received sequential chemotherapy in our
hospital with the pediatric CCCG-BNHL-2015 regimen for high-risk
patients (R4 group) (Table II),
which included rituximab, methotrexate (MTX), cytarabine (Ara-C)
and cyclophosphamide (CTX), intrathecal (IT) dexamethasone, IT
Ara-C and IT MTX. Recurrent pulmonary infections, oral mucosal
ulcers and venous thrombosis have been reported in children during
chemotherapy, either during the period of bone marrow suppression
or non-bone marrow recovery. Considering the genetic susceptibility
and immunological deficiency, whole-exome sequencing was performed,
according to the patient's history and clinical manifestations. The
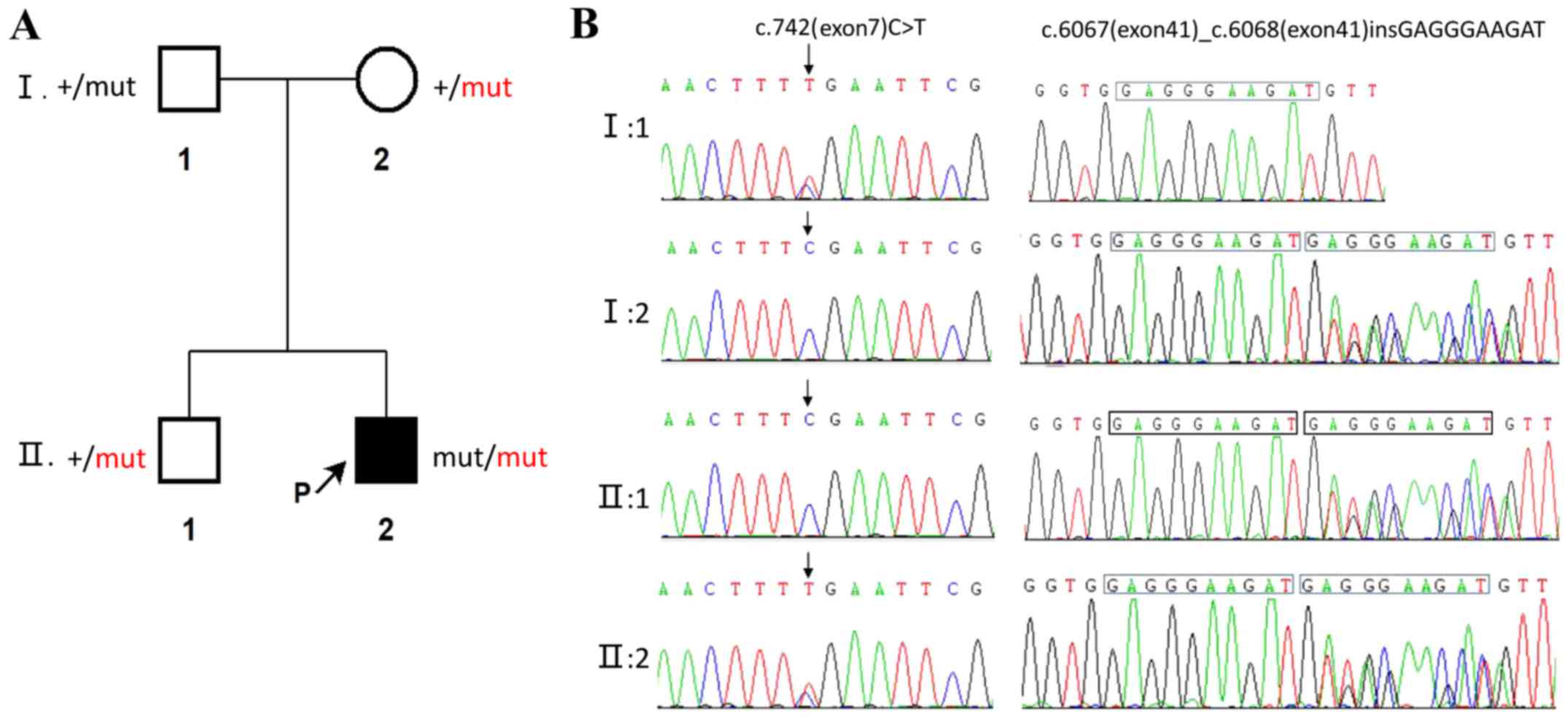
results demonstrated that two variants of the ATM gene were
associated with A-T, including c.742C>T (p.R248X, 2809;
rs730881336) in exon 7 and c.6067-c.6068 ins GAGGGAAGAT
(p.G2023Gfs*13) in exon 41. To verify the mutation, after obtaining
informed consent, we also sequenced the entire exome of the family
members (Table III) and
constructed a simplified pedigree chart (Fig. 3). Autosomal complex heterozygosity
inheritance was confirmed, as his parents were heterozygous
carriers. The patient achieved complete remission after 2 courses
of chemotherapy. Unfortunately, his parents refused follow-up
treatment and he succumbed to recurrent severe infections 4 months
after being diagnosed with Burkitt leukemia.
| Table II.Pediatric CCCG-BNHL-2015 regimen. |
Table II.
Pediatric CCCG-BNHL-2015 regimen.
|
| Dose | Administration | Days |
|---|
| AA (R4 +
rituximab) |
|
Cyclophosphamide | 800
mg/m2 | IV over 2 h | D1 |
|
Cyclophosphamide | 200
mg/m2 | IV over 1 h | D2, 3, 4 |
|
Vindesine | 3 mg/m2
(max 5 mg) | IV | D1 |
|
Doxorubicin | 20
mg/m2 | IV over 2 h | D2, D3 |
|
Cytarabine | 2,000
mg/m2 | IV over 3 h | D4 (q12 h) |
|
Prednisone | 60
mg/m2 | Per os | D1-7 |
| Triple sheath note
(dose by age) |
|
| D1, D8 |
| BB (R4 +
rituximab) |
|
Ifosfamide | 1,200
mg/m2 | IV over 2 h | D1-5 |
|
| Mesna 400
mg/m2, |
|
|
|
| 0, +4, +8 h |
|
|
|
Etoposide | 100
mg/m2 | IV over 2 h | D3-5 |
|
Methotrexate | 5,000
mg/m2 | IV over 24 h | D1 |
|
Vindesine | 3 mg/m2
(max 5 mg) | IV | D1 |
|
Prednisone | 60
mg/m2 | Per os | D1-7 |
| Triple
sheath note (dose by age) |
|
| D1, D8 |
| Table III.Results of gene detection in this
patient and his family members. |
Table III.
Results of gene detection in this
patient and his family members.
| Family member | Exon (ATM
gene) | Mutation type | NA changes | AA changes | Prediction |
|---|
| Patient | 7 | Heterozygous | c.742C>T | p.R248X | Harmful |
|
| 41 | Heterozygous | c.6067-c.6068 ins
GAGGGAAGAT | p.G2023Gfs*13 | Harmful |
| Father | 7 | Heterozygous | c.742C>T | p.R248X | Harmful |
|
| 41 | Normal | Normal | Normal | Normal |
| Mother | 7 | Normal | Normal | Normal | Normal |
|
| 41 | Heterozygous | c.6067-c.6068 ins
GAGGGAAGAT | p.G2023Gfs*13 | Harmful |
| Brother | 7 | Normal | Normal | Normal | Normal |
|
| 41 | Heterozygous | c.6067-c.6068 ins
GAGGGAAGAT | p.G2023Gfs*13 | Harmful |
Discussion
A-T is a rare autosomal recessive disorder
characterized by progressive cerebellar degeneration,
telangiectasia, immunodeficiency, recurrent respiratory infections,
radiation sensitivity, premature aging and a predisposition to
cancer development, particularly of lymphoid origin. Other
abnormalities include poor growth, gonadal atrophy, delayed
pubertal development and insulin-resistant diabetes. Laboratory
abnormalities in A-T commonly manifest as elevated and slowly
increasing serum α-fetoprotein levels after the age of 2 years, low
serum levels of immunoglobulins (IgA, IgG, IgG subclasses, IgE) and
lymphopenia (particularly affecting T-lymphocytes) (8). The majority of the patients manifesting
A-T usually succumb to a severe bronchopulmonary infection or
malignancy (9). A-T can present with
other mobility disorders in addition to ataxia, and a significant
proportion of cases do not present with telangiectasia. Thus, Teive
et al recommended re-naming this disease ATM syndrome in
2015 (10).
Our patient exhibited early neurological
manifestations, such as cerebellar ataxia, but without
telangiectasia, and he was not diagnosed with A-T until he
developed a hematological malignancy. The diagnosis of A-T can be
challenging due to the phenotype that may be incomplete during the
disease course. For example, ataxia is usually the earliest
clinical manifestation, whereas gait instability progresses slowly
and typically becomes obvious after 5 years of age (11). The clinical diagnosis becomes most
apparent after the age of 5 years, when ataxia, apraxia and
telangiectasia are fully manifested. By contrast, in very young
infants, the diagnosis may be more difficult and easily mistaken
for mild cerebral palsy, acute infectious or episodic ataxia, or
other rare genetic or mitochondrial disorders. Brain magnetic
resonance imaging can reveal cerebellar atrophy, but can also be
normal in the early stages of the disease (12). Characteristic telangiectasias are
rarely observed before the age of 3 years, whereas some patients do
not display any signs of capillary dilatation, such as of the eyes
and skin, throughout the course of the disease (10). Our patient had IgA deficiency,
recurrent infections and chemotherapeutic intolerance. As reported,
up to 71% of patients with A-T who have been investigated had some
form of immune deficiency, including low total immunoglobulin
levels (IgG, IgA or IgM), low IgG2, defective polysaccharide
antibody responses and lymphopenia (13). Furthermore, A-T patients with
elevated IgM levels constitute a distinct group with a severe
disease phenotype and worse prognosis (14); in our patient, IgM was within the
normal range. A-T patients usually do not display all the
characteristic clinical features, and may exhibit various subtypes
and severity of immune deficiency. Patients with gait disorders and
recurrent respiratory infections should immediately raise the
clinical suspicion of A-T. Early diagnosis can prevent exposure to
radiation and enable better management of infections and malignant
tumors.
A diagnosis of A-T can be confirmed by the finding
of an absence or deficiency of the ATM protein or its kinase
activity in cultured cell lines, and/or identification of the
pathological mutations in the ATM gene. According to the
pedigree chart (Fig. 3), two
mutations were found in the ATM gene (located on chromosome 11).
The c.742 (exon7) C>T mutation (nonsense mutation, p.R248X,
2809, chr11:108186610-108186611) was detected in patients with
capillary dilatation reported in the journal Ann Hum Genet in 2005
by Mitui et al (15), while
the ATM mutation c.6067-c.6068 ins GAGGGAAGAT in exon 41
(frameshift mutation, p.G2023Gfs*13, chr11:108115594-108115594) is
a novel mutation that was not found in the disease correlation
report based on OMIM, HGMD and Clinvar database. This case is very
rare, as each family member was a carrier for A-T. Unfortunately,
the illnesses in our patient were the result of their double
heterozygosity. The difficulties in clinical diagnosis may be
overcome by including genetic screening tests in the range of
available diagnostic tests, which may also reveal unexpected
results. Previous in vitro research has laid the foundation
for future mutation targeting therapy in A-T patients, although
there is currently no known cure for A-T patients (16).
A-T increases cancer susceptibility [~25% lifetime
risk of cancers (17,18)], which may be related to the
radiosensitivity of A-T resulting in chromosome and chromatid
breaks. Tumors are diverse, particularly those of lymphoid origin.
The patient's personal and family history is important for
identifying and diagnosing tumors. In a retrospective analysis of
279 patients with A-T, 69 had cancer and the majority were
hematological cancers, including 38 cases of non-Hodgkin's lymphoma
(18). In addition, the families of
17 patients had a history of malignant neoplasms, 11 patients had a
family history of recurrent infections and 35 patients had a family
history of immunodeficiency in a clinical and laboratory study of
104 A-T patients in Iran (19). In
addition, it has been found that different mutations express
different amounts of ATM proteins with different enzymatic
activities. The residual ATM kinase activity is the key to
carcinogenesis. Total absence of ATM kinase activity was almost
exclusively associated with the development of lymphoid tumors
during childhood (17). Therefore,
monitoring of ATM kinase activity in A-T patients may contribute to
disease prevention and management.
The treatment of A-T is mainly based on medical
management of immunodeficiencies and respiratory infections,
neurological dysfunction and malignant tumors, supplemented by
neurorehabilitation and nutritional counseling. Supportive
interventions include education on this disease, genetic
counseling, individual and family counseling (20). A-T increases cancer susceptibility.
Similarly, aberrancies of the ATM gene are among the most commonly
occurring somatic mutations in cancer and have generally been
associated with worse prognosis (21). Cancers with deficits in double-stand
DNA repair pathways are sensitive to platinum drugs, which are
known to act by inducing such double-strand DNA breaks.
Conventional chemotherapy regimens for Burkitt leukemia without
platinum drugs may not be suitable for such patients. Novel
therapies that may improve the response to therapy of patients with
ATM-deficient cancers are currently under development, including
PARP inhibitors, agents targeting ATR and nucleoside analogues,
such as sapacitabine, which induces synthetic lethality (21). The specific problems of managing
cancer with A-T are complicated, and treatment should therefore be
performed in academic oncology centers following consultation with
an A-T specialist. Patients with A-T experience increased toxicity
to radio- and chemotherapeutic treatment; therefore, chemotherapy,
radiation therapy and radiomimetic drugs should be administered
with caution (22). Overall, it is
critical that patients and professionals obtain appropriate expert
advice and recommendations from multidisciplinary teams on the
management of this rare condition, as A-T is a multisystemic
disease (23,24).
In conclusion, we herein report a case of A-T with a
hematological malignancy and describe two variants of the ATM gene
in the patient's family. The gene mutation c.6067-c.6068 ins
GAGGGAAGAT in exon 41 (frameshift mutation, p.G2023Gfs*13,
chr11:108115594-108115594) detected in this patient is a novel
mutation in the ATM gene and, to the best of our knowledge, this is
the first report that it may lead to A-T. Detailed medical history,
characteristic clinical manifestations and increasingly improved
exome sequencing techniques and detection techniques of kinase
activity may be beneficial in diagnosing this rare disease,
particularly since the initial phenotype of A-T may be incomplete.
Management of such diseases should be based on multidisciplinary
guidance and other treatment options must be investigated in the
future.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81770178 and
81601528) and the Natural Science Foundation of Hunan Province of
China (grant no. 2015J-J6110).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
FY: Data collection, biomedical discussion and
drafting of the article. LY: Clinical diagnosis and treatment and
follow-up of the patient, manuscript elaboration, revision and
final approval of the version to be submitted. WC:
Histopathological analysis and interpretation of data, revision and
final approval of the version to be submitted. MY: Analysis and
interpretation of data, revision and final approval of the version
to be submitted. MX: Analysis and interpretation of data, revision
and final approval of the version to be submitted. All authors have
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
The legal guardians of both patients provided
written informed consent to the publication of the case
details.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
A-T
|
ataxia-telangiectasia
|
|
MTX
|
methotrexate
|
|
IT
|
intrathecal
|
|
CTX
|
cyclophosphamide
|
|
Ara-C
|
cytosine arabinoside
|
|
PARP
|
poly ADP-ribose polymerase
|
|
ATR
|
ATM-related
|
References
|
1
|
Boder E and Sedgwick RP:
Ataxia-telangiectasia; a familial syndrome of progressive
cerebellar ataxia, oculocutaneous telangiectasia and frequent
pulmonary infection. Pediatrics. 21:526–554. 1958.PubMed/NCBI
|
|
2
|
McKinnon PJ: ATM and the molecular
pathogenesis of ataxia telangiectasia. Annu Rev Pathol. 7:303–321.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gatti RA: Ataxia-telangiectasia. Dermatol
Clin. 13:1–6. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gatti RA: The inherited basis of human
radiosensitivity. Acta Oncol. 40:702–711. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Swift M, Sholman L, Perry M and Chase C:
Malignant neoplasms in the families of patients with
ataxia-telangiectasia. Cancer Res. 36:209–215. 1976.PubMed/NCBI
|
|
6
|
Huang Y, Yang L, Wang J, Yang F, Xiao Y,
Xia R, Yuan X and Yan M: Twelve novel Atm mutations identified in
Chinese ataxia telangiectasia patients. Neuromolecular Med.
15:536–540. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jeddane L, Ailal F, Dubois-d'Enghien C,
Abidi O, Benhsaien I, Kili A, Chaouki S, Kriouile Y, El Hafidi N,
Fadil H, et al: Molecular defects in Moroccan patients with
ataxia-telangiectasia. Neuromolecular Med. 15:288–294. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rothblum-Oviatt C, Wright J, Lefton-Greif
MA, McGrath-Morrow SA, Crawford TO and Lederman HM: Ataxia
telangiectasia: A review. Orphanet J Rare Dis. 11:1592016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Su Y and Swift M: Mortality rates among
carriers of ataxia-telangiectasia mutant alleles. Ann Intern Med.
133:770–778. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Teive HA, Moro A, Moscovich M, Arruda WO,
Munhoz RP, Raskin S and Ashizawa T: Ataxia-telangiectasia-A
historical review and a proposal for a new designation: ATM
syndrome. J Neurol Sci. 355:3–6. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chun HH and Gatti RA:
Ataxia-telangiectasia, an evolving phenotype. DNA Repair (Amst).
3:1187–1196. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gatti RA, Becker-Catania S, Chun HH, Sun
X, Mitui M, Lai CH, Khanlou N, Babaei M, Cheng R, Clark C, et al:
The pathogenesis of ataxia-telangiectasia. Learning from a Rosetta
Stone. Clin Rev Allergy Immunol. 20:87–108. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nowak-Wegrzyn A, Crawford TO, Winkelstein
JA, Carson KA and Lederman HM: Immunodeficiency and infections in
ataxia-telangiectasia. J Pediatr. 144:505–511. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Krauthammer A, Lahad A, Goldberg L, Sarouk
I, Weiss B, Somech R, Soudack M and Pessach IM: Elevated IgM levels
as a marker for a unique phenotype in patients with Ataxia
telangiectasia. BMC Pediatr. 18:1852018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mitui M, Bernatowska E, Pietrucha B,
Piotrowska-Jastrzebska J, Eng L, Nahas S, Teraoka S, Sholty G,
Purayidom A, Concannon P and Gatti RA: ATM gene founder haplotypes
and associated mutations in Polish families with
ataxia-telangiectasia. Ann Hum Genet. 69:657–664. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nakamura K, Du L, Tunuguntla R, Fike F,
Cavalieri S, Morio T, Mizutani S, Brusco A and Gatti RA: Functional
characterization and targeted correction of ATM mutations
identified in Japanese patients with ataxia-telangiectasia. Hum
Mutat. 33:198–208. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Reiman A, Srinivasan V, Barone G, Last JI,
Wootton LL, Davies EG, Verhagen MM, Willemsen MA, Weemaes CM, Byrd
PJ, et al: Lymphoid tumours and breast cancer in ataxia
telangiectasia; substantial protective effect of residual ATM
kinase activity against childhood tumours. Br J Cancer.
105:586–591. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Suarez F, Mahlaoui N, Canioni D,
Andriamanga C, d'Enghien Dubois C, Brousse N, Jais JP, Fischer A,
Hermine O and Stoppa-Lyonnet D: Incidence, presentation, and
prognosis of malignancies in ataxia-telangiectasia: A report from
the French national registry of primary immune deficiencies. J Clin
Oncol. 33:202–208. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Moin M, Aghamohammadi A, Kouhi A,
Tavassoli S, Rezaei N, Ghaffari SR, Gharagozlou M, Movahedi M,
Purpak Z, Ghazi Mirsaeid B, et al: Ataxia-telangiectasia in Iran:
Clinical and laboratory features of 104 patients. Pediatr Neurol.
37:21–28. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Perlman S, Becker-Catania S and Gatti RA:
Ataxia-telangiectasia: Diagnosis and treatment. Semin Pediatr
Neurol. 10:173–182. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Choi M, Kipps T and Kurzrock R: ATM
mutations in cancer: Therapeutic implications. Mol Cancer Ther.
15:1781–1791. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schoenaker MH, Suarez F, Szczepanski T,
Mahlaoui N and Loeffen JL: Treatment of acute leukemia in children
with ataxia telangiectasia (A-T). Eur J Med Genet. 59:641–646.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
van Os NJH, Haaxma CA, van der Flier M,
Merkus PJFM, van Deuren M, de Groot IJM, Loeffen J, van de
Warrenburg BPC and Willemsen MAAP; A-T Study Group, :
Ataxia-telangiectasia: Recommendations for multidisciplinary
treatment. Dev Med Child Neurol. 59:680–689. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bhatt JM, Bush A, van Gerven M, Nissenkorn
A, Renke M, Yarlett L, Taylor M, Tonia T, Warris A, Zielen S, et
al: ERS statement on the multidisciplinary respiratory management
of ataxia telangiectasia. Eur Respir Rev. 24:565–581. 2015.
View Article : Google Scholar : PubMed/NCBI
|