Introduction
Hypoferraemia is part of the inflammatory response
(1). It is considered to be a
defence mechanism of the host that prevents pathogenic organisms
from gaining access to iron (2).
Tumour necrosis factor (TNF) has been viewed as being one of the
inflammatory cytokines that contributes considerably to the
generation of hypoferraemia and anaemia of inflammation (AI). TNF
has been shown to affect iron metabolism in murine models, in
humans and in vitro. The experimental administration of TNF
reportedly induced hypoferraemia (3) and anaemia with low serum iron and
preserved iron stores in mice (4).
A combination of interferon-γ (IFN-γ) and TNF was found to induce
hypoferraemia, decrease transferrin and soluble transferrin
receptor levels and increase ferritin levels in the sera of cancer
patients treated with these cytokines by isolated limb perfusion
(5). Iron release from macrophages
was reported to be inhibited by TNF in vitro(6). Previous findings demonstrated that
TNF regulates key molecules engaged in iron import and export in
the small bowel independently of hepcidin (3,7). TNF
stimulation was found to upregulate the cellular iron import
protein divalent metal transporter 1 (DMT1) and reduce the iron
exporter ferroportin 1 (Fp1) in a human monocytic cell line
(8), resulting in increased iron
sequestration.
The reported findings demonstrate that TNF, by
regulating several molecules crucial to iron absorption and iron
recycling, has a considerable impact on iron metabolism. In this
study, we investigated whether TNF contributes to the generation of
hypoferraemia during the early inflammatory response in a murine
model of protracted peritonitis in vivo. For this purpose,
we studied TNF-deficient and wild-type mice undergoing caecal
ligation and puncture (CLP).
Materials and methods
Mice
Female or male C57/BL6 mice (25–30 g) were bred at
the animal facility of the University of Regensburg, Germany.
TNF-deficient mice were generated by Körner et al(9), raised at the University of Erlangen,
Germany, and provided by H. Körner. The animals were kept under
standard conditions at the animal facility of the University of
Regensburg. Experiments were performed according to federal
guidelines for animal experimentation. Animals in the control and
treated groups were age- and gender-matched.
CLP
The mice were anaesthetised by i.p. injection of 75
mg/kg Ketanest® (Parke, Davis & Co., Munich,
Germany) and 16 mg/kg Rompun® (Bayer AG, Leverkusen,
Germany). The caecum was exteriorised and its distal end was
ligated and punctured to achieve a sublethal CLP as described in a
previous study (10).
Serum iron measurement and histology
The serum iron concentration was measured using the
ADVIA™ 1650 chemistry system (Bayer Diagnostics, Fernwald,
Germany). To obtain serum, groups of mice (n=5) were anaesthetised
and blood was drawn 8 h after CLP. The mice were subsequently
sacrificed by cervical dislocation and the spleen, liver and small
bowel were either harvested for histological analysis or snap
frozen in liquid nitrogen. For histology, mouse tissue was fixed in
4% buffered formalin overnight. Formalin-fixed tissue was
dehydrated and embedded in paraffin. Paraffin sections of 3–5 μm
were mounted on SuperFrost Plus slides, heated for 20 min at 72°C,
deparaffinised and rehydrated. Following antigen retrieval in a
citrate buffer (pH 7.3) with microwave irradiation for 30 min at
240 W, the slides were rinsed and endogenous peroxidase activity
was blocked with methanolic peroxide. The slides were rinsed again
and primary monoclonal antibodies against hemoxygenase 1 (Abcam,
Cambridge, UK), NRAMP (Alpha Diagnostic International, San Antonio,
TX, USA) or ferritin (Abcam), respectively, were applied in a
dilution of 1:100. The slides were incubated using a Ventana
machine (Ventana Medical Systems basic DAB detection kit; Ventana
Medical Systems Inc., Tucson, AZ, USA). The
streptavidin-biotin-peroxidase method was used for visualisation.
Immunohistochemical stainings for hepcidin (Alpha Diagnostic
International) and Fp1 (provided by M. Knutson) were applied in a
dilution of 1:200 following antigen retrieval in a citrate buffer.
To evaluate iron stores, the sections were stained by Perls’ acid
ferrocyanide reaction.
Isolation of RNA
Total cellular RNA was isolated using the RNeasy
Mini kit including an RNase-free DNase set according to the
manufacturer’s instructions (Qiagen, Hilden, Germany). RNA was
quantified using a fluorescence microplate reader and by following
the instructions of the RiboGreen® RNA quantitation
reagent and kit (Mobitec, Göttingen, Germany).
Quantification of mRNA expression by
real-time PCR
One-tenth of the RNA recovered from RNeasy spin
columns (Qiagen) was used for reverse transcription PCR. cDNA
fragments of hepcidin and Fp1 were amplified using the primers
designed by Laftah et al(3). Reverse transcription was performed in
a 20 μl reaction mix of 5 μl RNA, 4 μl 5X SuperScript™ buffer
(Invitrogen, Carlsbad, CA, USA), 2 μl DTT (0.1 M), 1 μl antisense
dN6 primer (1 μg/μl), 1 μl dNTPs (10 mM) and 1 μl reverse
transcriptase (SuperScript™, Invitrogen). The mixture was incubated
at 46°C for 45 min followed by 10 min at 70°C and RNase A digestion
at 37°C for 30 min. To precisely quantify the expression of mRNA,
the real-time PCR LightCycler® system (Roche
Diagnostics, Mannheim, Germany) was used. For PCR, 1–3 μl cDNA
preparation, 2.4 μl 25 mM MgCl2, 0.5 μM of forward and
reverse primer and 2 μl of SYBR Green LightCycler® Mix
in a total of 20 μl were applied. The following PCR programme was
performed: 60 sec at 95°C (initial denaturation); 20°C/sec
temperature transition rate up to 95°C for 15 sec, 10 sec at 58°C,
22 sec at 72°C and 10 sec at 82°C (acquisition mode single),
repeated 40 times (amplification). The MgCl2
concentration and annealing temperature were optimised for each
primer set. The PCR was evaluated by melting curve analysis
following the manufacturer’s instructions and checking the PCR
products on 1.8% agarose gels. Each quantitative PCR was performed
at least in duplicate for two sets of RNA preparations.
ELISA assay
The sera of 4 animals per time point were pooled and
maintained at −20°C until analysis. TNF concentrations were tested
using a sandwich ELISA assay (R&D Systems, Wiesbaden, Germany).
Absorption was measured at 405 nm and evaluated using
SoftMax® software.
Statistical analysis
P-values were determined using the Student’s t-test.
P<0.05 was considered to indicate a statistically significant
result.
Results
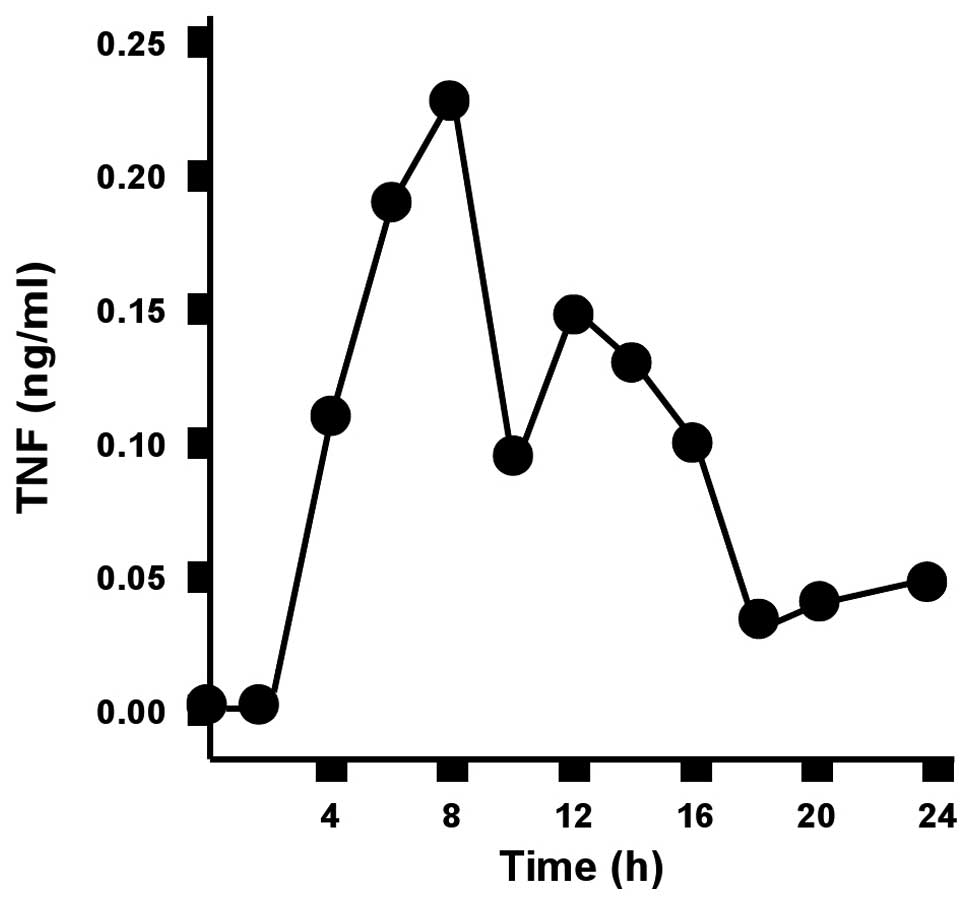
The serum concentration of TNF peaks 8 h
after CLP
The serum concentration of TNF in wild-type mice was
measured over a period of 24 h after CLP. The TNF concentration
increased rapidly within the first few hours and peaked 8 h after
CLP. The TNF concentration subsequently decreased to almost
baseline levels (Fig. 1).
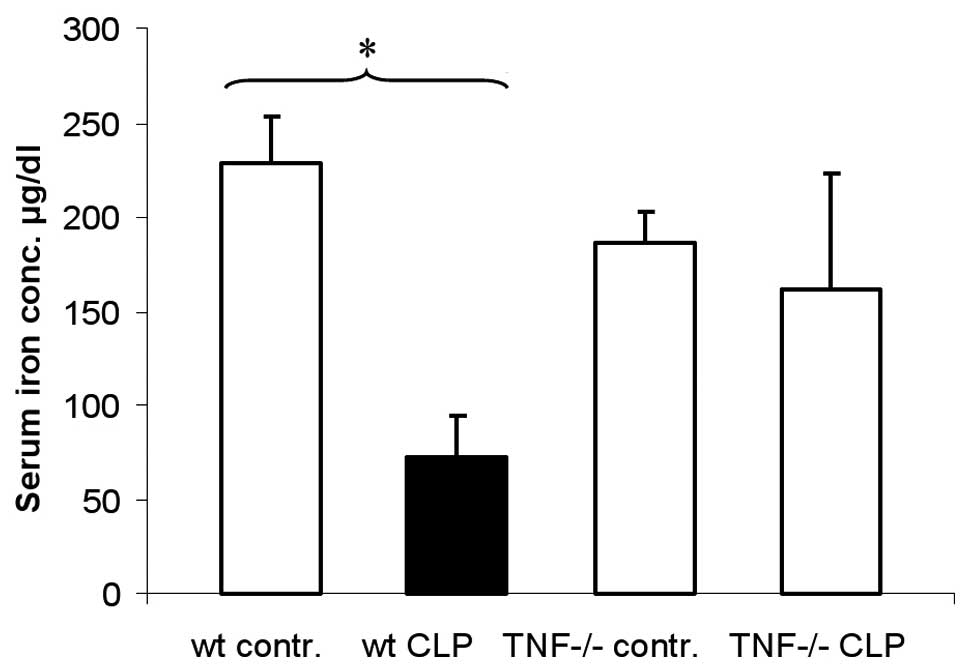
Hypoferraemia does not develop in
TNF-deficient mice 8 h after CLP
To test the role of TNF in the development of
hypoferraemia, TNF-deficient mice were used to study the serum iron
concentration following CLP. Wild-type mice showed a significant
decrease in serum iron concentration 8 h after CLP (Fig. 2). The serum iron concentration of
CLP-treated TNF-deficient animals did not differ significantly from
the serum iron concentrations of the untreated control and
untreated TNF-deficient animals.
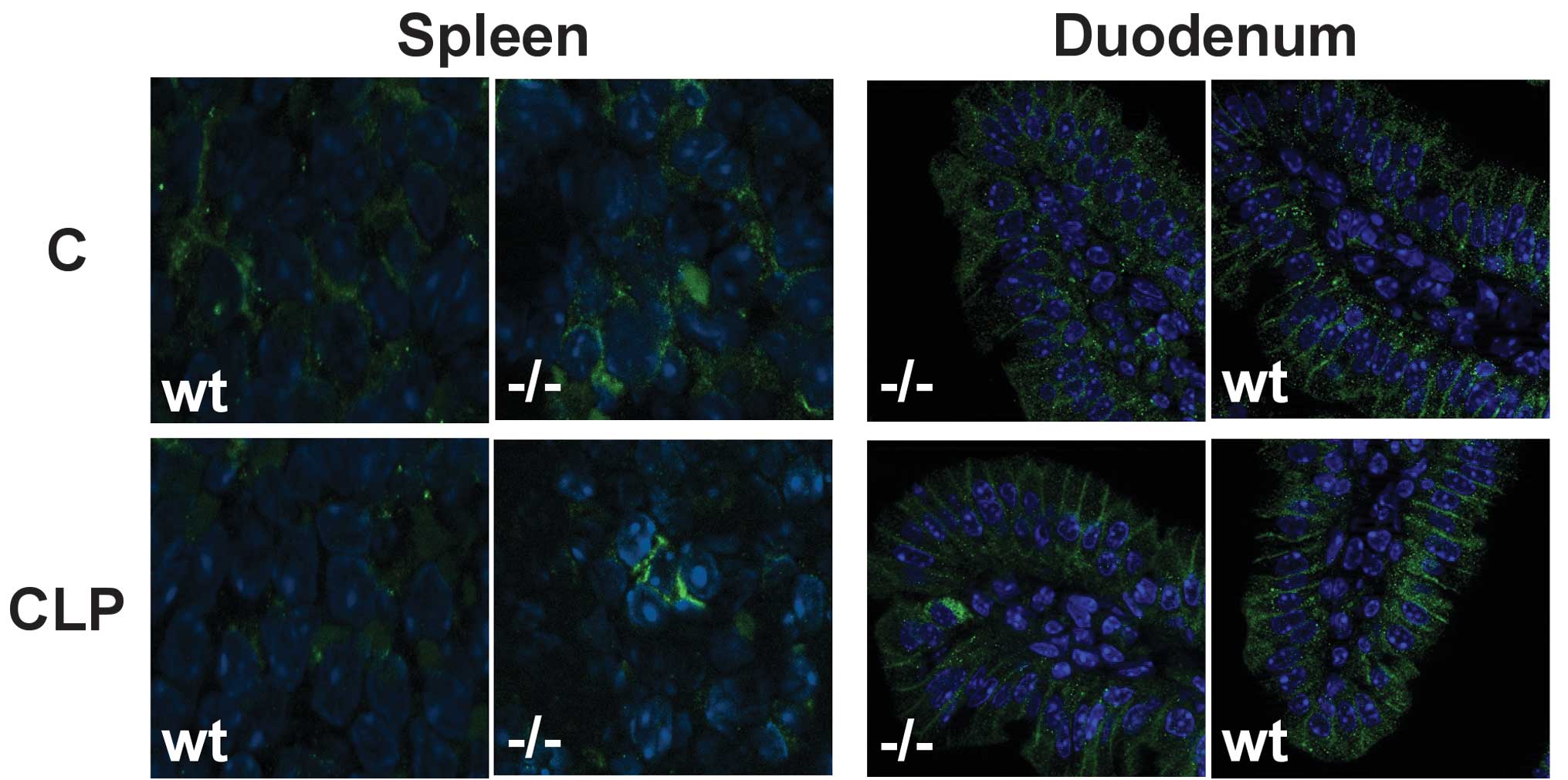
Hypoferraemia is accompanied by the
downregulation of Fp1 in wild-type mice
Immunofluorescent staining of hepatic and splenic
tissue revealed that the iron exporter Fp1 was downregulated in
Kupffer cells and splenic macrophages in wild-type mice 8 h after
CLP, whereas in enterocytes of the duodenum, the membrane staining
pattern for Fp1 was unaffected (Fig.
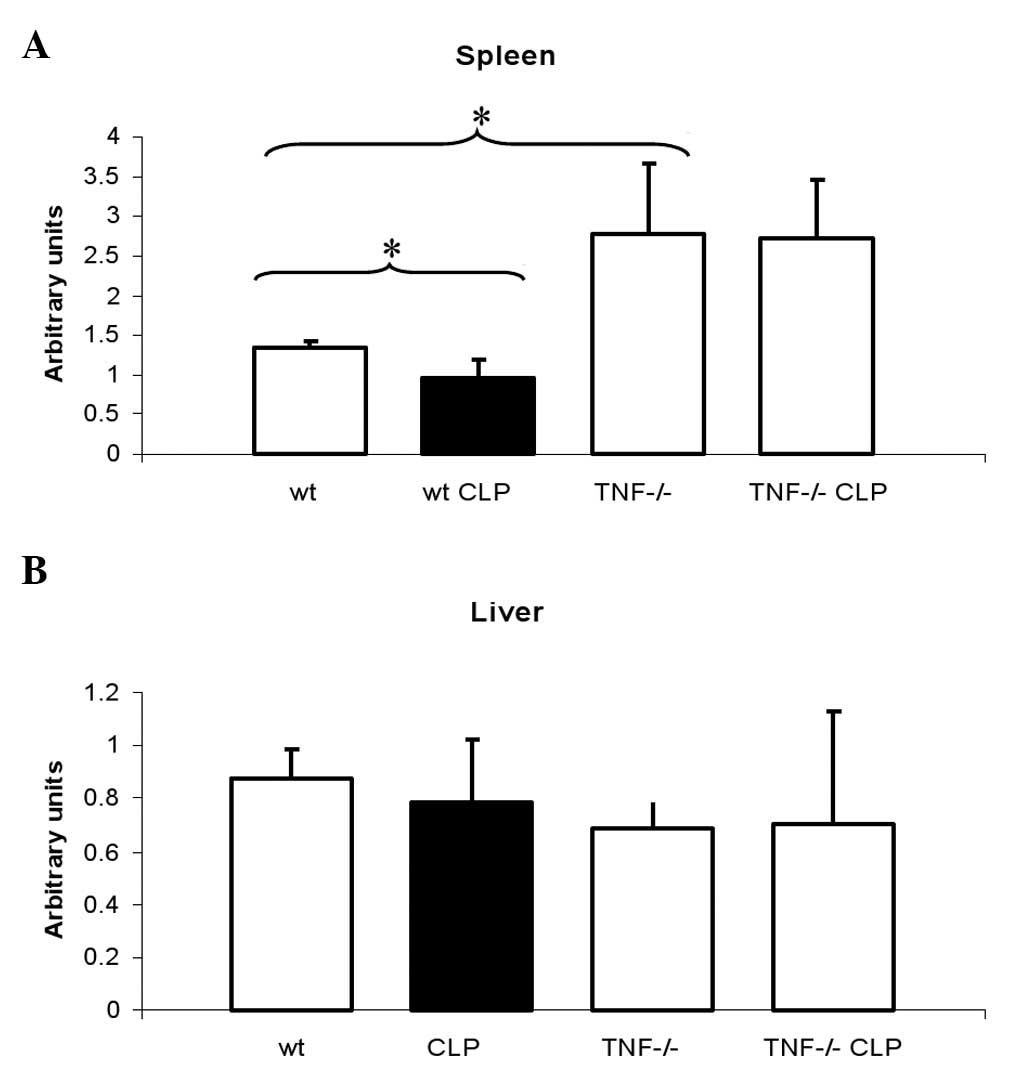
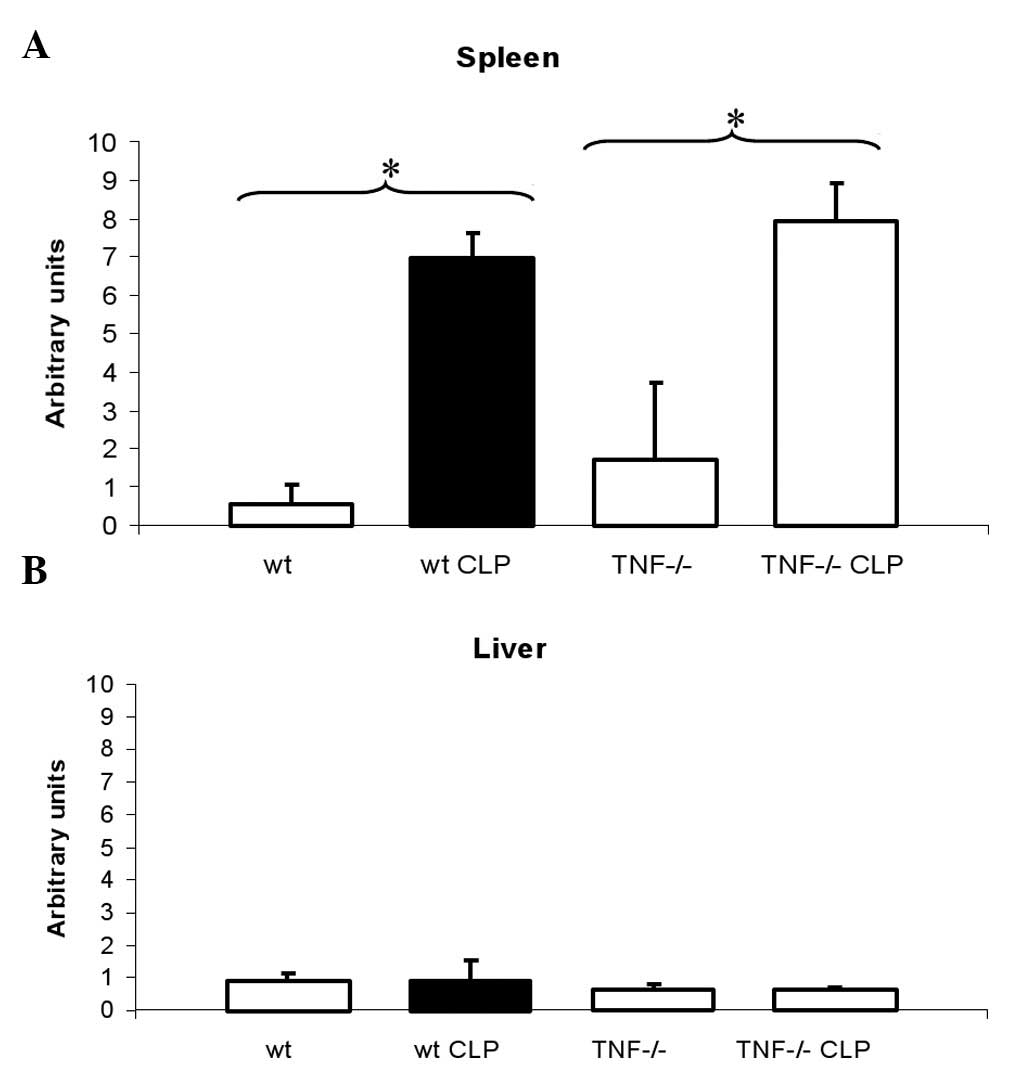
3). In agreement with this finding, the mRNA level of Fp1 was
decreased in mice following CLP compared with untreated animals
(Fig. 4A). These changes were
accompanied by an increase in cytoplasmic ferritin expression in
Kupffer cells and macrophages after CLP as revealed by the
immunohistochemical analysis (data not shown). Immunohistochemical
analysis for DMT1 and hemoxygenase 1 did not reveal any differences
between the treated and untreated animals or between the wild-type
and TNF-deficient mice (data not shown).
Fp1 is not downregulated in the absence
of TNF-activity following CLP
In TNF-deficient mice, immunofluorescent staining
for Fp1 after CLP exhibited membrane patterns similar to those of
untreated mice (Fig. 3).
Consequently, no changes in Fp1 mRNA expression were observed. Fp1
mRNA levels were equally increased in the untreated TNF-deficient
mice and in the TNF-deficient mice after CLP compared with the
wild-type mice (Fig. 4A).
No differences were found in the expression of Fp1
mRNA in the liver between the treated and untreated or wild-type
and TNF-deficient mice (Fig.
4B).
Hepatic hepcidin expression is not
increased 8 h after CLP in either wild-type or TNF-deficient
mice
The splenic and hepatic hepcidin expression was
determined in untreated wild-type and TNF-deficient mice 8 h after
CLP by quantitative RT-PCR. An initial increase in the splenic
hepcidin expression was observed in wild-type and TNF-deficient
mice following CLP (Fig. 5A). The
hepcidin mRNA levels were not increased 6 h after CLP (data not
shown). Hepatic hepcidin expression was equally low in all groups 8
h after CLP, indicating that hepatic hepcidin had not increased
(Fig. 5B). Immunohistochemically
stained sections of the liver revealed that hepcidin peptide was
not detectable in hepatocytes, Kupffer cells or the spleen (data
not shown).
Discussion
Various effects of TNF on iron metabolism have been
described in the literature. TNF, as a mediator of inflammation, is
thought to affect iron metabolism, similar to IL-6 and other
cytokines, in order to deprive pathogens of access to iron.
However, the precise mechanism by which TNF impacts iron metabolism
and the phase of inflammation in which this occurs are unknown.
Experimental administration of TNF to animals
results in a blockade of iron recycling, iron absorption and
hypoferraemia. High concentrations of TNF achieved by the
administration of TNF-secreting tumour cells were found to induce
anaemia, with a low serum iron concentration, and preserved iron
stores in nude mice (4). The
induction of hypoferraemia using low doses of TNF in mice has also
been described by other authors (6,11).
Laftah et al demonstrated that the administration of TNF
causes hypoferraemia associated with reduced intestinal iron
absorption in mice (3). The
concentrations used in this study equal the serum concentrations of
TNF that we measured after CLP (Fig.
1). After CLP, we observed a rapid increase in TNF serum
levels, peaking 8 h after CLP. At the same time point, marked
hypoferraemia was observed in wild-type mice. To test whether
hypoferraemia was mediated by TNF, we studied TNF-deficient mice
following CLP. The data demonstrated that TNF-deficient mice did
not develop hypoferraemia during the early inflammatory response.
During CLP-induced hypoferraemia in wild-type mice, the expression
levels of the iron importer DMT1 and hemoxygenase 1 were unchanged
compared with those of the untreated animals. The membrane
expression of the iron exporter Fp1 in splenic macrophages and
Kupffer cells was reduced in wild-type mice following CLP whereas
in the TNF-deficient mice, the membrane staining pattern of the
macrophages was maintained. Fp1 membrane expression was not reduced
in the duodenum of either the wild-type or TNF-deficient mice.
These findings indicate that hypoferraemia is achieved by a
TNF-induced downregulation of Fp1, the only known iron exporter
protein in macrophages.
It has previously been reported that hepcidin, an
iron regulatory peptide, regulates the degradation of Fp1 (12). During inflammatory conditions,
hepcidin is produced in the liver and secreted into the blood. It
binds to Fp1 located in the cell membranes of macrophages and
enterocytes. Subsequently, Fp1 is internalised and degraded
(12). The decrease of Fp1 in
macrophages and enterocytes results in reduced iron efflux and the
blockade of iron recycling as well as iron resorption (13). Macrophages and polymorphonuclear
leukocytes constitute other sources of hepcidin (14). The endogenous expression of
hepcidin in macrophages is mediated by TLR-4 and causes the
internalisation of Fp1 in an autocrine manner (14). Hepcidin expression in wild-type and
TNF-deficient mice after CLP was studied to determine whether the
TNF-induced downregulation of Fp1 is mediated by hepcidin. It
appears unlikely that systemically available hepcidin causes the
downregulation of Fp1 since the hepatic expression of hepcidin did
not increase after CLP. Furthermore, the internalisation of Fp1 was
not observed in duodenal enterocytes, indicating that the autocrine
production of hepcidin may be the cause of hypoferraemia rather
than a systemic hepcidin effect. The expression of hepcidin in
splenic macrophages and Kupffer cells 8 h after CLP was detectable
at the mRNA level but not at the protein level, negating the
possibility that hepcidin is involved in mediating the
downregulation of Fp1 by TNF. No difference was observed in
hepcidin expression between the wild-type and TNF-deficient mice.
TNF has marked hepcidin-independent effects on iron metabolism.
Laftah et al reported that the application of TNF suppresses
rather than stimulates hepcidin expression in the spleens and
livers of CD1 mice and that TNF induces iron sequestration in the
spleen and inhibits duodenal iron transfer independently of
hepcidin (3). The same authors
described a direct effect of TNF on the iron import and export of
enterocytes, which was associated with a decrease in DMT1 and Fp1
expression (7). The rapid action
of TNF on serum iron levels within 3 h after application reported
by Laftah et al(3)
apparently mediates hypoferraemia in the early inflammatory
response before hepcidin production in the liver increases
(Fig. 4). Moreover, for other
models of inflammation it has been reported that hepcidin
expression in the liver is not increased at this time point
(15), indicating that it is
unlikely that systemically available hepcidin is a mediator of
hypoferraemia during the early inflammatory response. This is in
line with our data, indicating that the TNF-induced immediate
hypoferraemia following CLP is not mediated by hepcidin and that
the downregulation of Fp1 must be achieved by a different
mechanism.
In untreated and CLP-treated TNF-deficient mice, an
enhanced Fp1 expression was observed at the mRNA and protein levels
compared with wild-type mice, indicating that TNF regulates the Fp1
expression by transcriptional control. In wild-type mice, a
decrease of Fp1 mRNA was observed after CLP. Downregulation of Fp1
by TNF was previously reported in human endothelial cells (16). LPS, a potent stimulator of TNF
production, also suppresses Fp1 expression in macrophages, cultured
splenic cells and in the mouse spleen (8,17,18).
The enhanced expression of Fp1 in untreated mice in the absence of
TNF suggests that TNF regulates the transcription of Fp1 not only
during inflammation, but also under physiological conditions.
Previously, we investigated the contribution of TNF
to the different aspects of AI in vivo in the CLP model
(19). Our results demonstrated
that AI may develop during protracted inflammation in the absence
of TNF two weeks after CLP. The role of TNF in the mediation of the
chronic phase of AI is therefore questionable. Since TNF does not
induce hepcidin production (3), it
is unlikely that TNF is a mediator of hypoferraemia in the later
inflammatory phase which is typically regulated by IL-6 and
hepcidin (20). This explains the
reason for hypoferraemia and AI developing in the absence of TNF
during the chronic phase of inflammation. However, in the early
inflammatory response TNF appears to exert its well-described
effects on iron metabolism and is necessary for the induction of
hypoferraemia prior to the onset of hepcidin production.
In summary, our findings suggest that TNF is a
mediator of hypoferraemia during the early inflammatory response by
regulating the expression of Fp1 in macrophages independently of
hepcidin.
Acknowledgements
The technical support of Simone Kaufmann is
gratefully acknowledged.
References
|
1
|
Weiss G: Modification of iron regulation
by the inflammatory response (Review). Best Pract Res Clin
Haematol. 18:183–201. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schaible UE and Kaufmann SH: Iron and
microbial infection. Nat Rev Microbiol. 2:946–953. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Laftah AH, Sharma N, Brookes MJ, McKie AT,
Simpson RJ, Iqbal TH and Tselepis C: Tumour necrosis factor alpha
causes hypoferraemia and reduced intestinal iron absorption in
mice. Biochem J. 397:61–67. 2006. View Article : Google Scholar
|
|
4
|
Johnson RA, Waddelow TA, Caro J, Oliff A
and Roodman GD: Chronic exposure to tumor necrosis factor in vivo
preferentially inhibits erythropoiesis in nude mice. Blood.
74:130–138. 1989.PubMed/NCBI
|
|
5
|
Feelders RA, Vreugdenhil G, Eggermont AM,
Kuiper-Kramer PA, van Eijk HG and Swaak AJ: Regulation of iron
metabolism in the acute-phase response: interferon gamma and tumour
necrosis factor alpha induce hypoferraemia, ferritin production and
a decrease in circulating transferrin receptors in cancer patients.
Eur J Clin Invest. 28:520–527. 1998. View Article : Google Scholar
|
|
6
|
Alvarez-Hernández X, Licéaga J, McKay IC
and Brock JH: Induction of hypoferremia and modulation of
macrophage iron metabolism by tumor necrosis factor. Lab Invest.
61:319–322. 1989.PubMed/NCBI
|
|
7
|
Sharma N, Laftah AH, Brookes MJ, Cooper B,
Iqbal T and Tselepis C: A role for tumour necrosis factor alpha in
human small bowel iron transport. Biochem J (Pt 2). 390:437–446.
2005.PubMed/NCBI
|
|
8
|
Ludwiczek S, Aigner E, Theurl I and Weiss
G: Cytokine-mediated regulation of iron transport in human
monocytic cells. Blood. 101:4148–4154. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Körner H, Cook M, Riminton DS, Lemckert
FA, Hoek RM, Ledermann B, Köntgen F, Fazekas de St Groth B and
Sedgwick JD: Distinct roles for lymphotoxin-alpha and tumor
necrosis factor in organogenesis and spatial organization of
lymphoid tissue. Eur J Immunol. 27:2600–2609. 1997.PubMed/NCBI
|
|
10
|
Schubert TE, Echtenacher B, Hofstädter F
and Männel DN: Failure of interferon-γ and tumor necrosis factor in
mediating anemia of chronic disease in a mouse model of protracted
septic peritonitis. Int J Mol Med. 16:753–758. 2005.
|
|
11
|
Tanaka T, Araki E, Nitta K and Tateno M:
Recombinant human tumor necrosis factor depresses serum iron in
mice. J Biol Response Mod. 6:484–488. 1987.PubMed/NCBI
|
|
12
|
Nemeth E, Tuttle MS, Powelson J, Vaughn
MB, Donovan A, Ward DM, Ganz T and Kaplan J: Hepcidin regulates
cellular iron efflux by binding to ferroportin and inducing its
internalization. Science. 306:2090–2093. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rivera S, Nemeth E, Gabayan V, Lopez MA,
Farshidi D and Ganz T: Synthetic hepcidin causes rapid
dose-dependent hypoferremia and is concentrated in
ferroportin-containing organs. Blood. 106:2196–2199. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Peyssonnaux C, Zinkernagel AS, Datta V,
Lauth X, Johnson RS and Nizet V: TLR4-dependent hepcidin expression
by myeloid cells in response to bacterial pathogens. Blood.
107:3727–3732. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nicolas G, Chauvet C, Viatte L, Danan JL,
Bigard X, Devaux I, Beaumont C, Kahn A and Vaulont S: The gene
encoding the iron regulatory peptide hepcidin is regulated by
anemia, hypoxia, and inflammation. J Clin Invest. 110:1037–1044.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nanami M, Ookawara T, Otaki Y, Ito K,
Moriguchi R, Miyagawa K, Hasuike Y, Izumi M, Eguchi H, Suzuki K and
Nakanishi T: Tumor necrosis factor-alpha-induced iron sequestration
and oxidative stress in human endothelial cells. Arterioscler
Thromb Vasc Biol. 25:2495–2501. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu XB, Nguyen NB, Marquess KD, Yang F and
Haile DJ: Regulation of hepcidin and ferroportin expression by
lipopolysaccharide in splenic macrophages. Blood Cells Mol Dis.
35:47–56. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yang F, Liu XB, Quinones M, Melby PC, Ghio
A and Haile DJ: Regulation of reticuloendothelial iron transporter
MTP1 (Slc11a3) by inflammation. J Biol Chem. 277:39786–39791. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schubert T, Echtenacher B, Hofstädter F
and Männel DN: TNF-independent development of transient anemia of
chronic disease in a mouse model of protracted septic peritonitis.
Lab Invest. 83:1743–1750. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ganz T and Nemeth E: Iron sequestration
and anemia of inflammation. Semin Hematol. 46:387–393. 2009.
View Article : Google Scholar : PubMed/NCBI
|