Introduction
Atrial fibrillation (AF) is the most common type of
cardiac arrhythmia encountered in clinical practice, responsible
for ~1/3 of hospitalizations for cardiac arrhythmias. This
condition shows a marked increase in prevalence with advancing age,
ranging from ~0.4% of the whole population to ~10% of the
octogenarian population (1,2).
According to the Framingham Heart Study (3), during the lifetime of subjects >40
years of age, there is a ~25% risk for the development of AF. The
chaotic heart rhythm is not merely associated with a variety of
symptoms, such as palpitations, dizziness, syncope or shortness of
breath, but is also accountable for significantly increased
morbidity and mortality (1). In
comparison with individuals in sinus rhythm, patients with AF have
a 6-fold increase in the risk of stroke, and >15% of all strokes
are ascribed to AF (4). Notably,
the risk of AF-related thromboembolism also significantly increases
with age, rising from 1.5% at the age of 50–59 years to 23.5% at
the age of 80–89 years (4). The
incidence of death is estimated to have doubled among patients with
AF compared with individuals with normal heart rhythm (5). AF also contributes to degraded
quality of life, compromised exercise performance, impaired
cognitive function or dementia, tachycardia-induced cardiomyopathy,
and left ventricular dysfunction or even congestive heart failure,
inflicting a large economic burden on the National Healthcare
Systems worldwide (6). Despite the
significant prevalence and therapeutic challenge, the molecular
mechanisms involved in the pathogenesis of AF remain poorly
understood.
Traditionally, AF has been considered as a
complication derived from miscellaneous adverse cardiac or systemic
conditions, including hypertension, coronary artery disease,
rheumatic heart disease, valvular heart disease, pulmonary heart
disease, cardiomyopathy, cardiac surgery, pericarditis, congestive
heart failure, type 2 diabetes mellitus, obstructive sleep apnea,
hyperthyroidism and electrolyte imbalance (1,6–10).
However, in 30–45% of AF patients, no underlying causes are
identified by routine procedures, where AF is termed ‘idiopathic’
or ‘lone’ (1), and ≥15% of AF
patients have a positive family history, a condition defined as
familial AF (11). Mounting
evidence has substantiated the familial aggregation of AF and
enhanced susceptibility to AF in the close relatives of patients
with AF, suggesting an important genetic basis for AF (12–18).
Genome-wide linkage analyses with polymorphic microsatellite
markers mapped susceptibility loci for AF on human chromosomes
10q22, 6q14–16, 11p15.5, 5p13 and 5p15, of which AF-causing
mutations in 2 genes, including KCNQ1 on chromosome 11p15.5
and NUP155 on chromosome 5p13, were identified and
functionally characterized (19–24).
Genetic scan of candidate genes unveiled a long list of
AF-associated genes, including KCNE2, KCNE3,
KCNE5, KCNH2, KCNJ2, KCNJ8,
KCNA5, SCN5A, NPPA, GATA4, GATA5
and GATA6(25–41). Nevertheless, AF is a genetically
heterogeneous disorder and the genetic determinants for AF in the
majority of patients remain to be identified (11).
A previous study has underscored the essential roles
of gap junction channels in heart electrophysiology, particularly
in cardiac action potential propagation (42). Gap junctions are intercellular
channels responsible for the exchange of ions and small molecules
between adjacent cells. The functional gap junction channel is
composed of two hemichannels, known as connexons, one provided by
each cell. Connexons are hexamers of membrane-spanning proteins
called connexins. At present, >20 connexin genes have been
identified in mouse and human (43). In the human heart, myocardial gap
junctions are constructed mainly by the connexin isoforms 40, 43
and 45. Connexin40, also designated gap junction protein α 5
(GJA5), is selectively expressed in the atrial myocytes,
atrioventricular node, His-bundle and ventricular conduction system
(Purkinje fibers), and is crucial in the electrical synchronization
of the atrium and the rapid conduction of impulses in the
His-Purkinje (44). In
GJA5-deficient mice, spontaneous or inducible arrhythmias as well
as conduction abnormalities have been observed (45). In the goat, alterations in
expression levels and the distribution pattern of atrial GJA5 may
constitute a cell substrate underlying susceptibility and
perpetuation of AF (46). In
human, cardiac GJA5 remodeling may lead to abnormal electrical
coupling, forming an electrophysiological matrix with potential
arrhythmogenic effect (47). By
reducing GJA5 protein levels, several closely linked polymorphisms
in the promoter region of the GJA5 gene have been strongly
associated with enhanced atrial vulnerability and increased risk
for lone AF (48–52). Furthermore, multiple somatic and
germline mutations in GJA5 have been reported to underlie AF
(53–55). These findings provide a rationale
to scan GJA5 as a logical candidate gene for AF.
In this study, sequence analysis of the GJA5
gene was performed in a cohort of 310 unrelated patients with lone
AF in contrast to a total of 200 ethnically matched, unrelated
healthy individuals, in order to evaluate the prevalence and
spectrum of GJA5 mutations associated with lone AF.
Materials and methods
Study subjects
A cohort of 310 unrelated patients with lone AF were
included in this study from the Chinese Han population. The
available relatives of the probands were also included. A total of
200 unrelated healthy individuals matched for age, gender and race
were included as controls. Peripheral venous blood specimens were
prepared and clinical data including medical records,
electrocardiogram and echocardiography reports were collected. The
study subjects were clinically classified using a consistently
applied set of definitions (11).
Briefly, diagnosis of AF was performed by a standard 12-lead
electrocardiogram demonstrating no P-waves and irregular R-R
intervals irrespective of clinical symptoms. Lone AF was defined as
AF occurring in individuals <60 years of age without other
cardiac or systemic diseases by physical examination,
electrocardiogram, transthoracic echocardiogram and extensive
laboratory tests. Relatives with AF occurring at any age in the
setting of structural heart disease (hypertensive, ischemic,
myocardial or valvular) were classified as ‘undetermined’ for
having an inherited form of AF. The ‘undetermined’ classification
was also used when documentation of AF on an electrocardiogram
tracing was absent in relatives with symptoms consistent with AF
(palpitations, dyspnea and light-headedness), or when a screening
electrocardiogram and echocardiogram were not performed, regardless
of the symptoms. Relatives were classified as ‘unaffected’ when
they were asymptomatic and had a normal electrocardiogram. In
addition, paroxysmal AF was defined as AF lasting >30 sec that
terminated spontaneously. Persistent AF was defined as AF lasting
>7 days and requiring either pharmacologic therapy or electrical
cardioversion for termination. AF that was refractory to
cardioversion or that was allowed to continue was classified as
permanent (1). The study protocol
was reviewed and approved by the local Institutional Ethics
Committee and written informed consent was obtained from all the
participants prior to investigation.
Genetic studies
Genomic DNA from the participants was extracted from
blood lymphocytes with the Wizard® Genomic DNA
Purification kit (Promega, Madison, WI, USA). The candidate gene
GJA5 was screened in 310 unrelated patients with lone AF and
genotyping of GJA5 in the relatives of mutation carriers and
200 unrelated control individuals was subsequently performed for
the presence of mutations identified in index patients. The
referential genomic DNA sequence of GJA5 was derived from
GenBank (accession number: NG_009369). With the aid of on-line
Primer3 software (http://frodo.wi.mit.edu), the primer pairs used to
amplify the complete coding region and splice junctions of
GJA5 by polymerase chain reaction (PCR) were designed as
previously described (54,55). PCR was performed using HotStar Taq
DNA Polymerase (Qiagen, Hilden, Germany) on a Veriti®
Thermal Cycler (Applied Biosystems, Foster, CA, USA) with standard
conditions and concentrations of reagents. Amplified products were
purified with the QIAquick® Gel Extraction kit (Qiagen).
Both strands of each amplicon were sequenced with a
BigDye® Terminator v3.1 Cycle Sequencing kit (Applied
Biosystems) under an ABI PRISM 3130 XL DNA Analyzer (Applied
Biosystems). Primer sequences were those previously designed for
specific region amplifications. DNA sequences were viewed and
analyzed with the DNA Sequencing Analysis Software v5.1 (Applied
Biosystems). The sequence variant was validated by resequencing of
an independent PCR-generated amplicon from the same subject and met
the quality control threshold with a call rate of >99%.
Additionally, an identified variant was searched in the single
nucleotide polymorphism (SNP) database from the National Center for
Biotechnology Information (NCBI, http://www.ncbi.nlm.nih.gov/SNP) to confirm the
novelty.
Alignment of multiple GJA5 protein
sequences across species
The multiple GJA5 protein sequences across various
species were aligned using the online program MUSCLE, version 3.6
(http://www.ncbi.nlm.nih.gov/).
Statistical analysis
Data were expressed as the means ± standard
deviation (SD). Continuous variables were tested for normality of
distribution and the Student’s unpaired t-test was used for
comparison of numeric variables between patient and control groups.
Comparison of the categorical variables between the 2 groups was
performed using Pearson’s χ2 or Fisher’s exact tests
when appropriate. A two-sided P-value of <0.05 was considered to
indicate statistically significant difference.
Results
Characteristics of the study
subjects
A cohort of 310 unrelated patients with lone AF were
included in this study and clinically evaluated in contrast to a
total of 200 matched, unrelated healthy individuals. None of the
subjects had documented traditional risk factors for AF. There were
no significant differences between lone AF and control groups in
baseline characteristics including age, gender, body mass index,
blood pressure, fasting blood glucose, serum lipid, left atrial
dimension, left ventricular ejection fraction, heart rate at rest,
as well as life style (data not shown). The basic clinical
characteristics of the 310 patients with lone AF are summarized in
Table I.
 | Table IClinical characteristics of the 310
unrelated patients with lone atrial fibrillation. |
Table I
Clinical characteristics of the 310
unrelated patients with lone atrial fibrillation.
|
Characteristics | No. or
quantity | Percentage or
range |
|---|
| Male:female | 142:168 | 71:84 |
| Age of onset
(years) | 45.2 | 18–59 |
| Paroxysmal AF on
presentation | 245 | 79 |
| Progression to
permanent AF | 54 | 17.4 |
| History of
cardioversion | 31 | 14 |
| History of
pacemaker | 18 | 5.8 |
| Resting heart rate
(bpm) | 72.5 | 50–158 |
| Systolic blood
pressure (mmHg) | 128.4 | 90–138 |
| Diastolic blood
pressure (mmHg) | 82.6 | 60–88 |
| Body mass index
(kg/m2) | 23.0 | 19–26 |
| Left atrial
dimension (mm) | 35 | 22–40 |
| Left ventricular
ejection fraction | 0.6 | 0.5–0.7 |
| Fasting blood
glucose (mmol/l) | 4.4 | 3.6–5.8 |
| Total cholesterol
(mmol/l) | 3.5 | 3.0–5.8 |
| Triglycerides
(mmol/l) | 1.3 | 0.5–1.7 |
| Medications |
| Aspirin | 86 | 27.7 |
| Warfarin | 115 | 37.1 |
| β-blocker | 102 | 32.9 |
| Calcium channel
blocker | 35 | 11.3 |
| Digoxin | 110 | 35.5 |
GJA5 mutations
A total of 310 unrelated patients with lone AF were
genetically evaluated. Direct sequencing of the entire coding
region and exon-intron boundaries of the GJA5 gene was
performed after PCR amplification of genomic DNA from the 310 AF
patients. Four heterozygous missense mutations in GJA5 were
identified in 4 of 310 unrelated index patients, respectively. The
total population prevalence of GJA5 mutations based on
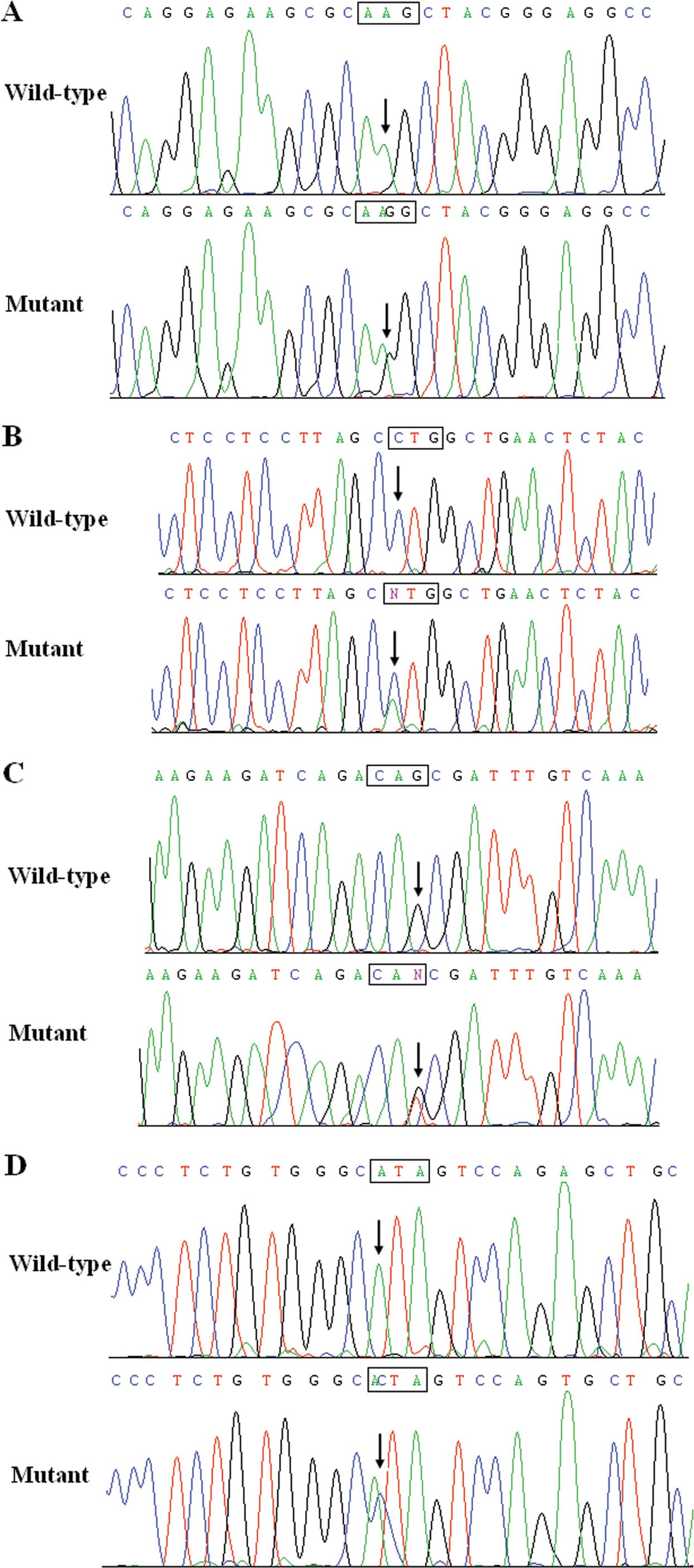
probands was ~1.29%. Specifically, a substitution of guanine for
adenine in the second nucleotide of codon 107 (c.320A>G),
predicting the transition of lysine into arginine at amino acid
position 107 (p.K107R) was detected in the proband from family 1. A
replacement of cytosine by adenine in the first nucleotide of codon
223 (c.667C>A), resulting in the transversion of leucine into
histidine (H) at amino acid 223 (p.L223H) was observed in the
proband from family 2. A change of guanine into thymine in the last
nucleotide of codon 236 (c.708G>T), corresponding to the
displacement of glutamine by H at amino acid 236 (p.Q236H) was
identified in the proband from family 3. An adenine-to-cytosine
conversion in the first nucleotide of codon 257 (c.769A>C),
equivalent to an isoleucine-to-leucine shift at amino acid 257
(p.I257L) was identified in the proband from family 4. The sequence
chromatograms showing the identified heterozygous GJA5
mutations of c.320A>G, c.667C>A, c.708G>T and c.769A>C
in comparison to corresponding control sequences are shown in
Fig. 1.
The missense mutations were not found in either the
400 control chromosomes or reported in the SNP database. Genetic
scanning of the families demonstrated that in each family, the gene
variant was present in all the affected living family members,
while it was absent in unaffected family members examined with the
exception of individuals III-2 in family 2 and III-9 in family 3,
suggesting that the long-term follow-up of asymptomatic subjects
harboring the variations is needed to confirm its clinical
significance. Analysis of the pedigrees showed that each mutation
co-segregated with AF transmitted in an autosomal dominant pattern
in the family with an incomplete penetrance. The pedigree
structures of the 4 families are shown in Fig. 2. The phenotypic characteristics and
results of genetic screening of the affected family members are
presented in Table II.
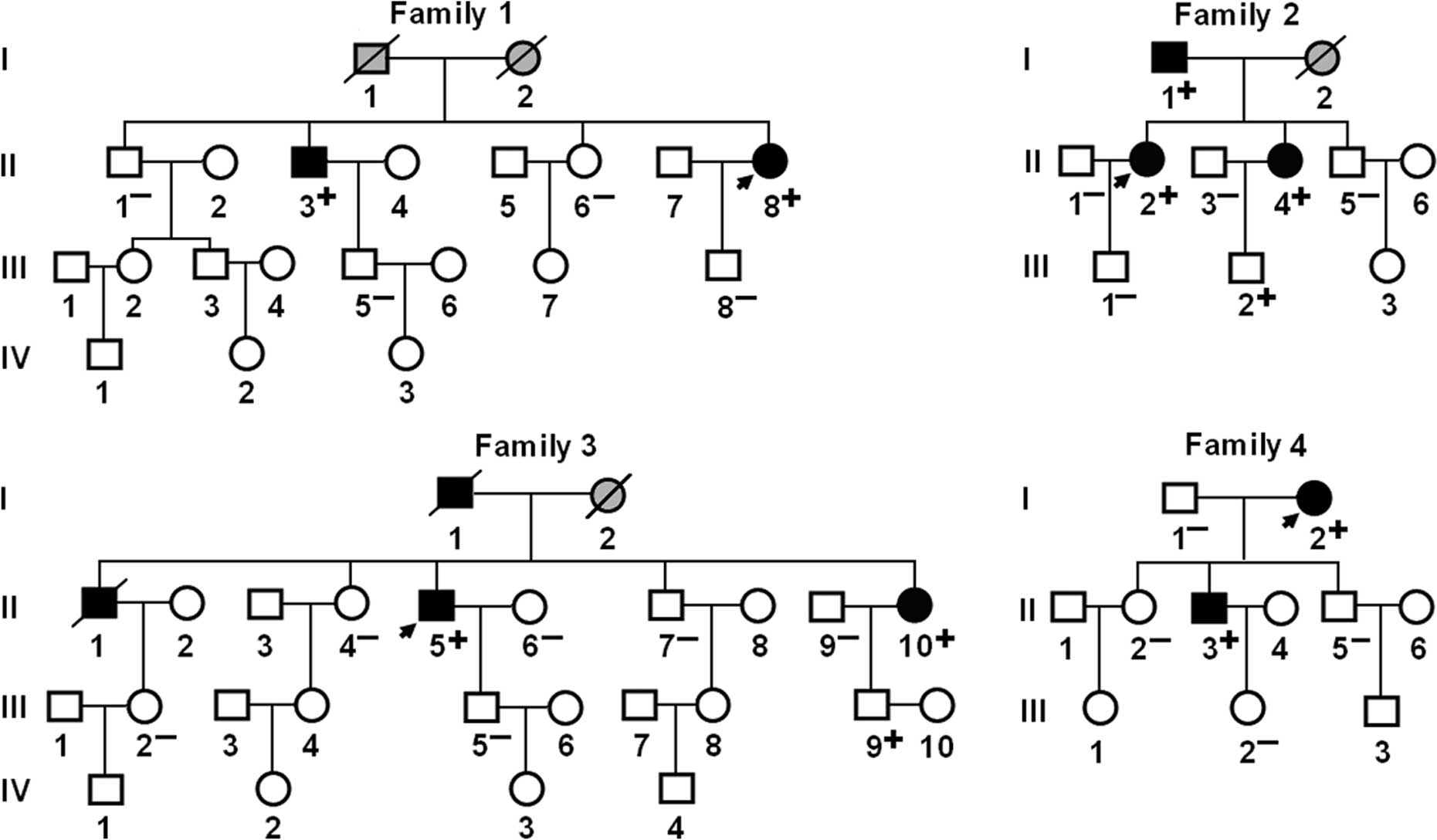 | Figure 2Pedigree structures of families with
atrial fibrillation (AF). Families are designated as 1, 2, 3 and 4,
respectively. Family members are identified by generations and
numbers. Squares, male family members; circles, female members;
symbols with a slash, the deceased members; closed symbols,
affected members; open symbols, unaffected members; stippled
symbols, members with phenotype undetermined; arrows, probands;
‘+’, carriers of the heterozygous mutations; and ‘−’,
non-carriers. |
| Table IIPhenotypic characteristics and status
of GJA5 mutations of the affected pedigree members. |
Table II
Phenotypic characteristics and status
of GJA5 mutations of the affected pedigree members.
| Subject
information | Phenotype |
Electrocardiogram | Echocardiogram | Genotype |
|---|
|
|
|
|
|
|---|
| Identity | Gender | Age at time of
study (years) | Age at diagnosis of
AF (years) | AF
(Classification) | P-wave (ms) | QRS interval
(ms) | LAD (mm) | LVEF (%) | GJA5 mutations |
|---|
| Family 1 | | | | | | | | | K107R |
| II-3 | M | 56 | 50 | Paroxysmal | 100 | 98 | 30 | 70 | +/− |
| II-8 | F | 48 | 42 | Persistent | 106 | 92 | 30 | 65 | +/− |
| Family 2 | | | | | | | | | L233M |
| I-1 | M | 65 | 50 | Permanent | 98 | 94 | 38 | 62 | +/− |
| II-2 | F | 41 | 38 | Paroxysmal | 92 | 90 | 36 | 56 | +/− |
| II-4 | F | 36 | 36 | Paroxysmal | 110 | 84 | 32 | 66 | +/− |
| Family 3 | | | | | | | | | Q236H |
| I-1 | M | 70a | 55 | Paroxysmal | N/A | 90 | N/A | N/A | N/A |
| II-1 | M | 64a | 53 | Paroxysmal | N/A | 92 | N/A | N/A | N/A |
| II-5 | M | 60 | 58 | Paroxysmal | 114 | 114.. | 38 | 64 | +/− |
| II-10 | F | 54 | 54 | Paroxysmal | 92 | 88 | 32 | 60 | +/− |
| Family 4 | | | | | | | | | I257L |
| I-2 | F | 64 | 45 | Paroxysmal | 102 | 90 | 36 | 65 | +/− |
| II-3 | M | 42 | 40 | Paroxysmal | 112 | 92 | 33 | 67 | +/− |
Multiple alignments of GJA5 protein
sequences across species
A cross-species alignment of GJA5 protein sequences
demonstrated that the altered amino acids were highly and
evolutionarily conserved with the exception of p.I257 (Fig. 3).
Discussion
In the present study, four novel heterozygous GJA5
mutations, p.K107R, p.L223M, p.Q236H and p.I257L, were identified
in four unrelated families with AF, respectively, with an estimated
mutational prevalence of 1.29%. In each family, the missense
mutation was present in all the affected family members examined,
while it was absent in the unaffected family members, with the
exception of individuals III-2 in family 2 and III-9 in family 3.
These mutations were absent in 400 normal chromosomes from an
ethnically-matched control population. A cross-species alignment of
GJA5 protein sequences demonstrated that the altered amino acids
were highly and evolutionarily conserved among species, with the
exception of p.I257. Therefore, it is likely that mutated GJA5
caused or conferred susceptibility to AF in these families.
Two carriers of GJA5 mutations, including individual
III-2 in family 2 who carried the p.L223M mutation and individual
III-9 in family 3 who harbored the p.Q236H mutation, did not have
AF during a 24-h electrocardiographic monitoring. This observation
may be explained by the following reasons. Firstly, AF occurs as
rarely as a few times in a lifetime for some patients with AF
(56); considering the performed
electrocardiographic monitoring for only 24 h, a longer duration of
monitoring may be required to record paroxysmal AF in these
patients. Secondly, AF occurs more commonly in older patients;
thus, these carriers may not be old enough to develop the disease.
Thirdly, familial AF caused by the mutation p.L223M or p.Q236H may
have a low or incomplete penetration. Additionally, p.L223M or
p.Q236H may be only a genetic risk factor predisposing to AF,
rather than a direct cause of AF, and environmental risk factors
may be required for the onset of AF.
Multiple GJA5 mutations or polymorphisms have
been previously involved in AF (48–55).
Similar to the present findings, Yang et al(54,55)
have previously performed a sequence analysis of the GJA5
gene in a total of 344 index patients with lone AF, and identified
four novel heterozygous missense mutations (p.Q49X, p.V85I, p.L221I
and p.L229M), with a mutational prevalence of ~1.16%. Gollob et
al(53)performed the first
scan of GJA5 in patients with lone AF and identified four novel
heterozygous missense mutations in 4 of 15 AF patients, of which 3
mutations (p.G38D, p.P88S and p.M163V) were found in the
cardiac-tissue specimens but not in the peripheral lymphocytes; one
mutation (p.A96S) was detected in both cardiac tissue and
lymphocytes, with a germline mutational prevalence of ~6.67%. The
p.A96S variant was absent in the patient’s 3 siblings and wife,
while it was present in the patient’s 2 sons without history of AF
and in 1 of 120 controls. Functional analysis of mutant GJA5
proteins demonstrated impaired intracellular transport or reduced
intercellular electrical coupling (53). By sequencing the 5′ untranslated
exon and the proximal promoter region of the GJA5 gene
(GenBank accession no. AF246295) in patients with familial atrial
standstill, Groenewegen et al(48) found two closely linked
polymorphisms, of which one was a G to A transition at 44
nucleotides upstream of the transcription start site (−44G>A),
and the other was a substitution of G for A in exon 1 at 71
nucleotides downstream of the transcription start site (+71A>G).
Luciferase reporter gene assays of the minor GJA5 haplotype
(−44A, +71G) in GJA5-expressing rat arterial smooth muscular cells
showed a >2-fold decrease in promoter activity compared with the
more common haplotype (−44G, +71A). The reduced GJA5 expression may
lead to a reduction of the total amount of GJA5 protein in
vivo, providing an atrial electrophysiological substrate
favoring arrhythmia (48).
Furthermore, the GJA5 polymorphisms have been strongly
associated with increased spatial dispersion of refractoriness as a
marker for enhanced atrial vulnerability and carriers of the −44AA
genotype had a significantly higher risk of AF compared with those
carrying the −44GG genotype (49).
In a larger case-control study, the rare haplotype frequency of
GJA5 (−44A, +71G) was statistically higher in the AF
compared with the control group, and also functional studies using
luciferase as the reporter have demonstrated that GJA5
(−44A, +71G) had significantly lower promoter activity compared
with GJA5 (−44G, +71A) in atrial myocytes from mice (50). A novel common GJA5 gene
promoter variant has recently been associated with reduced GJA5
expression in human atria and increased vulnerability to AF
(51). These results highlight the
pivotal role of GJA5 for atrial electrophysiology and indicate that
dysfunctional GJA5 may be an important molecular mechanism
involved in the pathogenesis of AF.
The association of abnormal GJA5 with
enhanced susceptibility to arrhythmias has been substantiated in
animal models. Targeted gene deletion of GJA5 in mice
produced multiple abnormalities including increased sinoatrial node
recovery time, decreased conduction velocity of atria,
atrioventricular node and bundle branch, and impaired sinoatrial
propagation with atrial ectopic pacemakers, which developed an
arrhythmogenic substrate prone to AF (57,58).
In a canine sterile pericarditis model, the gap junction
conduction-enhancing antiarrhythmic peptide, Gap-134, improved
conduction and reduced AF (59).
Similarly, in a dog model of AF due to myocardial ischemia,
administration of ZP123, a gap junction conductance-improving
modifier, prevented ischemia-induced conduction slowing and reduced
AF duration (60). Notably, in
experimental swine, gene therapy with adenovirus expressing GJA5
improved cardiac conduction and reduced AF, demonstrating the
viability of gene therapy for the prevention of atrial arrhythmias
(61).
It is well known that AF is a complex arrhythmia
ascribed to multiple possible mechanisms. Despite the presence of
an inherited defect, a favorable substrate for AF, within the
myocardial tissue of affected patients from birth, the onset of
genetically-based AF often requires a trigger for initiation,
presumably by exacerbating the already anomalous cardiac cellular
electrophysiology in the existence of mutant protein. One of the
most common triggers is the increased vagal tone mediated by
muscarinic receptors, causing uneven shortening of refractoriness
in the atria and, thus, electrophysiological heterogeneity
(62). The stimulation of
muscarinic receptors has been shown to impair the cell-cell
coupling mediated by gap junctions (63). Together with the data mentioned
above, this experimental result suggests a potential pathogenic
link between increased cardiac parasympathetic nerve activity,
impaired myocardial intercellular electrical coupling, and the
occurrence of AF.
Notably, GJA5 is an important determinant for
impulse propagation in the atrium as well as the specialized
conduction system and abnormal expression of GJA5 predisposes to
AF. However, functional changes in GJA5 alone may not be sufficient
for significantly prolonged P-wave duration, PQ interval, QRS
duration and QT duration in the surface electrocardiogram, as
observed in these AF families and other AF patients (48–55).
Additionally, full deficiency for GJA5 has been associated with
altered electrocardiographic parameters in GJA5 knockout
mice, in contrast to haploinsufficiency for GJA5 (57). These findings suggest that
additional factors combined with reduced coupling lead to AF.
In conclusion, the present investigation links novel
GJA5 mutations to AF, which provides novel insight into the
molecular mechanisms associated with the arrhythmogenesis and
ultimately may result in improved, patient-specific rhythm control
strategies.
Acknowledgements
The authors are greatly indebted to participants for
their dedication to the study. This study was supported by grants
from the National Natural Science Foundation of China (nos.
81070153, 81270161 and 30570768), the Natural Science Foundation of
Shanghai, China (10ZR1428000), the Personnel Development Foundation
of Shanghai, China (no. 2010019), and the Key Program of Basic
Research of Shanghai, China (nos. 10JC1414000, 10JC1414001 and
10JC1414002).
References
|
1
|
Fuster V, Rydén LE, Cannom DS, Crijns HJ,
Curtis AB, Ellenbogen KA, Halperin JL, Kay GN, Le Huezey JY, Lowe
JE, Olsson SB, Prystowsky EN, Tamargo JL, Wann LS, Smith SC Jr,
Priori SG, Estes NA III, Ezekowitz MD, Jackman WM, January CT, Lowe
JE, Page RL, Slotwiner DJ, Stevenson WG, Tracy CM, Jacobs AK,
Anderson JL, Albert N, Buller CE, Creager MA, Ettinger SM, Guyton
RA, Halperin JL, Hochman JS, Kushner FG, Ohman EM, Stevenson WG,
Tarkington LG and Yancy CW; American College of Cardiology
Foundation/American Heart Association Task Force. 2011 ACCF/AHA/HRS
focused updates incorporated into the ACC/AHA/ESC 2006 guidelines
for the management of patients with atrial fibrillation: a report
of the American College of Cardiology Foundation/American Heart
Association Task Force on practice guidelines. Circulation.
123:e269–e367. 2011.
|
|
2
|
Go AS, Hylek EM, Phillips KA, Chang Y,
Henault LE, Selby JV and Singer DE: Prevalence of diagnosed atrial
fibrillation in adults: national implications for rhythm management
and stroke prevention: the AnTicoagulation and Risk Factors in
Atrial Fibrillation (ATRIA) Study. JAMA. 285:2370–2375. 2001.
View Article : Google Scholar
|
|
3
|
Lloyd-Jones DM, Wang TJ, Leip EP, Larson
MG, Levy D, Vasan RS, D’Agostino RB, Massaro JM, Beiser A, Wolf PA
and Benjamin EJ: Lifetime risk for development of atrial
fibrillation: the Framingham Heart Study. Circulation.
110:1042–1046. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wolf PA, Abbott RD and Kannel WB: Atrial
fibrillation as an independent risk factor for stroke: the
Framingham Study. Stroke. 22:983–988. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Benjamin EJ, Wolf PA, D’Agostino RB,
Silbershatz H, Kannel WB and Levy D: Impact of atrial fibrillation
on the risk of death: the Framingham Heart Study. Circulation.
98:946–952. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nattel S: New ideas about atrial
fibrillation 50 years on. Nature. 415:219–226. 2002.PubMed/NCBI
|
|
7
|
Huang WJ, Zhou R, Zeng XR, Tan XQ, Cheng
ZH, Tang MH, Gou LT, Chen LJ, Tong AP, He Y and Yang JL:
Comparative proteomic analysis of atrial appendages from rheumatic
heart disease patients with sinus rhythm and atrial fibrillation.
Mol Med Rep. 4:655–661. 2011.PubMed/NCBI
|
|
8
|
Li H, Li S, Yu B and Liu S: Expression of
miR-133 and miR-30 in chronic atrial fibrillation in canines. Mol
Med Rep. 5:1457–1460. 2012.PubMed/NCBI
|
|
9
|
Cheng T, Wang XF, Hou YT and Zhang L:
Correlation between atrial fibrillation, serum amyloid protein A
and other inflammatory cytokines. Mol Med Rep. 6:581–584.
2012.PubMed/NCBI
|
|
10
|
Kim SM, Lee JH, Kim JR, Shin DG, Lee SH
and Cho KH: Female patients with atrial fibrillation have increased
oxidized and glycated lipoprotein properties and lower
apolipoprotein A-I expression in HDL. Int J Mol Med. 27:841–849.
2011.PubMed/NCBI
|
|
11
|
Darbar D, Herron KJ, Ballew JD, Jahangir
A, Gersh BJ, Shen WK, Hammill SC, Packer DL and Olson TM: Familial
atrial fibrillation is a genetically heterogeneous disorder. J Am
Coll Cardiol. 41:2185–2192. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fox CS, Parise H, D’Agostino RB Sr,
Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA and Benjamin EJ:
Parental atrial fibrillation as a risk factor for atrial
fibrillation in offspring. JAMA. 291:2851–2855. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ellinor PT, Yoerger DM, Ruskin JN and
MacRae CA: Familial aggregation in lone atrial fibrillation. Hum
Genet. 118:179–184. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Arnar DO, Thorvaldsson S, Manolio TA,
Thorgeirsson G, Kristjansson K, Hakonarson H and Stefansson K:
Familial aggregation of atrial fibrillation in Iceland. Eur Heart
J. 27:708–712. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Junttila MJ, Raatikainen MJ, Perkiömäki
JS, Hong K, Brugada R and Huikuri HV: Familial clustering of lone
atrial fibrillation in patients with saddleback-type ST-segment
elevation in right precordial leads. Eur Heart J. 28:463–468. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Christophersen IE, Ravn LS,
Budtz-Joergensen E, Skytthe A, Haunsoe S, Svendsen JH and
Christensen K: Familial aggregation of atrial fibrillation: a study
in Danish twins. Circ Arrhythm Electrophysiol. 2:378–383. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang YQ, Zhang XL, Wang XH, Tan HW, Shi
HF, Fang WY and Liu X: Familial aggregation of lone atrial
fibrillation in the Chinese population. Intern Med. 49:2385–2391.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lubitz SA, Yin X, Fontes JD, Magnani JW,
Rienstra M, Pai M, Villalon ML, Vasan RS, Pencina MJ, Levy D,
Larson MG, Ellinor PT and Benjamin EJ: Association between familial
atrial fibrillation and risk of new-onset atrial fibrillation.
JAMA. 304:2263–2269. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brugada R, Tapscott T, Czernuszewicz GZ,
Marian AJ, Iglesias A, Mont L, Brugada J, Girona J, Domingo A,
Bachinski LL and Roberts R: Identification of a genetic locus for
familial atrial fibrillation. N Engl J Med. 336:905–911. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ellinor PT, Shin JT, Moore RK, Yoerger DM
and MacRae CA: Locus for atrial fibrillation maps to chromosome
6q14–16. Circulation. 107:2880–2883. 2003.PubMed/NCBI
|
|
21
|
Chen YH, Xu SJ, Bendahhou S, Wang XL, Wang
Y, Xu WY, Jin HW, Sun H, Su XY, Zhuang QN, Yang YQ, Li YB, Liu Y,
Xu HJ, Li XF, Ma N, Mou CP, Chen Z, Barhanin J and Huang W: KCNQ1
gain-of-function mutation in familial atrial fibrillation. Science.
299:251–254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Oberti C, Wang L, Li L, Dong J, Rao S, Du
W and Wang Q: Genome-wide linkage scan identifies a novel genetic
locus on chromosome 5p13 for neonatal atrial fibrillation
associated with sudden death and variable cardiomyopathy.
Circulation. 110:3753–3759. 2004. View Article : Google Scholar
|
|
23
|
Darbar D, Hardy A, Haines JL and Roden DM:
Prolonged signal-averaged P-wave duration as an intermediate
phenotype for familial atrial fibrillation. J Am Coll Cardiol.
51:1083–1089. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang X, Chen S, Yoo S, Chakrabarti S,
Zhang T, Ke T, Oberti C, Yong SL, Fang F, Li L, de la Fuente R,
Wang L, Chen Q and Wang QK: Mutation in nuclear pore component
NUP155 leads to atrial fibrillation and early sudden cardiac death.
Cell. 135:1017–1027. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang Y, Xia M, Jin Q, Bendahhou S, Shi J
and Chen Y, Liang B, Lin J, Liu Y, Liu B, Zhou Q, Zhang D, Wang R,
Ma N, Su X, Niu K, Pei Y, Xu W, Chen Z, Wan H, Cui J, Barhanin J
and Chen Y: Identification of a KCNE2 gain-of-function mutation in
patients with familial atrial fibrillation. Am J Hum Genet.
75:899–905. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lundby A, Ravn LS, Svendsen JH, Hauns S,
Olesen SP and Schmitt N: KCNE3 mutation V17M identified in a
patient with lone atrial fibrillation. Cell Physiol Biochem.
21:47–54. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ravn LS, Aizawa Y, Pollevick GD,
Hofman-Bang J, Cordeiro JM, Dixen U, Jensen G, Wu Y, Burashnikov E,
Haunso S, Guerchicoff A, Hu D, Svendsen JH, Christiansen M and
Antzelevitch C: Gain of function in IKs secondary to a mutation in
KCNE5 associated with atrial fibrillation. Heart Rhythm. 5:427–435.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hong K, Bjerregaard P, Gussak I and
Brugada R: Short QT syndrome and atrial fibrillation caused by
mutation in KCNH2. J Cardiovasc Electrophysiol. 16:394–396. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xia M, Jin Q, Bendahhou S, He Y, Larroque
MM and Chen Y, Zhou Q, Yang Y, Liu Y, Liu B, Zhu Q, Zhou Y, Lin J,
Liang B, Li L, Dong X, Pan Z, Wang R, Wan H, Qiu W, Xu W, Eurlings
P, Barhanin J and Chen Y: A Kir2.1 gain-of-function mutation
underlies familial atrial fibrillation. Biochem Biophys Res Commun.
332:1012–1019. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Delaney JT, Muhammad R, Blair MA, Kor K,
Fish FA, Roden DM and Darbar D: A KCNJ8 mutation associated with
early repolarization and atrial fibrillation. Europace.
14:1428–1432. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Olson TM, Alekseev AE, Liu XK, Park S,
Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A and
Terzic A: Kv1.5 channelopathy due to KCNA5 loss-of-function
mutation causes human atrial fibrillation. Hum Mol Genet.
15:2185–2191. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang Y, Li J, Lin X, Yang Y, Hong K, Wang
L, Liu J, Li L, Yan D, Liang D, Xiao J, Jin H, Wu J, Zhang Y and
Chen YH: Novel KCNA5 loss-of-function mutations responsible for
atrial fibrillation. J Hum Genet. 54:277–283. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Darbar D, Kannankeril PJ, Donahue BS,
Kucera G, Stubblefield T, Haines JL, George AL Jr and Roden DM:
Cardiac sodium channel (SCN5A) variants associated with atrial
fibrillation. Circulation. 117:1927–1935. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hodgson-Zingman DM, Karst ML, Zingman LV,
Heublein DM, Darbar D, Herron KJ, Ballew JD, de Andrade M, Burnett
JC Jr and Olson TM: Atrial natriuretic peptide frameshift mutation
in familial atrial fibrillation. N Engl J Med. 359:158–165. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang JQ, Shen FF, Fang WY, Liu X and Yang
YQ: Novel GATA4 mutations in lone atrial fibrillation. Int J Mol
Med. 28:1025–1032. 2011.PubMed/NCBI
|
|
36
|
Yang YQ, Wang MY, Zhang XL, Tan HW, Shi
HF, Jiang WF, Wang XH, Fang WY and Liu X: GATA4 loss-of-function
mutations in familial atrial fibrillation. Clin Chim Acta.
412:1825–1830. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang J, Sun YM and Yang YQ: Mutation
spectrum of the GATA4 gene in patients with idiopathic atrial
fibrillation. Mol Biol Rep. 39:8127–8135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang YQ, Wang J, Wang XH, Wang Q, Tan HW,
Zhang M, Shen FF, Jiang JQ, Fang WY and Liu X: Mutational spectrum
of the GATA5 gene associated with familial atrial fibrillation. Int
J Cardiol. 157:305–307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang YQ, Wang XH, Tan HW, Jiang WF, Fang
WY and Liu X: Prevalence and spectrum of GATA6 mutations associated
with familial atrial fibrillation. Int J Cardiol. 155:494–496.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yang YQ, Li L, Wang J, Zhang XL, Li RG, Xu
YJ, Tan HW, Wang XH, Jiang JQ, Fang WY and Liu X: GATA6
loss-of-function mutation in atrial fibrillation. Eur J Med Genet.
55:520–526. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li J, Liu WD, Yang ZL and Yang YQ: Novel
GATA6 loss-of-function mutation responsible for familial atrial
fibrillation. Int J Mol Med. 4:783–790. 2012.PubMed/NCBI
|
|
42
|
Delmar M and Makita N: Cardiac connexins,
mutations and arrhythmias. Curr Opin Cardiol. 27:236–241. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jansen JA, van Veen TA, de Bakker JM and
van Rijen HV: Cardiac connexins and impulse propagation. J Mol Cell
Cardiol. 48:76–82. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Vozzi C, Dupont E, Coppen SR, Yeh HI and
Severs NJ: Chamber-related differences in connexin expression in
the human heart. J Mol Cell Cardiol. 31:991–1003. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hagendorff A, Schumacher B, Kirchhoff S,
Lüderitz B and Willecke K: Conduction disturbances and increased
atrial vulnerability in Connexin40-deficient mice analyzed by
transesophageal stimulation. Circulation. 99:1508–1515. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
van der Velden HM, Ausma J, Rook MB,
Hellemons AJ, van Veen TA, Allessie MA and Jongsma HJ: Gap
junctional remodeling in relation to stabilization of atrial
fibrillation in the goat. Cardiovasc Res. 46:476–486.
2000.PubMed/NCBI
|
|
47
|
Severs NJ, Coppen SR, Dupont E, Yeh HI, Ko
YS and Matsushita T: Gap junction alterations in human cardiac
disease. Cardiovasc Res. 62:368–377. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Groenewegen WA, Firouzi M, Bezzina CR,
Vliex S, van Langen IM, Sandkuijl L, Smits JP, Hulsbeek M, Rook MB,
Jongsma HJ and Wilde AA: A cardiac sodium channel mutation
cosegregates with a rare connexin40 genotype in familial atrial
standstill. Circ Res. 92:14–22. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Firouzi M, Ramanna H, Kok B, Jongsma HJ,
Koeleman BP, Doevendans PA, Groenewegen WA and Hauer RN:
Association of human connexin40 gene polymorphisms with atrial
vulnerability as a risk factor for lone atrial fibrillation. Circ
Res. 95:e29–e33. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Juang JM, Chern YR, Tsai CT, Chiang FT,
Lin JL, Hwang JJ, Hsu KL, Tseng CD, Tseng YZ and Lai LP: The
association of human connexin 40 genetic polymorphisms with atrial
fibrillation. Int J Cardiol. 116:107–112. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wirka RC, Gore S, Van Wagoner DR, Arking
DE, Lubitz SA, Lunetta KL, Benjamin EJ, Alonso A, Ellinor PT,
Barnard J, Chung MK and Smith JD: A common connexin-40 gene
promoter variant affects connexin-40 expression in human atria and
is associated with atrial fibrillation. Circ Arrhythm
Electrophysiol. 4:87–93. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chaldoupi SM, Hubens LE, Smit
Duijzentkunst DA, van Stuijvenberg L, Bierhuizen MF, van Aarnhem
EE, Nelen M, de Bakker JM, Hauer RN, van Rijen HV, Loh P and van
Veen TA: Reduced connexin40 protein expression in the right atrial
appendage of patients bearing the minor connexin40 allele (-44G→A).
Europace. 14:1199–1205. 2012.PubMed/NCBI
|
|
53
|
Gollob MH, Jones DL, Krahn AD, Danis L,
Gong XQ, Shao Q, Liu X, Veinot JP, Tang AS, Stewart AF, Tesson F,
Klein GJ, Yee R, Skanes AC, Guiraudon GM, Ebihara L and Bai D:
Somatic mutations in the connexin 40 gene (GJA5) in atrial
fibrillation. N Engl J Med. 354:2677–2688. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang YQ, Zhang XL, Wang XH, Tan HW, Shi
HF, Jiang WF, Fang WY and Liu X: Connexin40 nonsense mutation in
familial atrial fibrillation. Int J Mol Med. 26:605–610.
2010.PubMed/NCBI
|
|
55
|
Yang YQ, Liu X, Zhang XL, Wang XH, Tan HW,
Shi HF, Jiang WF and Fang WY: Novel connexin40 missense mutations
in patients with familial atrial fibrillation. Europace.
12:1421–1427. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chugh SS, Blackshear JL, Shen WK, Hammill
SC and Gersh BJ: Epidemiology and natural history of atrial
fibrillation: clinical implications. J Am Coll Cardiol. 37:371–378.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chaldoupi SM, Loh P, Hauer RN, de Bakker
JM and van Rijen HV: The role of connexin40 in atrial fibrillation.
Cardiovasc Res. 84:15–23. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Bagwe S, Berenfeld O, Vaidya D, Morley GE
and Jalife J: Altered right atrial excitation and propagation in
connexin40 knockout mice. Circulation. 112:2245–2253. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rossman EI, Liu K, Morgan GA, Swillo RE,
Krueger JA, Gardell SJ, Butera J, Gruver M, Kantrowitz J, Feldman
HS, Petersen JS, Haugan K and Hennan JK: The gap junction modifier,
GAP-134, improves conduction and reduces atrial
fibrillation/flutter in the canine sterile pericarditis model. J
Pharmacol Exp Ther. 329:1127–1133. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Shiroshita-Takeshita A, Sakabe M, Haugan
K, Hennan JK and Nattel S: Model-dependent effects of the gap
junction conduction-enhancing antiarrhythmic peptide rotigaptide
(ZP123) on experimental atrial fibrillation in dogs. Circulation.
115:310–318. 2007. View Article : Google Scholar
|
|
61
|
Igarashi T, Finet JE, Takeuchi A, Fujino
Y, Strom M, Greener ID, Rosenbaum DS and Donahue JK: Connexin gene
transfer preserves conduction velocity and prevents atrial
fibrillation. Circulation. 125:216–225. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zipes DP, Mihalick MJ and Robbins FT:
Effects of selective vagal stellate ganglion stimulation on atrial
refractoriness. Cardiovasc Res. 8:647–655. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Fritz S, Kunz L, Dimitrijevic N, Grunert
R, Heiss C and Mayerhofer A: Muscarinic receptors in human
luteinized granulosa cells: activation blocks gap junctions and
induces the transcription factor early growth response factor-1. J
Clin Endocrinol Metab. 87:1362–1367. 2002. View Article : Google Scholar
|