Introduction
Toll-like receptors (TLRs) are the most well-known
pattern recognition receptors, due to their role in the detection
of a variety of pathogen-associated molecular patterns (1). To date, 13 TLRs have been identified,
and each TLR has a specific ligand (2–4). In
particular, TLR2 heterodimerizes with TLR1 or TLR6, and recognizes
lipopeptides or lipoproteins (5).
TLR has been suggested to play a role in
atherosclerosis progression (6,7);
TLR2 is the most known receptor for the development of
atherosclesosis. The direct stimulation of TLR2 by the synthetic
ligand, Pam3CSK4, accelerates lesion
formation and, in TLR2 knockout (KO) mice, this phenomenon is
inhibited (8). The TLR2-associated
progression of atherosclerosis has been confirmed in various cells,
including endothelial cells, smooth muscle cells and macrophages.
For example, high expression of TLR2 in monocytes serves as an
important risk factor for arteriosclerotic disease (9,10).
In addition, increased expression of TLR2 in endothelial cells
causes interruption of blood flow, thus leading to more severe
early atherogenic events (11).
Apolipoprotein CIII regulates atherosclerosis-related gene
expression in adipocytes (12),
and even Chlamydia pneumonia-induced macrophage-form cell
formation is mediated by TLR2 (13). Therefore, the verification of
regulatory proteins in atherosclerosis induced by TLR2 may aid in
the prevention of the disease and could provide possibilities of
novel target medication.
Multiple regulators contribute towards the
progression of atherosclerosis. However, the recruitment of
monocytes on the injured sites of blood vessels is essential for
the onset of atherosclerosis, and the most essentially effective
chemokine is monocyte chemoattractant protein-1 (MCP-1) (14,15).
Monocytes activated by MCP-1 differentiate into macrophages and are
then converted into foam cells; however, each of the cells produce
more MCP-1, thus exasperating atherosclerosis progression. Although
MCP-1 has been shown to be associated with the formation of foam
cells and the progression of atherosclerosis, many questions remain
unanswered regarding the expression mechanism during TLR2
activation. There is very limited information on studies involved
in the TLR2-mediated MCP-1 production. At present, the function of
ERK and JNK has been suggested in MCP-1 production by TLR4
stimulation (16,17) and, recently, the existence of
multiple pathways, such as PKC, Akt and mitogen-activated protein
kinase (MAPK), have also been suggested (16,18,19).
We have previously reported the regulation of MCP-1 production via
TLR9 by cytosolic PLA2 and JNK pathway (20). However, not much information is
available regarding the MCP-1 production mechanism by TLR2
signaling. Although MAPK (ERK, p38, JNK) pathway is a well-known
mechanism with the involvement of the myeloid differentiation
primary response gene 88 (MyD88) complex in the TLR-mediated MCP-1
production, the participation of other types of kinases remains
obscure. Therefore, in our present study, we investigated the
participation of new signaling pathways, which regulate MCP-1
production by TLR2 stimulation.
Materials and methods
Cells lines and reagents
Raw264.7 cell lines were purchased from ATCC
(Manassas, VA, USA). Primary bone marrow-derived monocytes were
differentiated into bone marrow-derived macrophages (BMDM) for 5–7
days in DMEM supplemented with 10% L929 cell-conditioned medium [as
a source of macrophage-colony stimulating factor (M-CSF)]. Cell
culture reagents, including fetal bovine serum (FBS), were obtained
from Life Technologies (Grand Island, NY, USA). α-Akt, α-phospho
Akt (S473), α-phospho glycogen synthase kinase-3β (GSK3β) (S9)
antibodies were obtained from Cell Signaling Technology (Beverly,
MA, USA). α-β-actin, LiCl were purchased from Sigma-Aldrich (St.
Louis, MO, USA). Reverse transcription-polymerase chain reaction
(RT-PCR) kits were from Takara Bio (Kyoto, Japan). TLR2, MyD88, TIR
domain-containing adapter inducing IFN-β (TRIF) siRNAs and TRIzol
were purchased from Invitrogen (Carlsbad, CA, USA). Janus kinase
(JAK)1, JAK2, JAK3 and TYK2 siRNA were purchased from Bioneer
(Daejeon, Korea). α-GSK3β was purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). LY294002 and AG-490 were
purchased from Biomol (Plymouth Meeting, PA, USA). JAK inhibitor I
was purchased from Merck/Calbiochem (Darmstadt, Germany).
Pam3CSK4 was purchased from InvivoGen (San
Diego, CA, USA).
siRNAs and transfection
Stealth control and gene-specific siRNAs against the
following target genes were designed using the Block-IT Stealth
RNAi designer: i) TLR2, 5′-UUA AAG GGC GGG UCA GAG UUC UCC A-3′;
ii) MyD88, 5′-ACC ACC AUG CGG CGA CAC CUU UUC U-3′; iii) TRIF,
5′-UGU UGG UGG AGU GUA ACG UAU GUC C-3′; iv) JAK1, 5′-UCA CCG GGA
CUU AGC AGC AAG AAA U-3′; v) JAK2, 5′-CGG GUC GGC GCA ACC UAA GAU
UAA U-3′; vi) JAK3, 5′-CAC AUG ACU CGG AAG UCC UCC UGA A-3′; vii)
TYK2, 5′-GCG AGG AGG AGA UCC ACC ACU UUA A-3′. For transfection,
Raw264.7 cells were plated in 35-mm plates and transfected with
siRNA at a final concentration of 150–200 pM using nucleoporation
reagents from Lonza (Allendale, NJ, USA). Cells were nucleoporated
according to the manufacturer's protocol and incubated for 18–24 h
before Pam3CSK4 stimulation.
RT-PCR
Total RNA was extracted from cells using TRIzol.
First-strand of cDNA was synthesized from 1 μg total RNA in the
presence of random primers, oligo(dT) and reverse transcriptase
(Takara Bio). Cycling conditions for PCR were 95˚C for 5 min,
followed by 26–33 cycles at 95˚C for 1 min, 60–63˚C for 1 min and
72˚C for 1 min. For semi-quantitative PCR, target genes were
normalized for β-actin transcription. The sequences of PCR primers
used in the present study were as follows: i) MCP-1 forward, 5′-AGA
GAG CCA GAC GGG AGG AA-3′ and reverse, 5′-GTC ACA CTG GTC ACT CCT
AC-3′; ii) TLR2 forward, 5′-CGC CCT TTA AGC TGT GTC TC-3′ and
reverse, 5′-TTA TCT TGC GCA GTT TGC AG-3′; iii) MyD88 forward,
5′-CCA GTA TCC TGC GGT TCA TCA-3′ and reverse, 5′-GCT CCG CAT CAG
TCT CAT CTT-3′; iv) TRIF forward, 5′-GTA TGG GCC CTC TGA CTG AT-3′
and reverse, 5′-ATA GGT GTG GTC TTC CCT GC-3′; v) JAK1 forward,
5′-ATG GAA GAC GGA GGC AAT GGT-3′ and reverse, 5′-GGA ACT TTA GAG
GCA GAA TAC-3′; vi) JAK2 forward, 5′-AAG AGC AAC GGA AGA TTG C-3′
and reverse, 5′-CGT CAC AGT TTC TTC TGC CT-3′; vii) JAK3 forward,
5′-CAC ACC TGG CAT CCC GAA TC-3′ and reverse, 5′-AGC AGT AGG CGG
TCG GTG TG-3′; viii) TYK2 forward, 5′-CCT GGC CAT GAC CTG AAC AG-3′
and reverse, 5′-TGT GCC CTT CAC TGA CGG AG-3′; and ix) β-actin
forward, 5′-TCC TTC GTT GCC GGT CCA CA-3′ and reverse, 5′-CGT CTC
CGG AGT CCA TCA CA-3′.
Western blot analysis
Macrophages were cultured in 35-mm Petri dishes and
treated with Pam3CSK4 in the presence or
absence of inhibitor. Cell pellets were resuspended in lysis buffer
(50 mM Tris-HCl, pH 8.0; 5 mM EDTA; 150 mM NaCl; 0.5% Nonidet P-40;
1 mM phenylmethylsulfonyl fluoride; and protease inhibitor
cocktail). Proteins were separated on an 8% reducing SDS-PAGE gel
and transferred onto nitrocellulose membranes in 20% methanol, 25
mM Tris and 192 mM glycine. Membranes were then blocked with 5%
non-fat dry milk and incubated overnight with primary antibody at
4˚C before washing, followed by 1 h of incubation with horseradish
peroxidase-conjugated secondary antibody. Further washing steps
were followed with subsequent development with an enhanced
chemiluminescence system (GE Healthcare, Buckinghamshire, UK).
Results and Discussion
Pam3CSK4 stimulates
MCP-1 mRNA expression through TLR2-dependent, but MyD88-independent
pathway
Previously, we as well as others have reported on
the formation of foam cells by the TLR2 ligand; the foam cell is a
hallmark of early atherosclerosis lesions (13,20).
We studied the nature of molecules involved in TLR2-induced foam
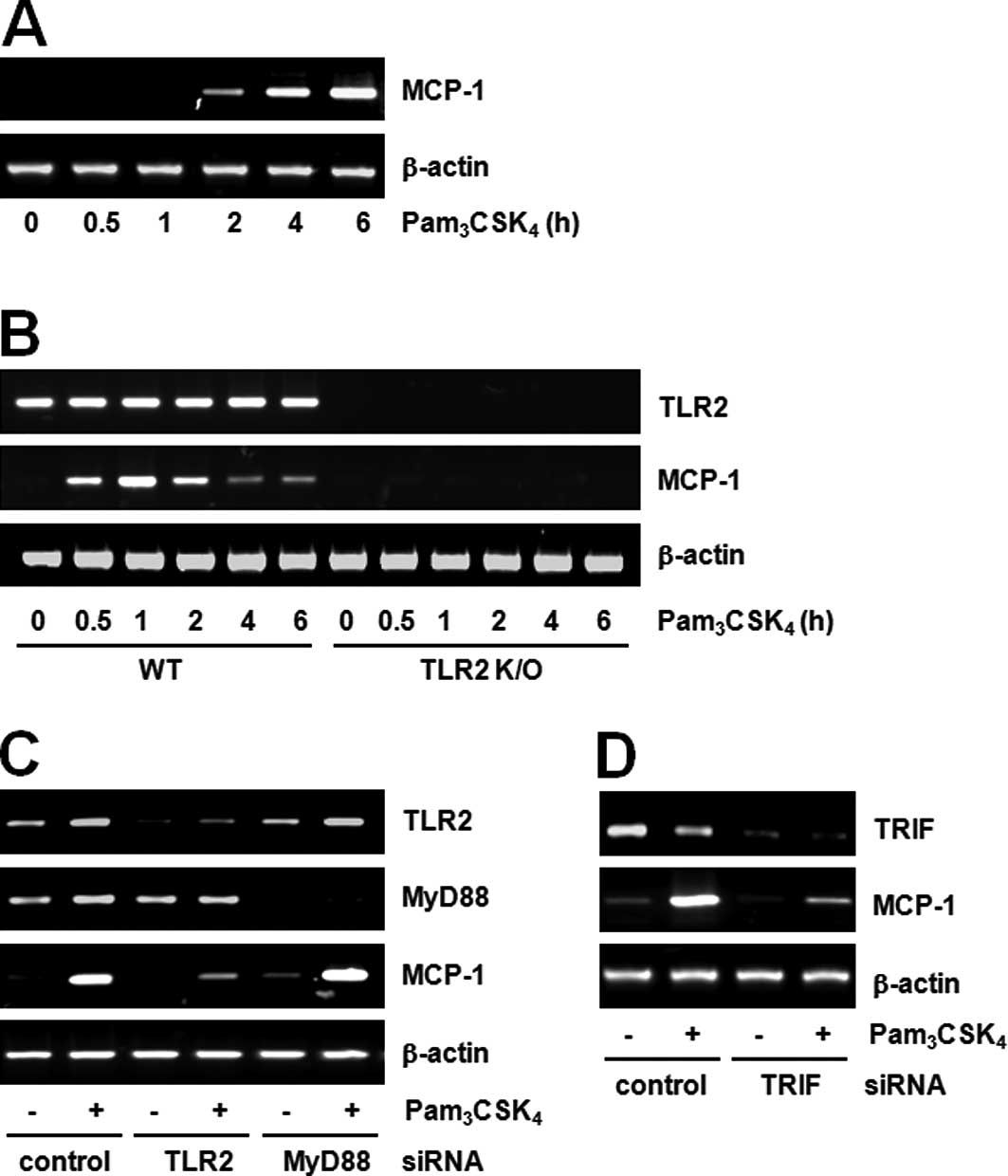
cell formation and found that MCP-1 plays a major role. RT-PCR
showed maximal induction of MCP-1 mRNA expression by
Pam3CSK4 in both cell lines, Raw264.7
(Fig. 1A) and BMDM (Fig. 1B). Subsequently, we investigated
whether the induction was TLR2-dependent using TLR2 KO mice. While
MCP-1 expression was induced by Pam3CSK4 in
BMDM from wild-type mice, its expression was not affected in BMDM
from mice with deletions in TLR2 (TLR2 KO; Fig. 1B). Therefore, we postulated that
MCP-1 expression by Pam3CSK4 is dependent
upon TLR2. Many types of TLR ligands (LPS, flagellin, FSL1 and
CpG-ODN) other than the TLR2 ligand have also been found to induce
the expression of MCP-1 (data not shown). These results suggest
that MCP-1 induction by TLR ligands is a general characteristic.
MCP-1 was found at higher expression levels within atherosclerotic
lesions and, thus, considered a key player in the recruitment of
monocytes into the arterial wall (21). Therefore, our data suggest that the
Pam3CSK4-induced MCP-1 expression is
associated with atherosclerosis.
We further investigated the involvement of MyD88 by
TLR2 stimulation in Raw264.7 cells. TLR2 or MyD88 siRNA transfected
into cells was capable of successfully downregulating their
respective target genes. While MCP-1 expression induced by
Pam3CSK4 decreased in TLR2 siRNA cells, there
were no changes in MCP-1 expression in MyD88 siRNA cells (Fig. 1C). These results suggest that MCP-1
expression induced by Pam3CSK4 is
TLR2-dependent, but MyD88-independent. Currently, MyD88 is the most
known adaptor molecule in the TLR2 signaling pathway (22). However, there are currently very
few studies on the MyD88-independent pathway. The TLR2 signaling
pathway is a well-known mechanism through which MyD88 activates the
NF-κB transcription factor and induces various genes, including
cytokines, chemokines and inflammatory mediators (23). However, as our results demonstrate,
the fact that MCP-1 production by TLR2 stimulation is
MyD88-independent is very unique.
The MyD88-independent pathway has mainly been
studied in association with the TLR3 or TLR4 signaling pathway, and
the functions of TRIF-related adaptor molecule (TRAM) and TRIF have
mainly been focused upon. Studies on the MyD88-independent pathway
in TLR2 signaling are very limited. Chen et al (24) reported the TRIF-dependent pathway
in Chlamydia pneumonia, TLR2 ligand-induced foam cell
formation. Our preliminary data also demonstrate the participation
of TRIF in TLR2 signaling. In other words, TRIF siRNA cells
exhibited decreased MCP-1 expression by TLR2 stimulation (Fig. 1D). These results suggest the
existence of the MyD88-independent pathway in TLR2 signaling and
the feasibility of the TRIF-dependent pathway.
JAK2 is involved in the TLR2-mediated
MCP-1 expression
We tried searching for the downstream molecules of
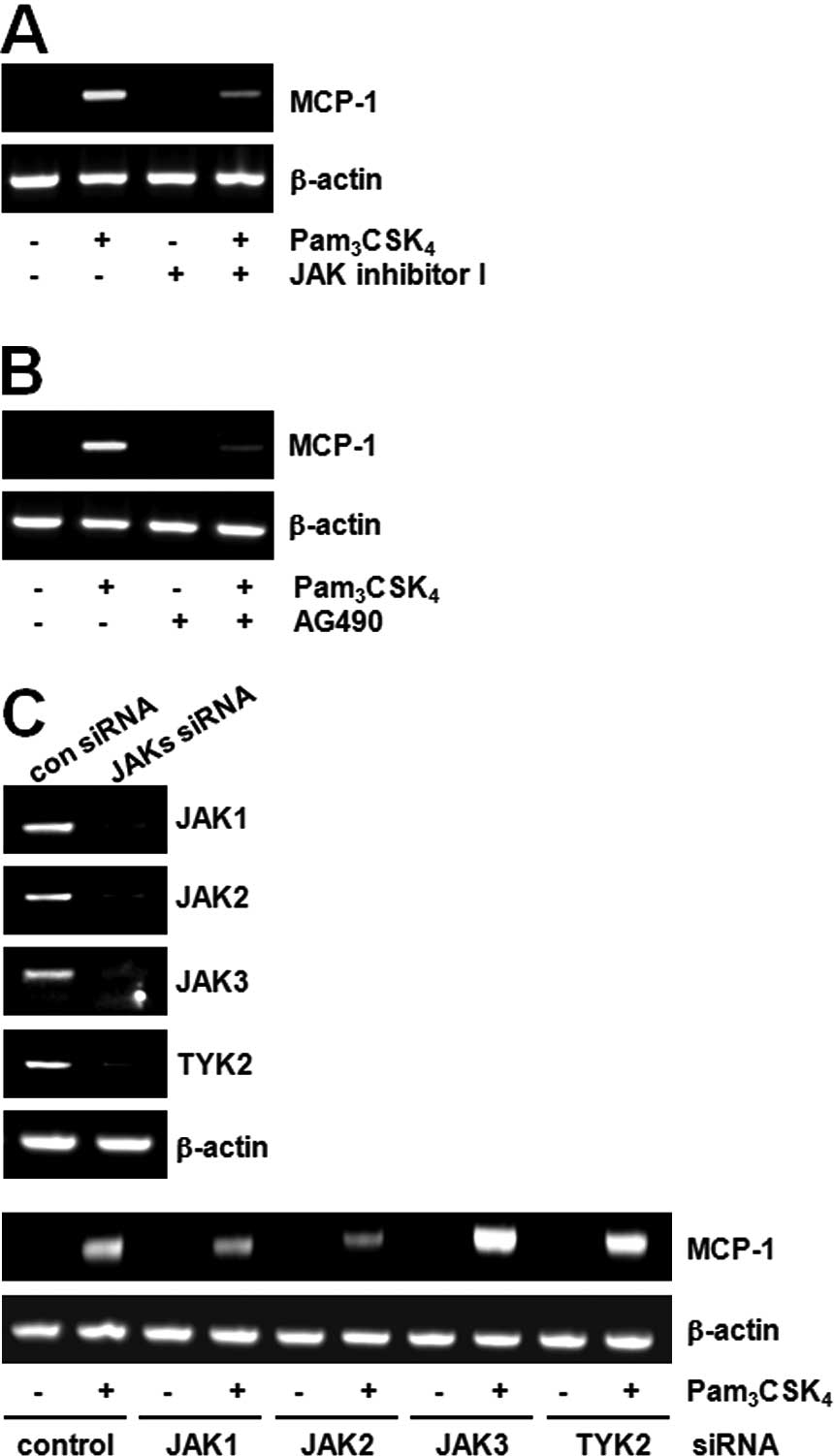
TLR2 in MCP-1 expression and found that JAK plays a role in MCP-1
induction. For example, pre-treatment with the JAK inhibitor
blocked MCP-1 expression induced by Pam3CSK4
(Fig. 2A). AG490, the JAK2
inhibitor, also inhibited MCP-1 expression induced by
Pam3CSK4 (Fig.
2B). To confirm the role of JAK2, the effects of
Pam3CSK4 were compared after decreasing JAK
expression using the siRNA method. The expression of JAK mRNA was
decreased to 60–80% in each siRNA-transfected cell. While
TLR2-mediated MCP-1 expression had no effects in JAK1, JAK3 or TYK2
siRNA cells, the expression of MCP-1 was decreased in the JAK2
siRNA cells (Fig. 2C).
Four types of JAKs have been identified and they
play an essential role in tyrosine kinase receptor signaling.
Although the participation of JAK2 in TLR2 signaling is of
interest, systematic study has not yet been performed. While not
much data is available on the correlation between TLR and JAK, the
function of JAK2 in TLR4-induced macrophage activation (25) and the function of TYK2 in
TLR4-induced endotoxin shock have been suggested (26). Currently, there are no reports on
the direct correlation between TLR2 and JAK2. In our data, the role
of JAK2 in TLR2-mediated MCP-1 production was highly feasible
through the MyD88-independent (TRIF) pathway. However, we still do
not know how TRIF regulates JAK2 activity for the MCP-1
expression.
Akt-GSK3β pathway is downstream of JAK2
for MCP-1 expression
We searched for the identity of the signaling
proteins regulated by JAK2 and found the involvement of the
Akt-GSK3β pathway. It is well-known that GSK3β is downstream kinase
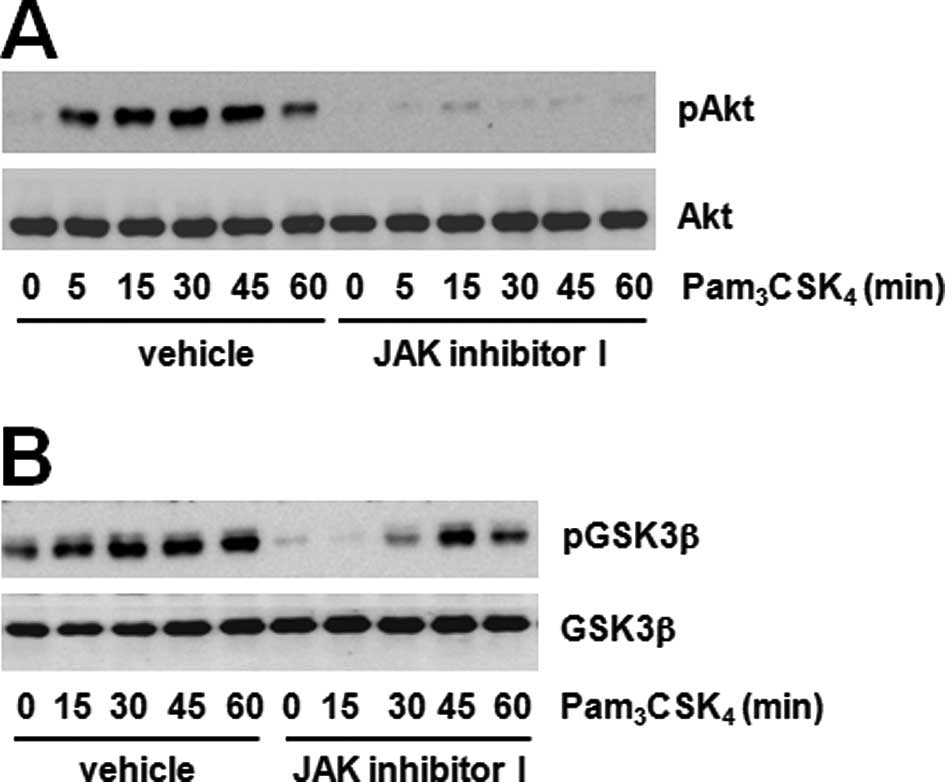
of Akt in many signaling pathways. We tested the phosphorylation of
two proteins using the JAK inhibitor to confirm the correlation
between JAK and the Akt-GSK3β pathway. The JAK inhibitor
dramatically decreased Akt and GSK3β phosphorylation induced by
Pam3CSK4 (Fig.
3A and B). These results suggest that JAK regulates the
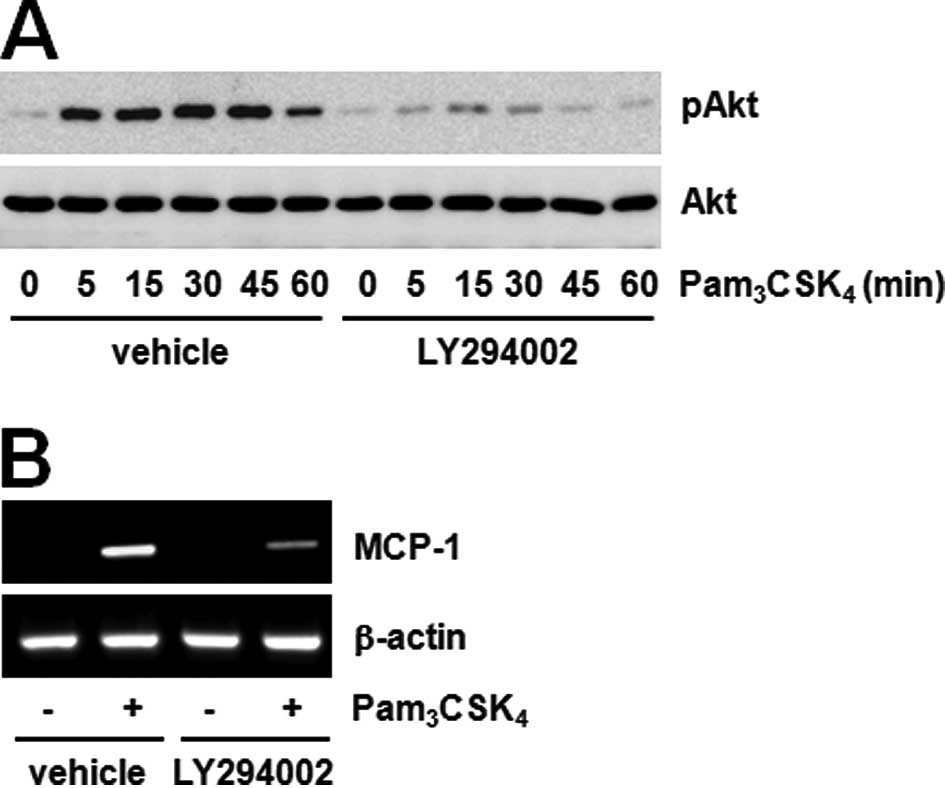
Akt-GSK3β pathway. Furthermore, LY294002 pre-treatment
significantly inhibited both Akt phosphorylation and MCP-1
expression induced by Pam3CSK4 (Fig. 4A and B). These results show that
Akt phosphorylation inhibited by JAK regulates MCP-1 expression
induced by TLR2. Previously, the possibility of PI3K-Akt pathway
activation by JAK in TLR2 signaling was assumed according to the
role played by IL-6 (27,28). The importance of the PI3K-Akt
pathway has also been suggested in TLR signaling. A number of
studies have demonstrated that the activation of the PI3K pathway
by a TLR2 agonist exerts effects on the production of inflammatory
mediators (29). The TLR4 agonist,
LPS, has been shown to induce Akt phosphorylation on both T308 and
S473, in which the blockade of PI3K attenuated phosphorylation
(30). However, the correlation
between the JAK and PI3K pathway has not been sufficiently studied
to date. Hirata et al (31)
suggested that IL-10 production by LPS plus imiquimod in dendritic
cells can be regulated by the JAK-PI3K axis. Our results also
showed the significant inhibition of TLR2-mediated Akt
phosphorylation by the JAK inhibitor, and therefore we believe that
the JAK-PI3K-Akt pathway is an essential step in MCP-1
regulation.
PI3K and Akt have been identified as kinases
involved in the ability of TLRs to mediate the regulation of GSK3β
activity (32).
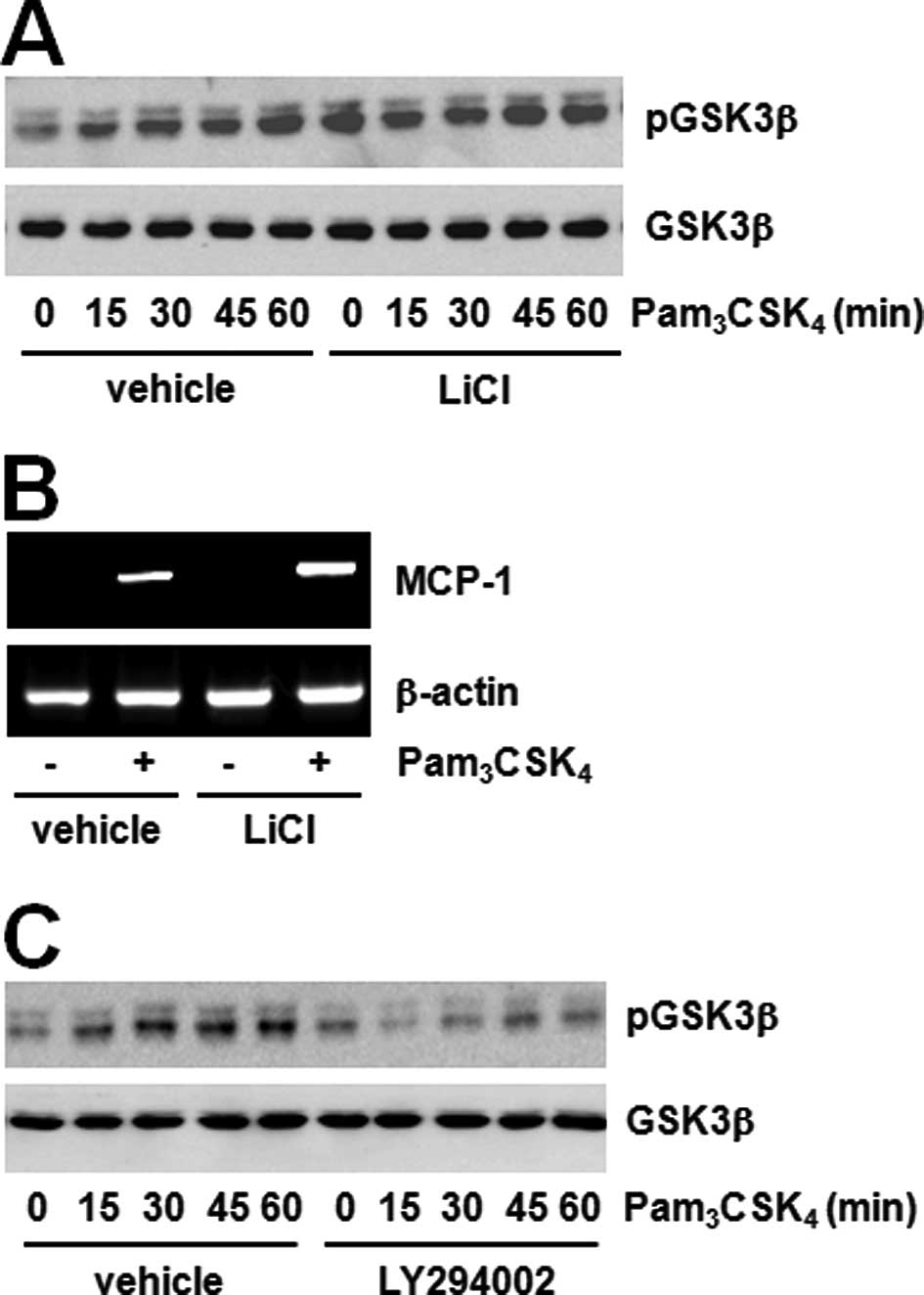
Pam3CSK4 increased GSK3β phosphorylation,
decreased activation and pre-treatment with the GSK3β inhibitor
LiCl potentiated GSK3β phosphorylation by stimulating
Pam3CSK4 (Fig.
5A). Additionally, LiCl pre-treatment amplified MCP-1
expression by stimulation of Pam3CSK4
(Fig. 5B). We then confirmed the
relationship between Akt and GSK3β. The PI3K-Akt inhibitor blocked
the TLR2-mediated GSK3β phosphorylation (Fig. 5C). These results suggest that the
JAK2-Akt-GSK3β pathway contributes to TLR2-mediated MCP-1
expression.
Overall, our results demonstrate a MyD88-independent
pathway in TLR2 signaling, thus providing a different mechanism
other than the already known MyD88-dependent pathway to regulate
MCP-1 expression, and thereby causing foam cell formation and
atherosclerosis. Additionally, TLR2-mediated GSK3β induced MCP-1
production in a negative manner through the JAK2-Akt signaling
pathway.
Acknowledgements
This study was supported by the Yeungnam University
research grants in 2010 (210-A-380-054).
Abbreviations:
|
TLRs
|
toll-like receptors
|
|
MCP-1
|
monocyte chemoattractant protein-1
|
|
JAK2
|
janus kinase 2
|
|
GSK3β
|
glycogen synthase kinase-3β
|
|
MyD88
|
myeloid differentiation primary
response gene 88
|
|
MAPKs
|
mitogen-activated protein kinases
|
|
BMDM
|
bone marrow-derived macrophage
|
|
TRIF
|
TIR domain-containing adapter inducing
IFN-β
|
References
|
1
|
A MencinJ KluweRF SchwabeToll-like
receptors as targets in chronic liver
diseasesGut58704720200910.1136/gut.2008.15630719359436
|
|
2
|
S AkiraToll-like receptor signalingJ Biol
Chem2783810538108200310.1074/jbc.R30002820012893815
|
|
3
|
S FrantzG ErtlJ BauersachsMechanisms of
disease: Toll-like receptors in cardiovascular diseaseNat Clin
Pract Cardiovasc Med4444454200710.1038/ncpcardio093817653117
|
|
4
|
S AkiraS UematsuO TakeuchiPathogen
recognition and innate
immunityCell124783801200610.1016/j.cell.2006.02.01516497588
|
|
5
|
JY KangX NanMS JinRecognition of
lipopeptide patterns by Toll-like receptor 2-Toll-like receptor 6
heterodimerImmunity31873884200910.1016/j.immuni.2009.09.01819931471
|
|
6
|
AH SchoneveldI HoeferJP SluijterJD LamanDP
de KleijnG PasterkampAtherosclerotic lesion development and Toll
like receptor 2 and 4
responsivenessAtherosclerosis19795104200810.1016/j.atherosclerosis.2007.08.00417888930
|
|
7
|
X LiuT UkaiH YumotoToll-like receptor 2
plays a critical role in the progression of atherosclerosis that is
independent of dietary
lipidsAtherosclerosis196146154200810.1016/j.atherosclerosis.2007.03.02517466307
|
|
8
|
AE MullickPS TobiasLK CurtissModulation of
atherosclerosis in mice by Toll-like receptor 2J Clin
Invest11531493156200510.1172/JCI2548216211093
|
|
9
|
S KuwahataS FujitaK OriharaHigh expression
level of Toll-like receptor 2 on monocytes is an important risk
factor for arteriosclerotic
diseaseAtherosclerosis209248254201010.1016/j.atherosclerosis.2009.08.04619766998
|
|
10
|
C MonacoN TerrandoKS MidwoodToll-like
receptor signaling: common pathways that drive cardiovascular
disease and rheumatoid arthritisArthritis Care Res
(Hoboken)63500511201110.1002/acr.2038221452263
|
|
11
|
AE MullickK SoldauWB KiossesTA Bell IIIPS
TobiasLK CurtissIncreased endothelial expression of Toll-like
receptor 2 at sites of disturbed blood flow exacerbates early
atherogenic eventsJ Exp
Med205373383200810.1084/jem.2007109618250194
|
|
12
|
Y AbeA KawakamiM OsakaApolipoprotein CIII
induces monocyte chemoattractant protein-1 and interleukin 6
expression via Toll-like receptor 2 pathway in mouse
adipocytesArterioscler Thromb Vasc
Biol3022422248201010.1161/ATVBAHA.110.210427
|
|
13
|
F CaoA CastrilloP TontonozF ReGI
ByrneChlamydia pneumoniae-induced macrophage foam cell
formation is mediated by Toll-like receptor 2Infect
Immun75753759200710.1128/IAI.01386-06
|
|
14
|
A ZerneckeE ShagdarsurenC WeberChemokines
in atherosclerosis: an updateArterioscler Thromb Vasc
Biol2818971908200810.1161/ATVBAHA.107.161174
|
|
15
|
RR KoenenC WeberTherapeutic targeting of
chemokine interactions in atherosclerosisNat Rev Drug
Discov9141153201010.1038/nrd304820118962
|
|
16
|
CW NiDL WangSC LienJJ ChengYJ ChaoHJ
HsiehActivation of PKC-epsilon and ERK1/2 participates in
shear-induced endothelial MCP-1 expression that is repressed by
nitric oxideJ Cell
Physiol195428434200310.1002/jcp.1025912704652
|
|
17
|
PG ArndtN SuzukiNJ AvdiKC MalcolmGS
WorthenLipopolysaccharide-induced c-Jun NH2-terminal kinase
activation in human neutrophils: role of phosphatidylinositol
3-Kinase and Syk-mediated pathwaysJ Biol
Chem2791088310891200410.1074/jbc.M30990120014699155
|
|
18
|
DW ParkK BaekJR KimResveratrol inhibits
foam cell formation via NADPH oxidase 1-mediated reactive oxygen
species and monocyte chemotactic protein-1Exp Mol
Med41171179200910.3858/emm.2009.41.3.02019293636
|
|
19
|
CS TsaiFY LinYH ChenCilostazol attenuates
MCP-1 and MMP-9 expression in vivo in LPS-administrated
balloon-injured rabbit aorta and in vitro in LPS-treated monocytic
THP-1 cellsJ Cell Biochem1035466200810.1002/jcb.2138817516547
|
|
20
|
JG LeeSH LeeDW ParkToll-like receptor
9-stimulated monocyte chemoattractant protein-1 is mediated via
JNK-cytosolic phospholipase A2-ROS signalingCell
Signal20105111200810.1016/j.cellsig.2007.09.00317939949
|
|
21
|
V BraunersreutherF MachS SteffensThe
specific role of chemokines in atherosclerosisThromb
Haemost97714721200717479181
|
|
22
|
R AhmadS El BassamP CordeiroJ
MenezesRequirement of TLR2-mediated signaling for the induction of
IL-15 gene expression in human monocytic cells by
HSV-1Blood11223602368200810.1182/blood-2008-02-13771118583567
|
|
23
|
JE ParkYI KimAK YiProtein kinase D1 is
essential for MyD88-dependent TLR signaling pathwayJ
Immunol18263166327200910.4049/jimmunol.080423919414785
|
|
24
|
S ChenR SorrentinoK ShimadaChlamydia
pneumoniae-induced foam cell formation requires MyD88-dependent
and -independent signaling and is reciprocally modulated by liver X
receptor activationJ
Immunol18171867193200810.4049/jimmunol.181.10.7186
|
|
25
|
S OkugawaY OtaT KitazawaJanus kinase 2 is
involved in lipopolysaccharide-induced activation of macrophagesAm
J Physiol Cell
Physiol285C399C408200310.1152/ajpcell.00026.200312686512
|
|
26
|
K KamezakiK ShimodaA NumataT MatsudaK
NakayamaM HaradaThe role of Tyk2, Stat1 and Stat4 in LPS-induced
endotoxin signalsInt
Immunol1611731179200410.1093/intimm/dxh11815226272
|
|
27
|
S RebouissouM AmessouG CouchyFrequent
in-frame somatic deletions activate gp130 in inflammatory
hepatocellular tumoursNature457200204200910.1038/nature07475
|
|
28
|
R RosellJ Bertran-AlamilloMA MolinaM
TaronIL-6/gp130/STAT3 signaling axis in cancer and the presence of
in-frame gp130 somatic deletions in inflammatory hepatocellular
tumorsFuture Oncol5305308200910.2217/fon.09.319374537
|
|
29
|
IT LeeCW LeeWH TungCooperation of TLR2
with MyD88, PI3K, and Rac1 in lipoteichoic acid-induced
cPLA2/COX-2-dependent airway inflammatory responsesAm J
Pathol17616711684201010.2353/ajpath.2010.09071420167866
|
|
30
|
GT BrownTM McIntyreLipopolysaccharide
signaling without a nucleus: kinase cascades stimulate platelet
shedding of proinflammatory IL-1beta-rich microparticlesJ
Immunol18654895496201110.4049/jimmunol.100162321430222
|
|
31
|
N HirataY YanagawaK IwabuchiK
OnoeSelective regulation of interleukin-10 production via Janus
kinase pathway in murine conventional dendritic cellsCell
Immunol258917200910.1016/j.cellimm.2009.03.00619361784
|
|
32
|
M MartinK RehaniRS JopeSM
MichalekToll-like receptor-mediated cytokine production is
differentially regulated by glycogen synthase kinase 3Nat
Immunol6777784200510.1038/ni122116007092
|