Introduction
Hypoxic-ischemic (HI) encephalopathy (HIE) is common
in clinical neonatal brain damage (1). However, knowledge of HIE has always
been limited. Most known treatments are symptomatic and prevent or
relieve the damaging effects of hypoxia-ischemia to neurons.
Prevention strategies are available (although limited), but
currently no strategies for the support of recovery/repair are on
the horizon, although hypothermia has been demonstrated in several
large multicenter studies as an essential treatment. However, at
present, no effective methods for promoting the regeneration of
injured neurons have been identified. Therefore, new strategies for
treating perinatal hypoxia-induced neuron injury are urgently
needed (2).
The failure of axonal regeneration in the central
nervous system (CNS) is largely attributed to the environment of
the injured axons. The inhibitory activity is principally
associated with CNS myelin components and the molecules in the
glial scar at the lesion site. Previous studies suggested that
three myelin proteins, i.e., the myelin-associated glycoprotein
(MAG), Nogo-A and oligodendrocyte myelin glycoprotein (OMgp),
collectively account for the majority of inhibitory activities in
CNS myelin (3).
The inhibitory activities of MAG, OMgp and the
extracellular domain of Nogo-A (Nogo-66) on HI-damaged neuron
regeneration may be mediated by common receptor complexes
consisting of the glycosylphosphatidylinositol-anchored Nogo
receptor (NgR) and its signaling coreceptors (4–7). A
number of studies have demonstrated that NgR may be involved in the
pathology observed following HI in neonatal rats and that the
inhibitory activity of NgR promotes axonal regeneration (8,9).
However, no difference has been observed in the neurite outgrowth
inhibition by the myelin inhibitors in the neurons of NgR-null and
wild-type mice. Moreover, no corticospinal-tract (10) regeneration has been reported. Thus,
a second receptor may mediate the inhibitory signaling of myelin
inhibitors.
The paired-immunoglobulin-like receptor B (PirB), a
second receptor, was originally identified in the cells of the
immune system and, more recently, in those of the nervous system
(11). Similar to NgR, PirB binds
Nogo66, MAG and OMgp. PirB is also capable of inhibiting neurite
outgrowth. The combined inhibition of NgR and PirB results in an
almost complete regeneration of damaged nerves. Moreover, neurons
of NgR-null mice which have been treated with PirB antibodies
partially block the axonal regeneration inhibition by Nogo66
(12). These findings strongly
suggest that compounds which target PirB and NgR enhance neuronal
regeneration in spinal cord injuries. However, certain studies have
indicated that PirB knockout in combination with NgR blockade did
not promote axonal regeneration and that blocking the function of
PirB is not sufficient to promote axonal reorganization or
functional recovery following cortical injury in vivo
(13,14). Therefore, it is possible that the
PirB and NgR inhibitory signaling pathways have some unknown
contacts. We aimed to investigate whether there were differences in
the HI-damaged neurons of newborn rats.
In the present study, we hypothesized that the
inhibition of PirB stimulates the regeneration of damaged nerves
through the inhibition of the signal transduction of myelin
inhibitors. PirB and NgR are functional receptors of myelin
inhibitors and the inhibitory activity of NgR on axonal growth is
mediated by the Rho-ROCK signaling pathways. Thus, we also
hypothesized that the inhibitory activity of PirB is mediated by
the Rho-ROCK signaling pathways. In the present study, we
investigated the effects of PirB on axonal regeneration and
explored the underlying signaling pathway in order to initiate the
search for new methods that promote nerve regeneration.
Materials and methods
Animals
Two-day-old Sprague-Dawley rats were used in the
experiments. The rats were obtained from the West China Medical
Laboratory Animal Center and maintained in a conventional mouse
facility. Protocols were approved by the Animal Care and the
Guidelines of Animal Use and Protection of Sichuan University were
followed. All possible efforts were made to minimize unnecessary
suffering of the animals.
Primary cultures of cortical neurons
Sprague-Dawley rats were sacrificed by cervical
dislocation. The cortices of rat were immersed in an ice-cold
calcium- and magnesium-free Hank’s Balanced Salt Solution. The
tissues were digested with trypsin (0.125% trypsin maintained at
37°C; HyClone Laboratories, Logan, UT, USA) for 30 min. The trypsin
solution was decanted and the tissues were washed three times with
serum-free Dulbecco’s minimal essential media (DMEM) (10 ml)
(HyClone Laboratories) supplemented with 20% fetal bovine serum
(HyClone Laboratories). The tissues were dispersed in this medium
with a screen at room temperature. The cells were placed at
1.5×105 cells/cm2 in polylysine-coated 6-well
plates (Becton Dickinson Labware, Franklin Lakes, NJ, USA)
containing DMEM supplemented with 10% heat-inactivated horse serum,
10% heat-inactivated fetal bovine serum, 2 mM glutamine and 25 mM
glucose without antibiotics. The cultures were maintained in a
humidified 5% CO2 incubator at 37°C. On the second day,
10 μM Ara-C was added to the cultures to inhibit non-neuronal cell
proliferation. Twice a week thereafter, half of the medium was
replaced with fresh fetal bovine serum-free culture medium. The
experiments on the cultured neurons were performed after 10
days.
HI neuron model
The neurons were randomly divided into four groups:
negative control, deprivation of oxygen and glucose, anti-PirB
antibody treatment and anti-PirB antibody-treated deprivation of
oxygen and glucose. For the deprivation of oxygen and glucose
group, the neurons were washed twice with sugar-free Earle’s
culture medium, maintained in the same medium and placed in a
humidified 5% CO2/95% N2 incubator at 37°C
for 4 h. For the anti-PirB antibody treatment group, the cells were
cultured in Earle’s culture medium supplemented with 50 μg/ml of
6C1, a specific anti-PirB antibody (Santa Cruz Biotechnology, Santa
Cruz, CA, USA) targeting the PirB extracellular region. For the
anti-PirB antibody-treated deprivation of oxygen and glucose group,
the cells were cultured in Earle’s culture medium supplemented with
50 μg/ml of 6C1 and placed in a humidified 5% CO2/95%
N2 incubator at 37°C for 4 h.
Immunocytochemistry
Following treatment, the cortical neurons were fixed
with 4% paraformaldehyde for 10 min, washed three times with 0.01 M
PBS for 5 min and blocked with 10% goat serum dissolved in PBS at
room temperature for 1 h. The cells were then incubated together
with anti-PirB antibodies overnight at 4°C, washed three times with
0.01 M PBS for 5 min, incubated together with goat anti-rabbit
antibodies and visualized using DAB. The Metamorph software was
used for capturing the images and quantification of the cell area.
To measure the staining intensities, we graphically highlighted
each individual neuron, including the nuclear region.
Neurite outgrowth assay
The isolated cortical neurons were placed in
polylysine-coated 6-well plates. Following the treatments, the
cultures were fixed for 30 min with 4% paraformaldehyde,
permeabilized with ice-cold methanol and immunostained with rabbit
polyclonal antibodies against βIII-tubulin (1:400; Santa Cruz
Biotechnology). The slides were viewed under an inverted
fluorescence microscope. The length of the longest neurite for each
βIII-tubulin-positive neuron for the first 180–200 neurons
encountered upon a systematic scanning of the slide was determined
using the Simple PCI image analysis program. Neurite length
quantitation and statistical analyses were performed as described
previously (15).
Fluorescent staining
The cells were washed with PBS and fixed with 4%
paraformaldehyde at room temperature for 10 min. After being washed
twice with ice-cold PBS, the fixed cells were blocked with 5% BSA
(Sigma, St. Louis, MO, USA) at room temperature for 30 min,
followed by incubation with the primary antibodies and a specific
anti-PirB antibody (Santa Cruz Biotechnology) for 1 h at room
temperature. The cells were then treated with the secondary
antibody conjugated with fluoresceinisothiocyanate (FITC; 1:100
dilution) in PBS for 1 h at room temperature. The images were
visualized using a Zeiss Imager Z1 fluorescence microscope (Zeiss,
Oberkochen, Germany).
Reverse transcription-polymerase chain
reaction (RT-PCR)
In a previous experiment (16), 6×105 cells from the
control and HI-damaged groups were used. After being centrifuged
and washed twice with 1 ml of PBS, the cell pellets were lysed
using ice-cold TRIpure TRIzol reagent. The total RNA was extracted
using chloroform, isopropanol and absolute ethyl alcohol
sequentially. The RNA pellets were dissolved in DEPC water. The
quality and concentration of the extracted RNA were confirmed using
1% agarose gel and an ultraviolet spectrophotometer. The qualified
RNA was stored at −80°C until use. Reverse transcription was
performed using 10 μl of reaction mixture containing 1 μl of 10X
PCR buffer, 1 μl of dNTP mix, 2 μl of negative sense primer, 0.25
μl of RNasin, 0.5 μl of AMV-RT, 2.75 μl of DEPC water and 2 μl of
the extracted RNA at 42°C for 30 min. The cDNA was stored at −20°C
until use. PCR was then performed in 50 μl of reaction mixture
containing 10 μl of 5X PCR buffer, 0.25 μl of Taq DNA polymerase,
0.5 μl of targeted gene premier (sense and reverse), 0.5 μl of
reference gene premier (sense and reverse) and 10 μl of cDNA. The
thermocycler program included denaturation at 94°C for 2 min; 35
cycles at 94°C for 30 sec and 58°C for 40 sec for the PirB gene and
56°C for 40 sec for the β-actin gene; annealing at 72°C for 40 sec;
and a final extension at 72°C for 30 sec. The RT-PCR products were
analyzed using 1% agarose gel electrophoresis followed by
densitometric scanning. The optical density of the gene bands was
analyzed using Quantity One software (Bio-Rad Laboratories,
Hercules, CA, USA) and the ratios of the targeted gene products to
β-actin products were calculated.
The primer pairs used for the RT-PCR with β-actin
and PirB were: β-actin forward: 5′-TCCGCTGCAGACAGACTGGCCAG-3′ and
reverse: 5′-AGTAACAGTCCGCCTAGAAGC-3′ (295 bp); PirB forward:
5′-TCGGGGAAAATTCAGGAA-3′ and reverse: 5′-GAGAAATCTCTAGCTTTATTT-3′
(215 bp).
ROCK II kinase activation
The cortical neurons were placed in
polylysine-coated 100 mm dishes. The cells were collected, washed
twice with PBS and then lysed in immune precipitation buffer (50 mM
of Tris-HCl, pH 7.4, 150 mM of NaCl, 5 mM of EDTA, 1% Nonidet P-40,
0.5% sodium deoxycholate, 0.1% SDS and proteinase inhibitors) for 1
h on ice. The receptor proteins were then immunoprecipitated with
50 μl of protein A-agarose beads preloaded with 10 μl anti-ROCK II
antibodies. The immunocomplexes were suspended in 2X SDS loading
buffer [125 mmol/l Tris-HCl (pH 6.8), 20% glycerol, 0.01%
bromophenol blue, 4% SDS and 200 mmol/l DTT], boiled for 5 min and
separated using SDS polyacrylamide gel electrophoresis with 30 μg
for each lane. Following electrophoresis, the proteins were
transferred to a PVDF membrane and infected with anti-β-actin
antibodies (Santa Cruz Biotechnology) or anti-PirB antibodies
(Upstate Biotechnology, Billerica, MA, USA), followed by
HRP-conjugated goat anti-mouse or anti-rabbit antibodies (Santa
Cruz Biotechnology). Enhanced chemiluminescence (Santa Cruz
Biotechnology) was employed. The density of the protein bands was
analyzed using the Quantity One software (Bio-Rad Laboratories).
The ratios of the targeted protein bands to GAPDH bands were
calculated.
Statistical analysis
Results were presented as the means ± standard
errors. The Student’s t-test was used to determine the levels of
difference among the groups. P<0.05 was considered to indicate a
statistically significant result.
Results
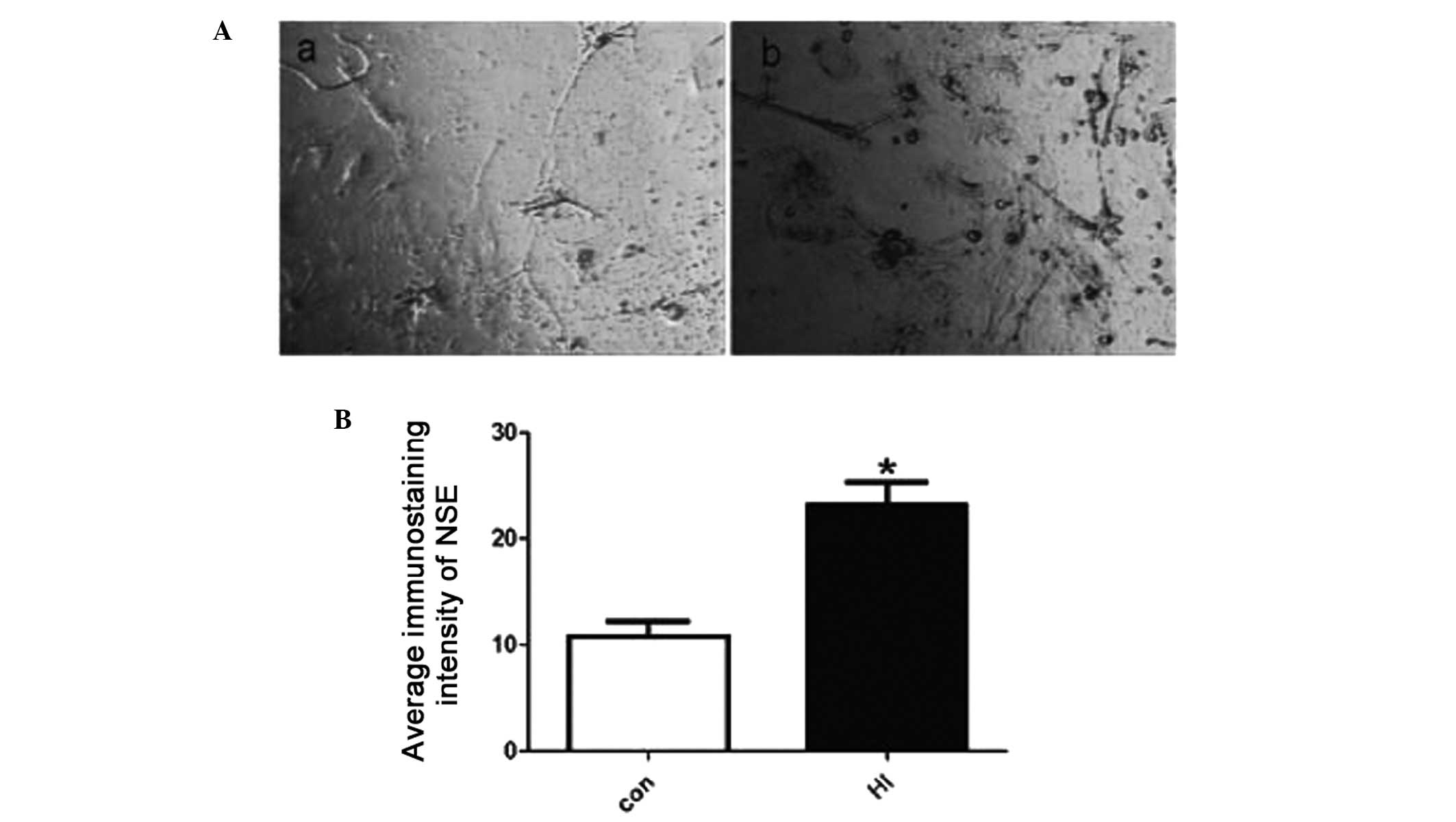
Cultured cortical neurons of newborn
rats
The cortical neurons of newborn rats were cultured
for eight days and randomly divided into two groups: control and
HI-damaged. Neuron injury was determined by neuron-specific enolase
immunostaining, which was predominantly and exclusively localized
in neuron cytoplasm and significantly increased following HI
treatment (Fig. 1).
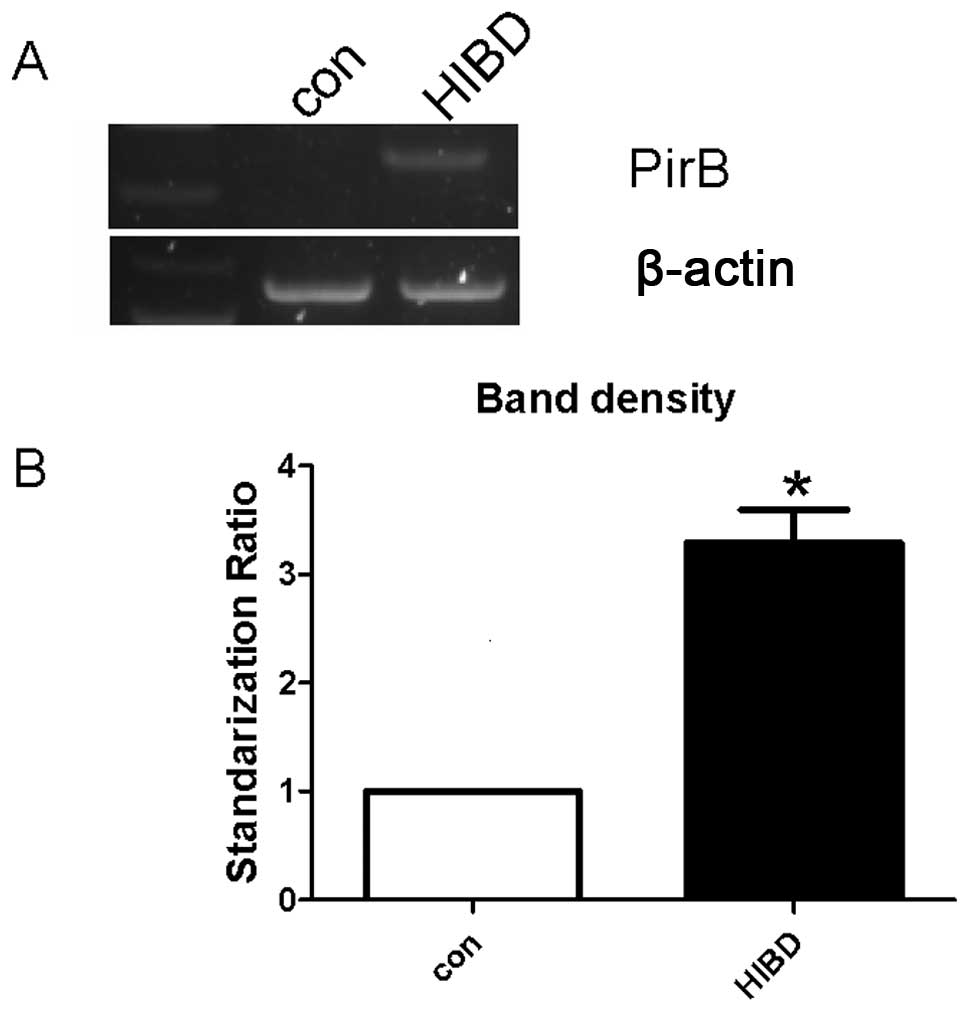
Cortical neurons
The PirB mRNA levels of the cortical neurons were
first measured using RT-PCR following HI injury. The level of PirB
mRNA increased in HI-damaged cortical neurons (Fig. 2A and B). Immunocytochemistry
confirmed the increased PirB protein expression in the HI-damaged
group (Fig. 2C and D). These
results demonstrate that the PirB mRNA and protein expression
increased in the cortical neurons of newborn rats following HI
damage.
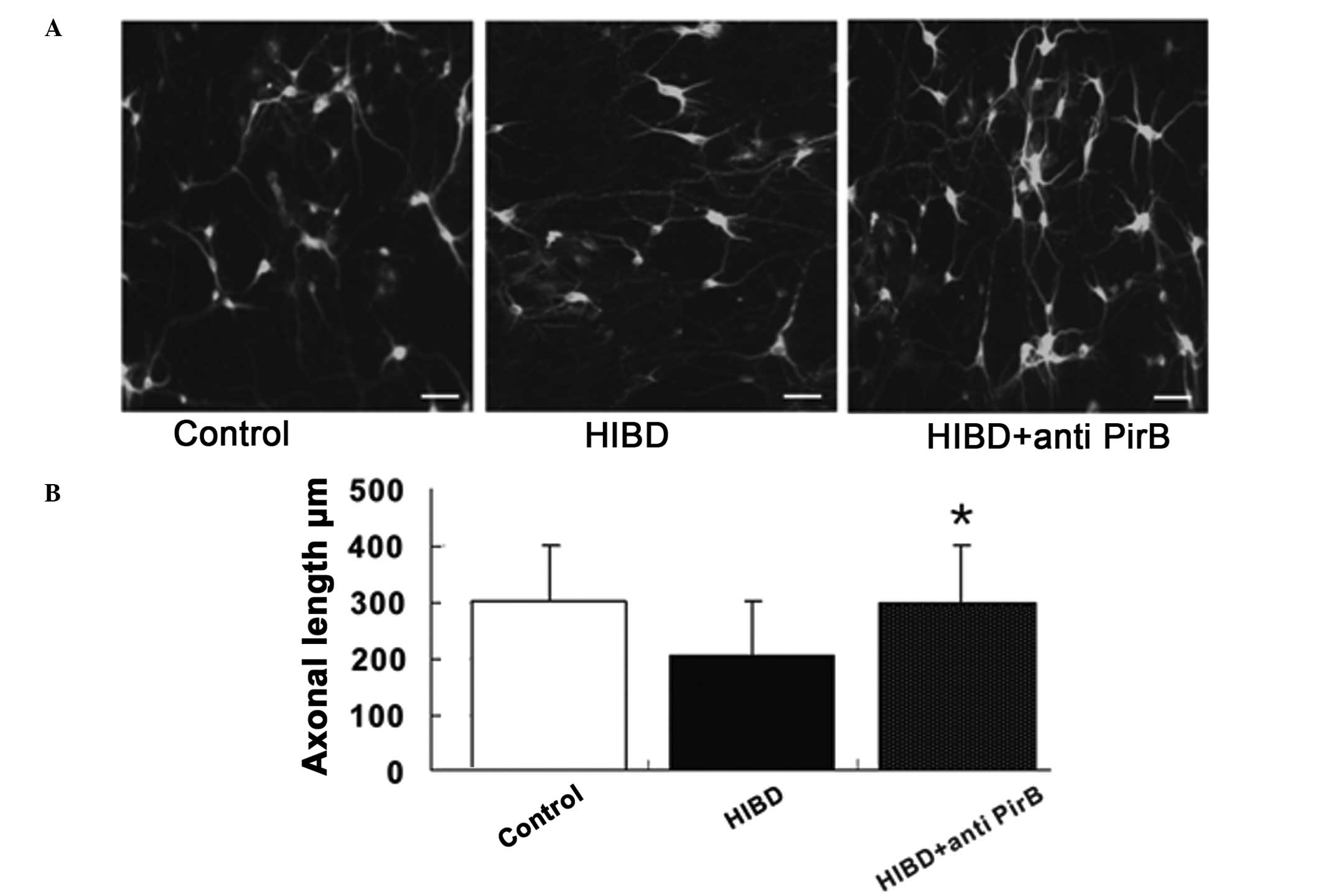
Axonal regeneration
The PirB mRNA and protein expression levels
increased following HI damage and findings of a previous study
revealed that PirB is able to bind myelin inhibitors (12). Therefore, the blocking of PirB
protein expression was investigated to determine whether it was
able to enhance axonal regeneration in HI-damaged cortical neurons.
The axonal length was evaluated using immunofluorescence inverted
microscopy in control neurons treated with PirB antibodies,
HI-damaged neurons and HI-damaged neurons treated with PirB
antibodies. PirB antibody treatment was not able to promote axonal
growth in normal control neurons but significantly induced
regeneration in HI-damaged neurons (Fig. 3), indicating that PirB is able to
improve axonal regeneration following HI damage.
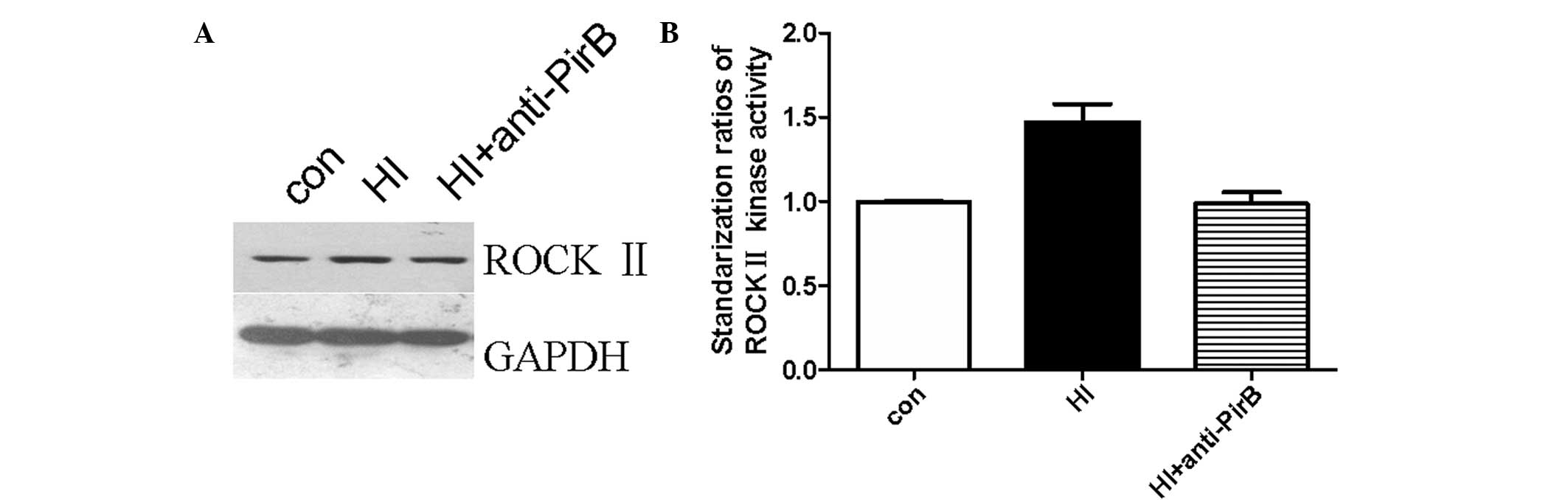
ROCK II kinase expression
ROCK II plays a significant role in the inhibition
of signal transduction when myelin inhibitors bind NgR. Thus, we
performed a ROCK II activation assay to investigate whether ROCK II
activity changed in HI-injured neurons and mediated the promoting
effect of PirB antibodies on axonal regeneration. First, we
observed that ROCK II expression increased following HI injury.
Second, we showed that the increased ROCK II expression was blocked
following the anti-PirB antibody treatment. The representative
western blot analysis result is shown in Fig. 4A and the standardization ratio
statistics of three tests are shown in Fig. 4B. The average intensities of the
bands significantly increased in the HI-damaged group. This result
suggests that ROCK II expression increased in HI-damaged cortical
neurons and was blocked by PirB antibodies.
Discussion
HI brain damage (HIBD) due to peri- or postnatal
asphyxia is a major cause of disability. Although various
neuroprotective strategies have been studied, the options for the
management of HI brain injuries are currently limited. Previous
studies have revealed that the prognosis of HIE is correlated with
the extent of neuronal damage. Thus, the successful regeneration of
HI-injured neurons is key in HIBD treatment.
Several myelin proteins are believed to account for
the inhibitory activity in CNS myelin. The most recognized of these
proteins include MAG, Nogo-A and OMgp. Inhibitory signaling is
transduced via their receptors, NgR and PirB. The trimeric receptor
complexes stimulate the RhoA-ROCK pathway when myelin-associated
inhibitors bind with Nogo-A. Nogo-A binds with NgR-P75NTR and PirB
binds not only with Nogo66 but also with MAG and OMgp with, at
least for MAG, the same affinity as that when it binds with
NgR-P75NTR (10).
The identification of PirB as a receptor for three
major myelin inhibitors of regeneration not only expands our
understanding but also adds to the complexity of preventing axonal
regeneration following brain injury. Thus, PirB is also a possible
target for therapeutic intervention to promote in vivo
regeneration. However, certain studies have indicated that PirB
knockout in combination with NgR blockade did not promote axonal
regeneration and that blocking the function of PirB is not
sufficient to promote axonal reorganization or functional recovery
following cortical injury in vivo (13,14).
Therefore, it is possible that the PirB and NgR inhibitory
signaling pathways have some unknown contacts and PirB may have a
role in inhibiting axonal regeneration in specific PirB-expressing
neurons in the CNS.
In the present study, we first examined PirB
expression in the cortical neurons of newborn rats following HI
injury. Then, we blocked the PirB protein expression using PirB
antibodies to determine whether it was possible to improve axonal
regeneration.
The results of the immunohistochemistry assay using
a specific antibody targeting the PirB intracellular segment
demonstrated that the distribution and content of PirB increased in
newborn rats following HIBD. Most of the PirB-positive neurons were
localized in the cerebral cortex, hippocampus and cerebellum
regions in which the MHC1 mRNA and protein were mainly located.
PirB, with a molecular weight of 130 kDa, was
identified in the immunoprecipitate from the brains of newborn
rats. This molecular weight is consistent with that of the PirB
found in the immune system. Our previous results demonstrate that
PirB protein expression significantly increased in the brain
tissues of newborn rats following HIBD using western blot analysis.
RT-PCR and immunohistochemistry also revealed that PirB mRNA and
the protein expression increased in the cortical neurons of newborn
rats following HI injury. The anti-PirB antibodies specifically
targeting the PirB extracellular region promoted the axonal
regeneration of the cortical neurons following HI injury,
indicating that the inhibition of PirB is a potential therapeutic
tool for improving axonal regeneration following HIBD.
As a functional receptor for myelin inhibitors, the
underlying signaling pathways that transduce the inhibitory signal
after PirB binds with the myelin inhibitors need to be identified.
Considering that Rho-ROCK plays a significant role in NgR-mediated
inhibitory effects and that Rho expression and activity were
elevated following HIBD, we hypothesized that the PirB-mediated
inhibitory effects are realized through the Rho-ROCK signaling
pathway. ROCK is a serine/threonine protein kinase and two isoforms
of ROCK encoded by two different genes have been described: ROCK I
(also known as ROKβ or p160 ROCK) and ROCK II (also known as ROKα).
ROCK II is preferentially expressed in the brain (17), whereas ROCK I has the highest
expression levels in non-neuronal tissues. Our results demonstrate
that ROCK II activity increased in the cortical neurons of newborn
rats following HI damage and this increase could be inhibited by
anti-PirB antibodies. This finding suggests that ROCK II is
regulated by PirB and that Rho-ROCK may be the signaling pathway
that transduces inhibitory signaling from PirB to the intracellular
part of neurons.
Acknowledgements
This study was supported by the grants from the
National Natural Scientific Fundation of China (No. 81070521;
30822039).
References
|
1
|
Roland EH and Hill A: Clinical aspects of
perinatal hypoxic-ischemic brain injury. Semin Pediatr Neurol.
2:57–71. 1995.
|
|
2
|
Lai MC and Yang SN: Perinatal
hypoxic-ischemic encephalopathy. J Biomed Biotechnol.
2011:6098132011.
|
|
3
|
Schwab ME: Nogo and axon regeneration.
Curr Opin Neurobiol. 14:118–124. 2004.
|
|
4
|
Fournier AE, GrandPre T and Strittmatter
SM: Identification of a receptor mediating Nogo-66 inhibition of
axonal regeneration. Nature. 409:341–346. 2001.
|
|
5
|
Wang KC, Koprivica V, Kim JA, Sivasankaran
R, Guo Y, Neve RL and He Z: Oligodendrocyte-myelin glycoprotein is
a Nogo receptor ligand that inhibits neurite outgrowth. Nature.
417:941–944. 2002.
|
|
6
|
Domeniconi M, Cao Z, Spencer T, et al:
Myelin-associated glycoprotein interacts with the Nogo66 receptor
to inhibit neurite outgrowth. Neuron. 35:283–290. 2002.
|
|
7
|
Shao Z, Browning JL, Lee X, et al:
TAJ/TROY, an orphan TNF receptor family member, binds Nogo-66
receptor 1 and regulates axonal regeneration. Neuron. 45:353–359.
2005.
|
|
8
|
Wang H, Yao YJ and Mu DZ: Expression of
Nogo-A and NgR in the developing rat brain after hypoxia-ischemia.
Brain Res. 1114:212–220. 2006.
|
|
9
|
GrandPré T, Li S and Strittmatter SM:
Nogo-66 receptor antagonist peptide promotes axonal regeneration.
Nature. 417:547–551. 2002.
|
|
10
|
Zheng B, Atwal J, Ho C, He XL, Garcia KC,
Steward O and Tessier-Lavigne M: Genetic deletion of the Nogo
receptor does not reduce neurite inhibition in vitro or promote
corticospinal tract regeneration in vivo. Proc Natl Acad Sci USA.
102:1205–1210. 2005.
|
|
11
|
Syken J, GrandPre T, Kanold PO and Shatz
CJ: PirB restricts ocular-dominance plasticity in visual cortex.
Science. 313:1795–1800. 2006.
|
|
12
|
Atwal JK, Pinkston-Gosse J, Syken J,
Stawicki S, Wu Y, Shatz C and Tessier-Lavigne M: PirB is a
functional receptor for myelin inhibitors of axonal regeneration.
Science. 322:867–870. 2008.
|
|
13
|
Nakamura Y, Fujita Y, Ueno M, Takai T and
Yamashita T: Paired immunoglobulin-like receptor B knockout does
not enhance axonal regeneration or locomotor recovery after spinal
cord injury. J Biol Chem. 286:1876–1883. 2011.
|
|
14
|
Omoto S, Ueno M, Mochio S, Takai T and
Yamashita T: Genetic deletion of paired immunoglobulin-like
receptor B does not promote axonal plasticity or functional
recovery after traumatic brain injury. J Neurosci. 30:13045–13052.
2010.
|
|
15
|
Cohen-Cory S and Fraser SE: Effects of
brain-derived neurotrophic factor on optic axon branching and
remodelling in vivo. Nature. 378:192–196. 1995.
|
|
16
|
Cohen-Cory S and Fraser SE: Effects of
brain-derived neurotrophic factor on optic axon branching and
remodelling in vivo. Nature. 378:192–196. 1995.
|
|
17
|
Hashimoto R, Nakamura Y, Kosako H, Amano
M, Kaibuchi K, Inagaki M and Takeda M: Distribution of Rho-kinase
in the bovine brain. Biochem Biophys Res Commun. 263:575–579.
1999.
|