Introduction
Previous epidemiological studies have hypothesized
an association between diabetes and cancer. Firstly, the incidence
of several types of cancer, including pancreatic, liver, breast,
colorectal, urinary tract and of the female reproductive organs, is
increased in diabetic patients (1). Secondly, cancer patients with
diabetes have been reported to respond poorly to cancer
chemotherapy and have a poorer prognosis than non-diabetic patients
(2).
Previous studies investigating patients with type 2
diabetes have reported that metformin significantly reduces cancer
incidence and improves the survival of these cancer patients
(3). Metformin is the most widely
used antidiabetic drug, functioning to reduce circulating glucose
levels, increase insulin sensitivity and reduce insulin
resistance-associated hyperinsulinemia (4). A number of studies have reported that
metformin is a novel anticancer drug that may be used to treat
specific types of cancer, including breast, ovarian, endometrial
and pancreatic (5–8). However, Bodmer et al reported
that metformin did not decrease the risk of lung cancer and colon
cancer (9,10). This may be ascribed to the various
genetic backgrounds of the numerous tumor types analyzed. For
example, Buzzai et al reported that metformin selectively
impairs p53-deficient tumor cell growth (11). To date, there has been extensive
epidemiological and retrospective studies that support the
antineoplastic effects of metformin (4). However, the precise mechanisms by
which metformin exerts its anti-tumor effects remain unclear.
Ras proteins are prototypical G-proteins that have
been shown to play key roles in signal transduction, proliferation
and differentiation (12). Under
normal conditions, Ras is activated transiently in a
stimulus-dependent fashion. By contrast, mutated Ras proteins are
constitutively active and thus, persistently stimulate growth or
differentiation (13). Therefore,
mutationally activated Ras genes are hypothesized to
represent important targets for cancer therapy. K-, H-and N-Ras of
the Ras family are central in regulating diverse cellular functions
important for growth, differentiation and survival. Of the Ras
proteins, K-ras is the most frequently mutated (14). A number of strategies have been
developed to target K-ras and specific agents have been
demonstrated to effectively treat cancer (15). However, agents that specifically
target K-ras with no toxicity in normal cells have not yet been
developed (16).
A previous study found that insulin induces
activation of the Ras gene (4). In addition, other studies have
reported that metformin reduces the levels of circulating insulin
(17–19,20).
Thus, we hypothesized that metformin exerts its antitumor effects
by reducing activation of the Ras gene. In the current
study, metformin was found to effectively inhibit the growth of
K-ras-mutated tumors but not K-ras wild-type tumors. In addition,
the antitumor effects of metformin were found to be mediated via
induction of apoptosis and inhibition of proliferation.
Downregulation of K-ras downstream signaling pathways contributed
to the inhibition of proliferation and induction of apoptosis. In
conclusion, observations of the current study hypothesize the use
of metformin to inhibit the growth of tumors with various molecular
signatures and in particular with different K-ras backgrounds.
Materials and methods
Cells and reagents
K-ras mutant tumor cells [human A549 lung
adenocarcinoma (21) and human
PANC-1 pancreatic (22)] and K-ras
wild-type tumor cells [human A431 epidermoid adenocarcinoma
(23)] were purchased from
American Type Culture Collection (Manassas, VA, USA) and were
cultured according to the manufacturer’s instructions. Metformin
was purchased from Sigma-Aldrich (St. Louis, MO, USA) and epidermal
growth factor (EGF) was obtained from Shanghai PrimeGene Bio-tech
Co., Ltd., (Shanghai, China).
Tumor growth assay
Seven-week-old female athymic (BALB/c, nu/nu) mice
used for the in vivo NSCLC xenograft study were purchased
from Beijing HFK Bioscience Co., Ltd. (Beijing, China). A549
(107), PANC-1 (107) and A431 cells
(107) were injected subcutaneously into the flanks of
the female nude mice and allowed to grow to a density of 100
mm3. Animals were randomly divided into two groups of
six mice. Control mice received daily intragastric administration
of the vehicle solution for metformin, H2O, while
treated mice received daily intragastric administration of
metformin (250 mg/kg).
Detection of Ki67 expression within
tumors
Tumor tissue was removed from tumor-bearing nude
mice following the final treatment and immunohistochemical analysis
was performed with an anti-Ki67 antibody (1:100; Thermo Fisher
Scientific Inc., Waltham, MA, USA) to determine the expression of
Ki67.
Terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labeling (TUNEL) assay in tumors
in situ
Tumor tissue was removed from tumor-bearing nude
mice following the final treatment and apoptosis levels in
vivo were determined using a TUNEL assay according to the
manufacturer’s instructions (Promega Corporation, Madison, WI,
USA).
Lactic acid assay
A549, PANC-1 and A431 cells were seeded at
2.5×105 cells/well in 6-well plates in the appropriate
media overnight and treated with metformin at four concentrations
(0, 1, 5 and 10 mM) for an additional 48 h. Supernatants of the
cell culture media were harvested and the concentration of lactic
acid was analyzed using a lactic acid assay according to the
manufacturer’s instructions (Nanjing Jiancheng Bioengineering
Institute, Nanjing, China). Viable cells were counted and the
lactic acid release/cell was calculated as follows: concentrations
of lactic acid/cell densities.
Cell cycle analysis by flow
cytometry
A549, PANC-1 and A431 cells were seeded at
2.5×105 cells/well in 6-well plates in the appropriate
media overnight. Subsequently, cells were starved for 24 h and
treated with 10% serum and four concentrations of metformin for 24
h. Cells were harvested, washed twice with PBS, fixed in a 70%
methanol solution and stored at 4°C overnight. Following fixation,
cells were resuspended and stained with PI and analyzed using flow
cytometry (Beckman Coulter Inc., Brea, CA, USA).
Methylthiazolyldiphenyl-tetrazolium
bromide (MTT) cell proliferation assay
A549, PANC-1 and A431 cells were treated with
varying doses of metformin (0, 0.5, 1, 2.5, 5 and 10 mM) for 72 h.
MTT (5 mg/ml; Sigma-Aldrich) was added to the 96-well plates at 10
μl/well and the plates were incubated for an additional hour. The
MTT reaction was terminated by the addition of 100 μl DMSO.
Subsequently, MTT assay results were obtained by measuring the
absorption of each well at 490 nm.
Apoptosis analysis by flow cytometry
A549, PANC-1 and A431 cells were seeded at
2.5×105 cells/well in 6-well plates in the appropriate
media overnight and were treated with 10% serum and four
concentrations of metformin for an additional 24 h. Cells were
harvested, washed twice with PBS, resuspended and stained with PI
and apoptosis levels were analyzed using flow cytometry (Beckman
Coulter, Inc.).
Western blot analysis
A549, PANC-1 and A431 cells were seeded at
2×105 cells/well in 6-well plates in the appropriate
media and treated with two concentrations of metformin (0 and 5 mM)
for 24 h. Cell lysates were prepared and analyzed by western blot
analysis as previously described (7). Antibodies against phospho-T172
adenosine monophosphate activated protein kinase (AMPK),
mitogen-activated protein kinase (MAPK), phospho-T202/204 MAPK, AKT
(protein kinase B), phospho-T308 AKT and p53 were purchased from
Cell Signaling Technology, Inc. (Danvers, MA, USA). The antibody
against AMPK was purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). Peroxidase-labeled antibodies against mouse
and rabbit were used as secondary antibodies (Beijing Zhongshan
Golden Bridge Biotechnology Co., Ltd., Beijing, China).
Statistical analysis
Data are presented as the mean ± SEM from a minimum
of three independent experiments, unless otherwise noted.
Statistical analyses were performed by Student’s t-test. P<0.05
was considered to indicate a statistically significant
difference.
Results
Differential inhibition of xenograft
tumor growth induced by metformin on various K-ras backgrounds
To gain an improved understanding of the effects of
metformin treatment on cancer cell xenografts in vivo, mice
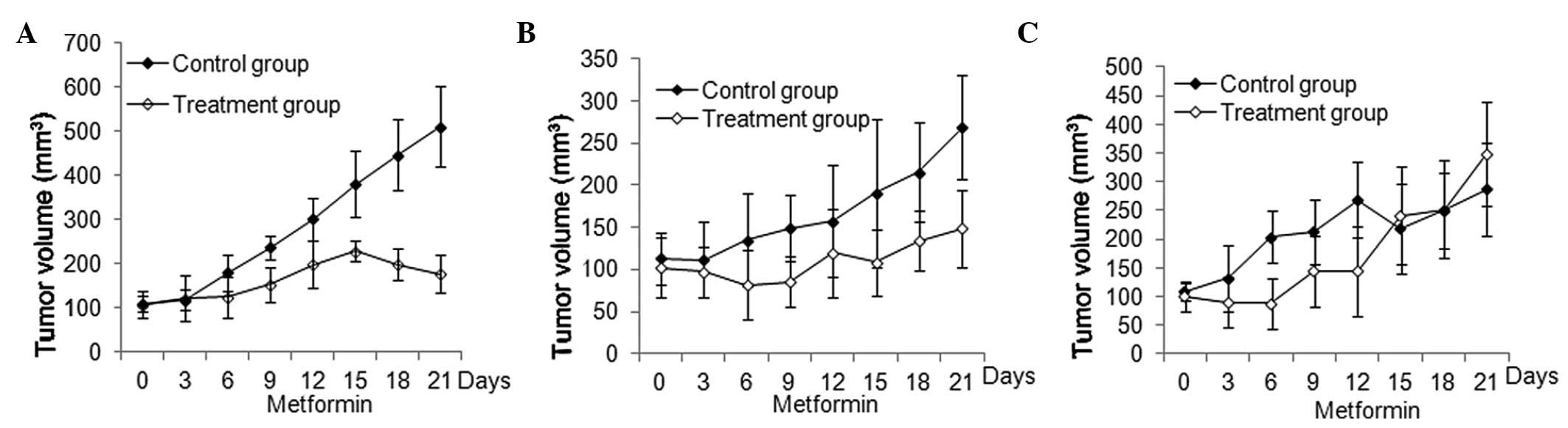
were administered metformin orally. As shown in Fig. 1, treatment with metformin resulted
in significant inhibition of human A549 and PANC-1 (both P<0.05)
but not A431 xenograft tumor growth (P>0.05).
Metformin induces apoptosis and inhibits
the proliferation of K-ras-mutated tumor cells in vivo
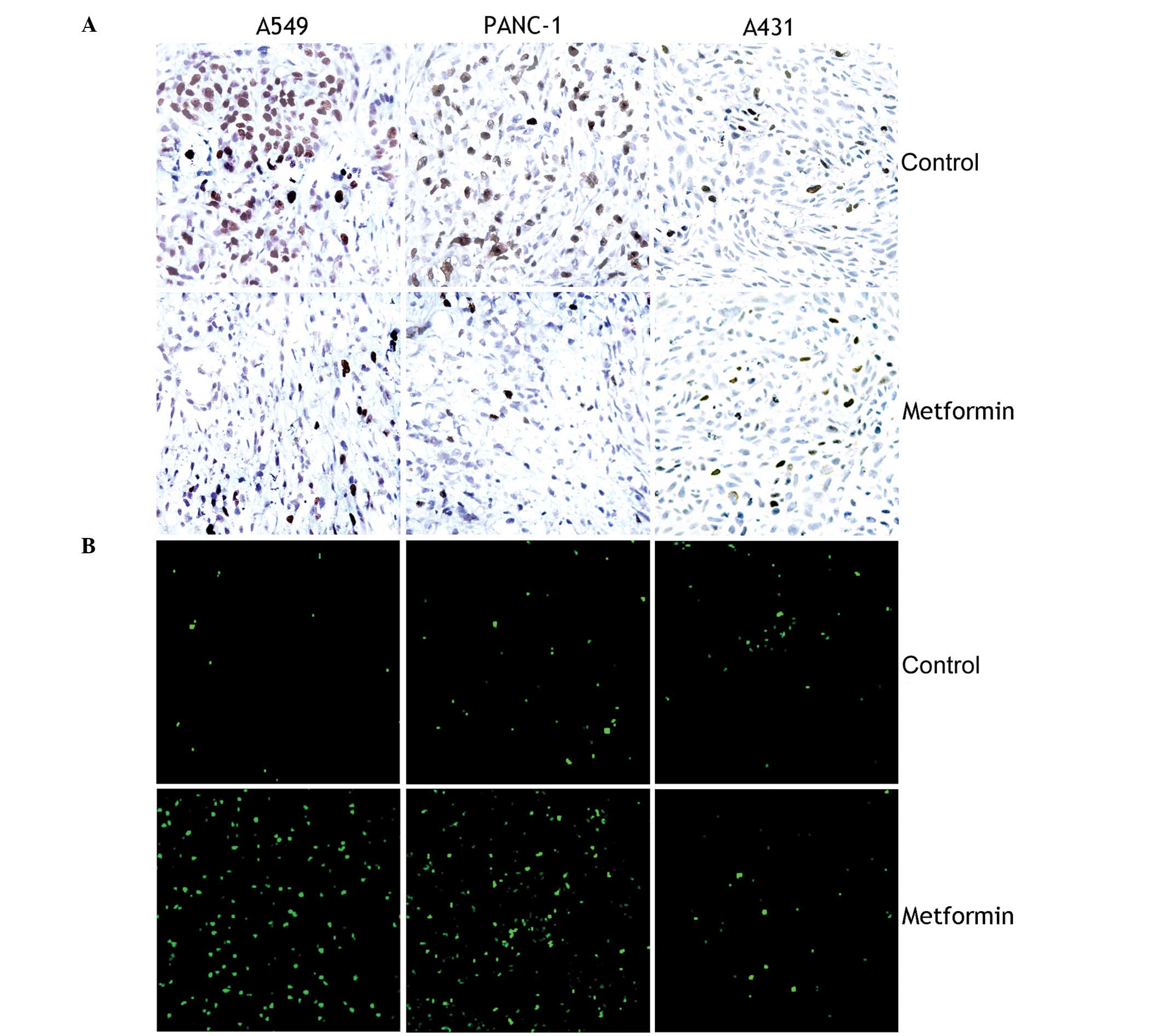
The effects of metformin on cell proliferation in
vivo were determined by staining sections from each group with
an anti-Ki67 antibody. As presented in Fig. 2A, treatment with metformin resulted
in significant inhibition of cell proliferation in A549 and PANC-1
cell xenografts (both P<0.05) but not in A431 cell xenografts
(P>0.05). Next, a TUNEL assay was performed to examine the
effects of metformin on apoptosis in vivo. As shown in
Fig. 2B, treatment with metformin
resulted in significant induction of apoptosis in A549 and PANC-1
cell xenografts (both P<0.05) but not in A431 cell xenografts
(P>0.05).
Metformin increases glycolysis and
induces cell cycle arrest and apoptosis while inhibiting
proliferation ex vivo
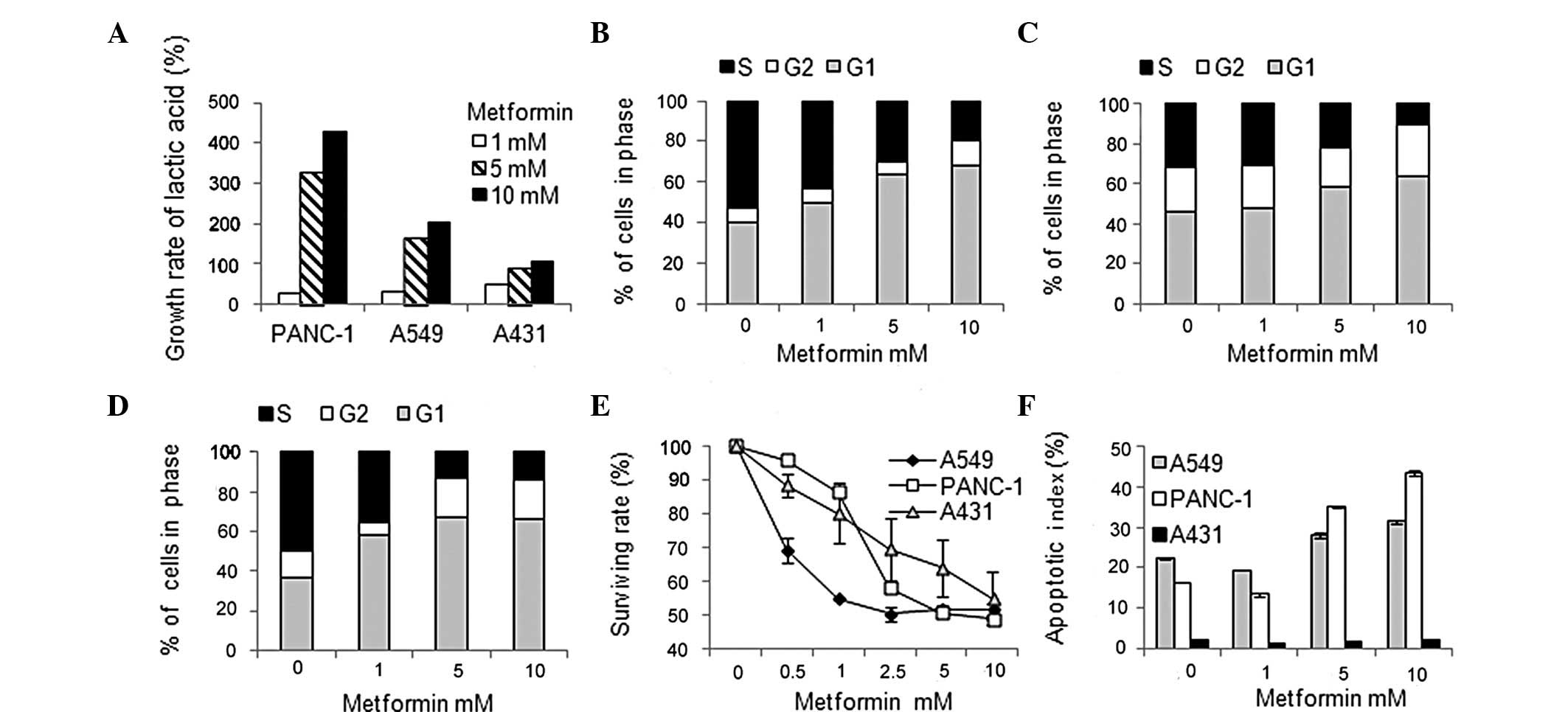
To understand the effects of metformin treatment on
glycolysis, the concentration of lactic acid in the supernatants of
the cell culture media was quantified and the lactic acid
release/cell was analyzed following treatment with metformin. As
presented in Fig. 3A, treatment
with metformin resulted in elevated levels of glycolysis in the
three cancer cell lines.
Effects of metformin treatment on cell cycle
progression were determined by flow cytometry analysis following
treatment with metformin. As presented in Fig. 3B–D, metformin significantly blocked
serum-induced entry into S phase in a dose-dependent manner,
resulting in cell cycle arrest in G1 phase in the three cancer cell
lines following 24 h metformin treatment.
To determine the effects of metformin treatment on
cell proliferation, an MTT assay was performed following treatment
with metformin (24). As presented
in Fig. 3E, metformin inhibited
cell growth in a dose-dependent manner in all three cancer cell
lines following 72 h metformin treatment. The mean IC50
value was ~5 mM for A549 and PANC-1 cells and ~10 mM for A431
cells.
The effects of metformin treatment on apoptosis were
investigated by flow cytometry analysis following treatment with
metformin. As presented in Fig.
3F, metformin induced apoptosis in A549 and PANC-1 cells. The
percentage change ranged between 9.2 and 27.2%, depending on the
cell line. However, metformin did not induce apoptosis in A431
cells.
Metformin inhibits the downstream
signaling effectors of K-ras
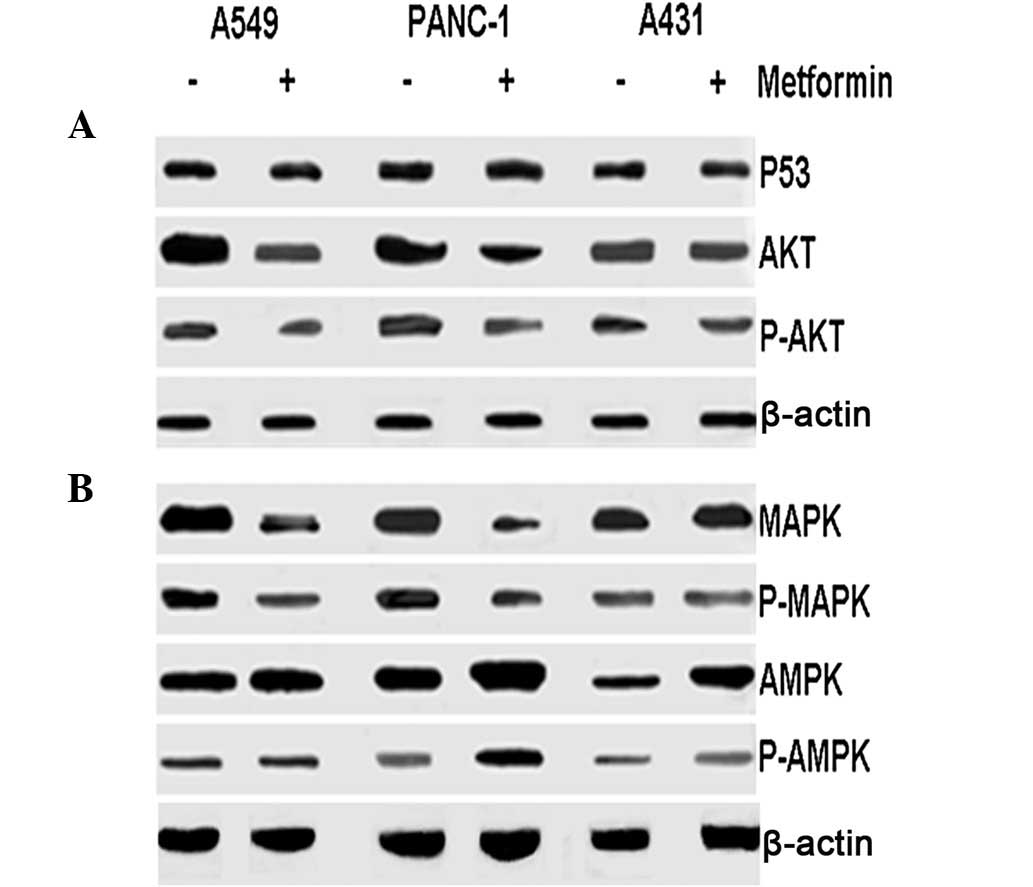
Western blotting was performed on the three cell
lines following treatment with metformin to determine its
underlying anti-tumor mechanisms. As presented in Fig. 4, expression of p53 was not affected
by metformin in A549, PANC-1 and A431 cells. Treatment with
metformin resulted in decreases in MAPK, p-MAPK, AKT and p-AKT
levels in A549 and PANC-1 cells but not in A431 cells. However,
metformin treatment increased AMPK and p-AMPK in PANC-1 cells and
A431 cells, but not in A549 cells.
Discussion
An increasing number of studies have implicated
metformin as a potentially effective anticancer drug; however, the
mechanism of action remains unclear. Observations of the current
study indicate that metformin specifically targets K-ras to elicit
its anticancer effects.
Using an in vivo mouse model, metformin was
observed to only exert anticancer effects in K-ras mutant tumors,
but not in K-ras wild-type tumors. To understand the mechanisms of
these effects in vitro, the effects of metformin on
glycolysis were investigated. As described by Warburg et al,
tumor cells maintain a high glycolytic rate even in conditions of
adequate oxygen supply (25).
Therefore, interfering with aerobic glycolysis represents a
potentially effective strategy to selectively target cancer cells.
Metformin increased the rate of glycolysis in K-ras mutant and
wild-type cell lines, indicating that the anticancer effects of
metformin are mediated through pathways other than aerobic
glycolysis.
Next, the effects of metformin on cell cycle
progression and proliferation were investigated. Metformin was
observed to significantly arrest the cells in G1 phase and inhibit
the proliferation of K-ras mutant and wild-type tumors. These
results did not highlight an explanation for the differential
efficacy of metformin on K-ras mutant and wild-type tumors in
vivo. Therefore, the effects of metformin on apoptosis were
investigated and it was observed that metformin induces apoptosis
in K-ras mutant tumors but not in K-ras wild-type tumors. This
indicates that metformin is likely to inhibit tumor growth of K-ras
mutant tumors by increasing apoptosis and inhibiting proliferation,
which is consistent with the in vivo results. The results
indicate that the anticancer activity of metformin is primarily due
to the induction of apoptosis and that metformin is cytostatic and
cytotoxic to K-ras mutant tumors. To gain a full understanding of
the underlying mechanisms by which metformin induces apoptosis, p53
expression, an essential component of apoptosis induction was
investigated. Expression of p53 was not affected by metformin
treatment in all three cancer cell lines and observations made by
Buzzai et al support the hypothesis that p53 is unnecessary
for apoptosis induction by metformin (11).
In the current study, metformin was observed to
target important downstream effectors of the Ras signaling pathway
in cancer therapy, including AKT. Akt/PKB is a downstream target of
PI3-K that plays a significant role in the regulation of apoptosis
by activating proapoptotic proteins, including Bad and caspase 9
(26). Following metformin
treatment, AKT levels were identified to decrease in A549 and
PANC-1 cells but not in A431 cells. These observations indicate
that metformin induces apoptosis by inhibiting AKT rather than via
the activation of p53. A previous study reported that metformin may
exert antitumor effects via the downregulation of AKT (27).
An additional important downstream effector of the
Ras signaling pathway is MAPK, which mediates the anticancer
effects of metformin. MAPK activation results in the transcription
of a number of genes associated with proliferation (28). Metformin treatment was observed to
reduce MAPK and p-MAPK levels in A549 and PANC-1 cells. However,
although metformin did not downregulate MAPK in A431 cells, the
proliferation of these cells was inhibited, indicating that
alternative mechanisms underlie the anti-proliferative effect of
metformin in these cells.
It has been widely reported that metformin may
function as an AMPK activator to target the LKB1/AMPK signaling
pathway in cancer therapy (29).
Thus, the impacts of metformin on AMPK activity in K-ras mutant and
wild-type cell lines were determined. AMPK is a cellular energy
sensor that inhibits tumorigenesis via the regulation of cell
growth, cell proliferation, autophagy, stress responses and cell
polarity (30). Treatment with
metformin was observed to activate AMPK in PANC-1 and A431 cells
but not in A549 cells. This indicates that activation of AMPK is a
key mediator of the anti-proliferative mechanisms of metformin in
PANC-1 and A431 cells. Therefore, the current study demonstrates an
inhibition of proliferation by metformin via the downregulation of
MAPK or the activation of AMPK. Once the LKB1 gene is
mutated, metformin may only inhibit proliferation by downregulating
MAPK. These observations are consistent with previous studies
(31–33).
In summary, the present study indicates that
metformin targets K-ras and may therefore represent a drug target
for the treatment of cancer. Mutational activation of Ras
genes is associated with 33% of human cancers, hence, Ras is
considered to be an important target for cancer therapy (34). However, an ideal agent that
specifically targets K-ras has not yet been developed (14,16).
Metformin is orally active, with a relatively favorable toxicity
profile and is low in cost. The promising results of our
preclinical study are consistent with previous studies analyzing
metformin treatment in clinical settings (35). Since the current observations are
preliminary, in depth molecular profiling of the patient’s tumor
must be performed prior to cancer treatment with metformin.
Acknowledgements
This study was supported by a grant from the
National Major Project (no. 2011ZX09302-001-01).
References
|
1
|
Vigneri P, Frasca F, Sciacca L, Pandini G
and Vigneri R: Diabetes and cancer. Endocr Relat Cancer.
16:1103–1123. 2009. View Article : Google Scholar
|
|
2
|
Gallagher EJ and LeRoith D: Insulin,
insulin resistance, obesity, and cancer. Curr Diab Rep. 10:93–100.
2010. View Article : Google Scholar
|
|
3
|
Martin-Castillo B, Vazquez-Martin A,
Oliveras-Ferraros C and Menendez JA: Metformin and cancer: doses,
mechanisms and the dandelion and hormetic phenomena. Cell Cycle.
9:1057–1064. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gonzalez-Angulo AM and Meric-Bernstam F:
Metformin: a therapeutic opportunity in breast cancer. Clin Cancer
Res. 16:1695–1700. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zakikhani M, Dowling R, Fantus IG,
Sonenberg N and Pollak M: Metformin is an AMP kinase-dependent
growth inhibitor for breast cancer cells. Cancer Res.
66:10269–10273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jones RG, Plas DR, Kubek S, et al:
AMP-Activated protein kinase induces a p53-dependent metabolic
checkpoint. Mol Cell. 18:283–293. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cantrell LA, Zhou C, Mendivil A, Malloy
KM, Gehrig PA and Bae-Jump VL: Metformin is a potent inhibitor of
endometrial cancer cell proliferation - implications for a novel
treatment strategy. Gynecol Oncol. 116:92–98. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kisfalvi K, Sinnett-Smith J, Eibl G and
Rozengurt E: Metformin inhibits growth of human pancreatic cancer
cells in vitro and in vivo. Pancreas. 38:1016–1017. 2009.
|
|
9
|
Bodmer M, Becker C, Jick SS and Meier CR:
Metformin does not alter the risk of lung cancer: a case-control
analysis. Lung Cancer. 78:133–137. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bodmer M, Becker C, Meier C, Jick SS and
Meier CR: Use of metformin is not associated with a decreased risk
of colorectal cancer: a case-control analysis. Cancer Epidemiol
Biomarkers Prev. 21:280–286. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Buzzai M, Jones RG, Amaravadi RK, et al:
Systemic treatment with the antidiabetic drug metformin selectively
impairs p53-deficient tumor cell growth. Cancer Res. 67:6745–6752.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Reuter CW, Morgan MA and Bergmann L:
Targeting the Ras signaling pathway: a rational, mechanism-based
treatment for hematologic malignancies? Blood. 96:1655–1669.
2000.PubMed/NCBI
|
|
13
|
Cox AD and Der CJ: Ras family signaling:
therapeutic targeting. Cancer Biol Ther. 1:599–606. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Friday BB and Adjei AA: K-ras as a target
for cancer therapy. Biochim Biophys Acta. 1756:127–144.
2005.PubMed/NCBI
|
|
15
|
Legaspi A, Jeevanandam M, Starnes HF Jr
and Brennan MF: Whole body lipid and energy metabolism in the
cancer patient. Metabolism. 36:958–963. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Blum R and Kloog Y: Tailoring Ras-pathway
- inhibitor combinations for cancer therapy. Drug Resist Updat.
8:369–380. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Skolnik EY, Batzer A, Li N, et al: The
function of GRB2 in linking the insulin receptor to Ras signaling
pathways. Science. 260:1953–1955. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Noguchi T, Matozaki T, Horita K, Fujioka Y
and Kasuga M: Role of SH-PTP2, a protein-tyrosine phosphatase with
Src homology 2 domains, in insulin-stimulated Ras activation. Mol
Cell Biol. 14:6674–6682. 1994.PubMed/NCBI
|
|
19
|
Desbois-Mouthon C, Cadoret A, Blivet-Van
Eggelpoël MJ, et al: Insulin and IGF-1 stimulate the beta-catenin
pathway through two signalling cascades involving GSK-3beta
inhibition and Ras activation. Oncogene. 20:252–259. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Medema RH, de Vries-Smits AM, van der Zon
GC, Maassen JA and Bos JL: Ras activation by insulin and epidermal
growth factor through enhanced exchange of guanine nucleotides on
p21ras. Mol Cell Biol. 13:155–162. 1993.PubMed/NCBI
|
|
21
|
Lehman TA, Bennett WP, Metcalf RA, et al:
p53 mutations, ras mutations and p53-heat shock 70 protein
complexes in human lung carcinoma cell lines. Cancer Res.
51:4090–4096. 1991.PubMed/NCBI
|
|
22
|
Watanabe M, Nobuta A, Tanaka J and Asaka
M: An effect of K-ras gene mutation on epidermal growth factor
receptor signal transduction in PANC-1 pancreatic carcinoma cells.
Int J Cancer. 67:264–268. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Oliveras-Ferraros C, Cufi S, Queralt B, et
al: Cross-suppression of EGFR ligands amphiregulin and epiregulin
and de-repression of FGFR3 signalling contribute to cetuximab
resistance in wild-type KRAS tumour cells. Br J Cancer.
106:1406–1414. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Iivanainen E, Lauttia S, Zhang N, et al:
The EGFR inhibitor gefitinib suppresses recruitment of pericytes
and bone marrow-derived perivascular cells into tumor vessels.
Microvasc Res. 78:278–285. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bartrons R and Caro J: Hypoxia, glucose
metabolism and the Warburg’s effect. J Bioenerg Biomembr.
39:223–229. 2007.
|
|
26
|
Feig LA and Buchsbaum RJ: Cell signaling:
life or death decisions of ras proteins. Curr Biol. 12:R259–R261.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ferla R, Haspinger E and Surmacz E:
Metformin inhibits leptin-induced growth and migration of
glioblastoma cells. Oncol Lett. 4:1077–1081. 2012.PubMed/NCBI
|
|
28
|
Adjei AA: Blocking oncogenic Ras signaling
for cancer therapy. J Natl Cancer Inst. 93:1062–1074. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang W and Guan KL: AMP-activated protein
kinase and cancer. Acta Physiol (Oxf). 196:55–63. 2009. View Article : Google Scholar
|
|
30
|
Carling D: AMP-activated protein kinase:
balancing the scales. Biochimie. 87:87–91. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mahoney CL, Choudhury B, Davies H, et al:
LKB1/KRAS mutant lung cancers constitute a genetic subset of NSCLC
with increased sensitivity to MAPK and mTOR signalling inhibition.
Br J Cancer. 100:370–375. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ben Sahra I, Regazzetti C, Robert G, et
al: Metformin, independent of AMPK, induces mTOR inhibition and
cell-cycle arrest through REDD1. Cancer Res. 71:4366–4372.
2011.PubMed/NCBI
|
|
33
|
Kalender A, Selvaraj A, Kim SY, et al:
Metformin, independent of AMPK, inhibits mTORC1 in a rag
GTPase-dependent manner. Cell Metab. 11:390–401. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cox AD and Der CJ: Farnesyltransferase
inhibitors: promises and realities. Curr Opin Pharmacol. 2:388–393.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li D: Metformin as an antitumor agent in
cancer prevention and treatment. J Diabetes. 3:320–327. 2011.
View Article : Google Scholar : PubMed/NCBI
|