Introduction
Two-dimensional gel electrophoresis (2-DE), a
technical tool possessing the ability to separate complex protein
mixtures with high resolution, has been widely employed for protein
profiling studies since the mid-1970s (1,2). It
is also the major method used in comparative proteomic studies to
screen biomarkers and identify molecular targets of drug actions,
as it enables differentially expressed proteins between different
groups to be readily visualized. However, optimization studies of
the 2-DE method have not yet been conducted, particularly regarding
the analytical conditions and procedures of protein extraction,
duration of isoelectric focusing (IEF) and reduction and alkylation
reaction.
The 2-DE experiment was divided into three major
steps: The extraction of proteins from tissues or cells;
first-dimension (1D) separation based on protein isoelectric points
(pI) using IEF; and the second-dimension (2D) separation
based on protein molecular weight using sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (3). Of these, protein extraction and 1D
separation are the most crucial steps for optimizing 2-DE analysis.
During protein extraction, a number of proteins may be
non-specifically lost or degraded, leading to variability of
results in 2-DE analysis. In 1D separation, immobilized pH gradient
(IPG) strips with different lengths are used according to the
specific purposes of the study. For instance, the shorter strips
(7, 11 and 13 cm) usually result in faster and cost-effective
screening, but the protein loading capacity is limited and the
quantitation and identification of particular spots may be
compromised due to spot overlapping (4). The longer strips (17, 18 and 24 cm),
which are designed for maximizing resolution and loading capacity,
allow the improved detection of spots and easier selection and
identification of the proteins in the map. In addition, the
alkylation of amino acid residues by cross-linkers during IEF
processes may be non-specifically induced (5). Therefore, conditions in these
procedures require optimization.
In the 2-D gel analysis of tumor tissue samples, it
is particularly difficult to achieve acceptable resolution using
the existing 2-DE protocols due to the multicellularity of tumor
tissues. To date, 2-D gel methods performed on tumor tissue
proteomes have been, generally, based on the existing procedures.
In the present study, the key steps of sample preparation,
reduction and alkylation, and the 1D separation process in 2-DE
analysis on 24-cm strips were optimized for the 2-DE images of
proteins in mouse tumor tissue samples. The optimized conditions
provide a valuable tool for identifying novel biomarkers of
diseases and the underlying molecular mechanisms and targets of
drugs.
Materials and methods
Establishment of a lung cancer model in
mice
The LLC-1 cells were obtained from the American Type
Culture Collection (Rockville, MD, USA). Male C57BL/6J mice (n=12)
at the age of 6–8 weeks were obtained from The Chinese University
of Hong Kong (Hong Kong, China). Animal care and treatment
procedures conformed to the Institutional Guidelines and Animal
Ordinance (Department of Health, HKSAR). The LLC-1 cells harvested
from in vitro culture were adjusted to a concentration of
1.5×107 cells/ml and 0.1 ml cell suspension was injected
subcutaneously into the dorsal region of the male C57BL/6J mice.
After 21 days, the mice were sacrificed and their tumor tissues
were dissected and isolated for frozen storage at −80ºC. The animal
experiment was approved by the ethics committee of Macau University
of Science and Technology (Macau, China).
Protein extraction from tumor
tissues
Tumor tissues (50 mg) were washed with distilled
water three times and protein extraction was performed by one of
the following four procedures. i) Bead mill-based protein
extraction using a TissueLyser LT (Qiagen, Hilden, Germany):
Proteins in the mouse tumor tissues were extracted using
TissueLyser LT (Qiagen): Proteins in the mouse tumor tissues were
extracted using TissueLyser for 2 min at 50 Hz with two metal beads
with urea/thiourea lysis buffer [7 M urea, 2 M thiourea, 4% (w/v)
CHAPS, 30 mM Tris/HCl, pH 9.0; 1:10 w/v] and urea lysis buffer [8 M
urea, 4 % (w/v) CHAPS, 30 mM Tris/HCl, pH 9.0; 1:10 w/v],
respectively. ii) Homogenization-assisted protein extraction: The
tumor tissues were cut into fine pieces and homogenized with
urea/thiourea lysis buffer (1:10 w/v). iii) Sonication-assisted
protein extraction: The tumor tissues were sonicated (10 strokes,
low amplitude) with urea/thiourea lysis buffer (1:10 w/v) on ice.
iv) Grinding-assisted protein extraction: The frozen tumor tissues
were ground to a fine powder under liquid nitrogen and then
immediately suspended in urea/thiourea lysis buffer (1:10 w/v).
The lysates obtained by each of the four techniques
were incubated on ice for 5 min and centrifuged at 17,000 × g at
4ºC for 1 h. The supernatants were collected and 200 μl of the
extracts was precipitated with a 2-D Clean Up kit (GE Healthcare,
Chalfont St. Giles, UK) according to the manufacturer’s
instructions, or with ice-cold acetone. The pellets were
resuspended in urea/thiourea lysis buffer and the protein
concentration was determined using a 2-D Quant kit (GE Healthcare).
The protein concentration was adjusted to 5 μg/μl.
2-DE
Rehydration
For rehydration, the existing procedure (3) and the modified methods of the present
study were used as below.
The existing procedures (
3)
The protein sample was made up to 5 μg/μl in 450 μl
with rehydration buffer [8 M urea, 2% (w/v) CHAPS, 0.5% (v/v) of pH
3.0–10.0 NL IPG buffer (GE Healthcare)] containing 1% or 2% DTT.
Additionally, an extra paper soaked with 1% DTT was placed near the
cathode during 1D IEF. The mixtures were applied to Immobiline
DryStrip gels (IPG strips; pH 3–10 NL, 24 cm; GE Healthcare) by
in-gel rehydration for 16 h at room temperature in an immobiline
DryStrip Reswelling Tray (GE Healthcare).
Modified methods
Reduction and alkylation of proteins was performed
prior to IEF. The protein samples (5 μg/μl) was pre-reduced and
alkylated by one of the six methods shown in Table I. The mixtures were made up to 450
μl with rehydration buffer and applied to the IPG strips using the
same procedure as previously described.
 | Table IDifferent combinations of conditions
for 2-DE. |
Table I
Different combinations of conditions
for 2-DE.
| Treatment per 5 μg/μl
protein | | | |
|---|
|
| | | |
|---|
| Combination | DTT | IAA | DTT | Temperature | Time | Quality of image |
|---|
| I | 10 mM | 40 mM | 40 mM | On ice | 0.5 h | Low resolution and
horizontal streaks |
| II | 10 mM | 40 mM | 40 mM | On ice | 1 h | High resolution and
more protein spots |
| III | 10 mM | 40 mM | 40 mM | On ice | 1.5 h | High resolution and
more protein spots |
| IV | 10 mM | 40 mM | 40 mM | 24ºC | 1 h | Low resolution and
horizontal streaks |
| V | 10 mM | 10 mM | 10 mM | On ice | 1 h | Low resolution and
horizontal streaks |
| VI | 20 mM | 80 mM | 80 mM | On ice | 1 h | Low resolution;
horizontal streaks; strip was burned |
1D IEF
The IPG strips were transferred to an Ettan IPGphor
II Manifold (GE Healthcare). IEF was initiated at a low voltage
(200 V, 1 h; step-n-hold, 500 V, 1 h; step-n-hold, 1,000 V, 1 h;
step-n-hold), and then raised to 10,000 V for 6 h, with the current
limited to a maximum of 75 μA/strip throughout the procedure. The
strips were focused for 40,000, 50,000, 70,000 and 80,000 Vhr,
respectively.
2D SDS-PAGE
The focusing IPG strips were immediately
equilibrated in SDS equilibration buffer [6 M urea, 75 mM Tris-HCl
(pH 8.8), 30% glycerol (v/v), 2% SDS (w/v), 0.002% (w/v)
bromophenol blue] containing 10 mg/ml DTT for 15 min, and
thereafter in the SDS equilibration buffer containing 25 mg/ml IAA
for 15 min. Following equilibration, proteins were separated by
12.5% SDS-PAGE, which was run until the bromophenol blue dye
reached the end of the gel.
Image analysis
Following 2-DE separation, the gels were fixed for
>30 min in 40% ethanol and 10% acetic acid, and stained using a
PlusOne Silver Staining kit (GE Healthcare). The stained gels were
then scanned with an Image Scanner III (GE Healthcare) and images
of the spots were automatically analyzed using ImageMaster 2D Elite
software (GE Healthcare).
Protein identification by
MALDI-TOF/TOF
The protein spots of interest were excised from the
silver-stained gels and subjected to trypsin digestion, according
to the methods of Shevchenko et al (6). The digested peptides were extracted
from the excised gel pieces and analyzed using an autoflex™ speed
MALDI-TOF/TOF system (Bruker Daltonics, Bremen Germany).
For the acquisition of mass spectra, a thin layer of
matrix (40 mg/ml α-cyano-4-hydroxycinnamic acid in 98% acetone) was
deposited on an MTP AnchorChip™ 384 MALDI target (800 μm diameter;
Bruker Daltonics). One microliter of the extracted peptides was
spotted onto a thin layer of the matrix solution on the target
plate and allowed to air dry. MALDI TOF/TOF MS data were acquired
using FlexControl 2.4 (Bruker Daltonics) and analyzed using
FlexAnalysis 2.4 (Bruker Daltonics). The generated peak list was
searched against the SwissProt Mus musculus protein database
(SwissProt 57.1; 462,764 sequences, 163,773,385 residues) using
in-house MASCOT Server software, version 2.3 (Matrix Science,
London, UK) with the following parameters: cysteine
carbaminomethylation and methionine oxidation as fixed modification
and variable modification, respectively; precursor mass tolerance,
50 ppm; MS/MS mass tolerance, 0.5 Da; and a maximum of one missed
cleavage was allowed. Proteins were only identified when the ion
score was above the sequence identity threshold.
Results
Optimization of the protein extraction
methods
The quality of 2-DE analysis largely depends on the
quality of sample preparation during protein extraction.
Unnecessary procedures should be avoided to increase the
reproducibility and minimize modifications that may result in
artifactual spots on 2D gels. The present study compared four
protein extraction methods in terms of protein yield and processing
time (Table II). In general,
protein extraction by grinding in liquid nitrogen, sonication or
homogenization was highly time-consuming, with a low throughput and
low protein yield. Extraction using a TissueLyser yielded the
highest amount of protein within a minimal processing time. In
addition, 12 samples were processed simultaneously. Therefore, this
optimized method was adapted for the subsequent 2-DE analysis.
| Table IIComparison of protein yield and
processing time for four protein extraction conditions with
urea/thiourea lysis buffer. |
Table II
Comparison of protein yield and
processing time for four protein extraction conditions with
urea/thiourea lysis buffer.
| Conditions | Processing time
(min/no. of samples) | Protein yield
(μg/mg) |
|---|
| TissueLyser | 2/12 | 100 |
| Homogenization | 2/1 | 20 |
| Grinding | 1/1 | 70 |
| Sonication | 2/1 | 80 |
Clean-up of protein samples
Following protein extraction, contaminants such as
salts, lipids, nucleic acids and detergents must be removed to
prevent horizontal streaking during IEF. Proteolytic enzymes must
also be inhibited to prevent non-specific protein degradation
(7). The present study used a 2-D
Clean-Up kit and acetone precipitation for contaminant removal
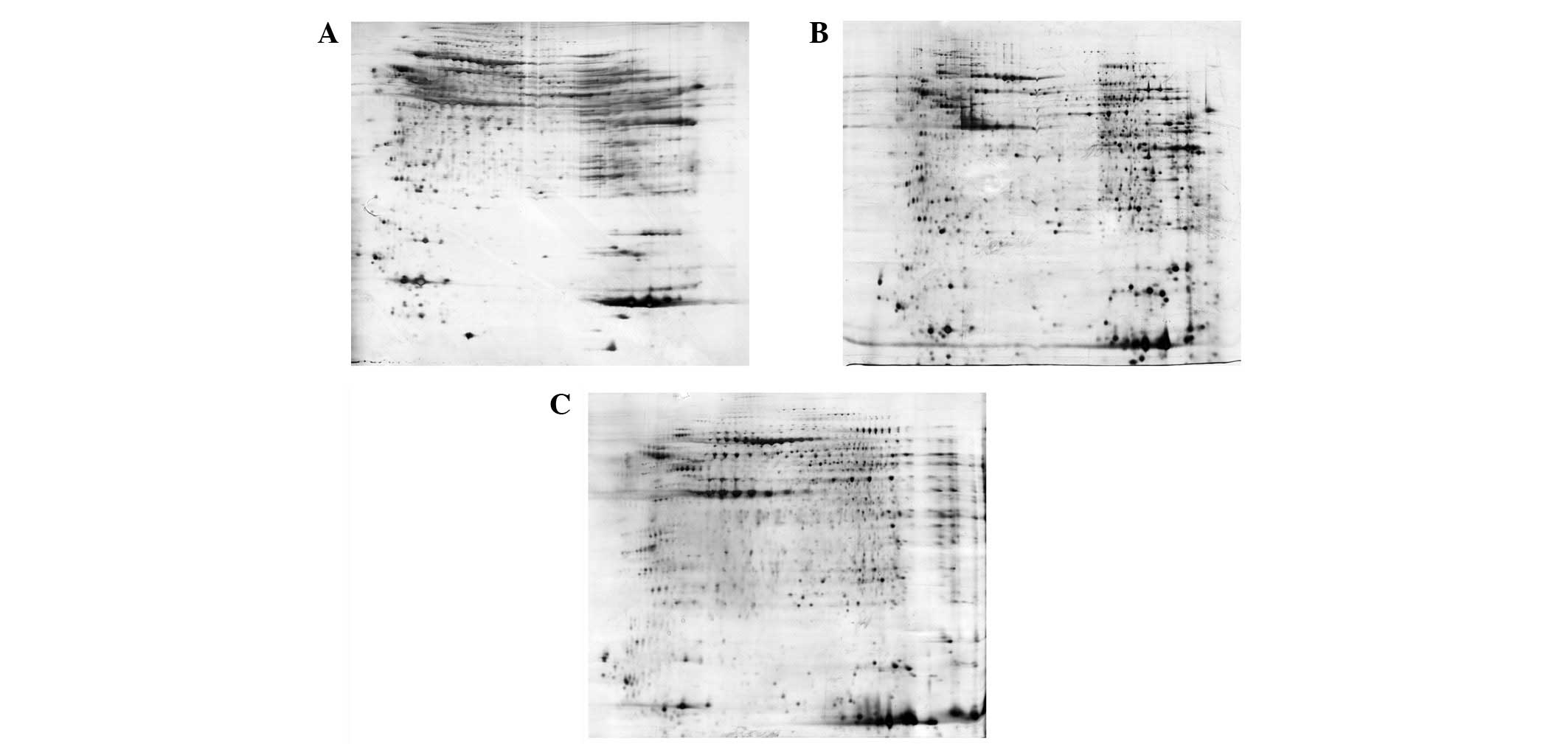
(Fig. 1). Prior to sample
clean-up, 1,478 protein spots were detected and extensive
horizontal streaking was clearly observed (Fig. 1A). Following purification with the
2-D Clean-Up kit, 2,000 protein spots were detected and the
horizontal streaking was markedly reduced. The gel image also
provided higher resolution when compared with the raw sample
(Fig. 1B). On the other hand,
acetone precipitation minimizes protein degradation by denaturing
most of the proteolytic enzymes and removes contaminants contained
in the tissue samples. However, the incomplete precipitation and
resolubilization of proteins results in non-specific loss of
proteins (3). As such, only 1,657
protein spots were detected and the resolution was decreased by
using ice-cold acetone precipitation in the present study (Fig. 1C). Therefore, the 2-D Clean-Up kit
was used in the subsequent 2-DE analysis, which may achieve 20%
more protein spots.
Influences of the buffer used for
solubilization of proteins
The buffer used for the extraction and
solubilization of proteins in the tested samples also influenced
the quality and reproducibility of the 2-D gel images. Urea lysis
buffer has been reported to be insufficient for solubilizing highly
hydrophobic proteins, including membrane proteins (8). By contrast, the addition of thiourea
to the lysis buffer improves the solubilization of proteins
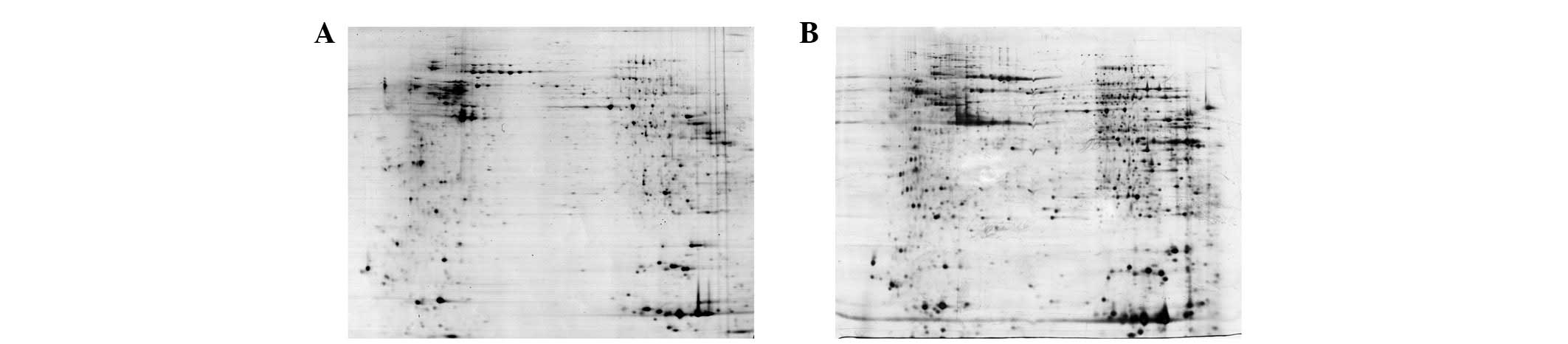
(8). As shown in Fig. 2, using urea/thiourea lysis buffer
in the sample preparation allowed more protein spots to be
identified and provided higher gel resolution than when using urea
lysis buffer. When using urea lysis buffer, 2,000 protein spots
were detected (Fig. 2A), while
2,500 protein spots were detected when urea/thiourea lysis buffer
was used (Fig. 2B). Therefore,
urea/thiourea lysis buffer was used in the subsequent analysis,
which may achieve 20% more protein spots.
Optimization of the conditions during
electrophoresis
In the IEF procedures of electrophoresis, the
focusing time and pre-reduction processes are the pivotal factors
in protein separation and detection by 2-D gel analysis.
Focusing time
The focusing time strongly affects the attainment of
steady-state IEF patterns. It is also an essential factor in
achieving high quality and reproducible 2-D gel images.
Insufficient focusing induces horizontal and vertical streaking,
while overfocusing may result in distorted protein patterns and
horizontal streaking at the basic gel end, leading to loss of
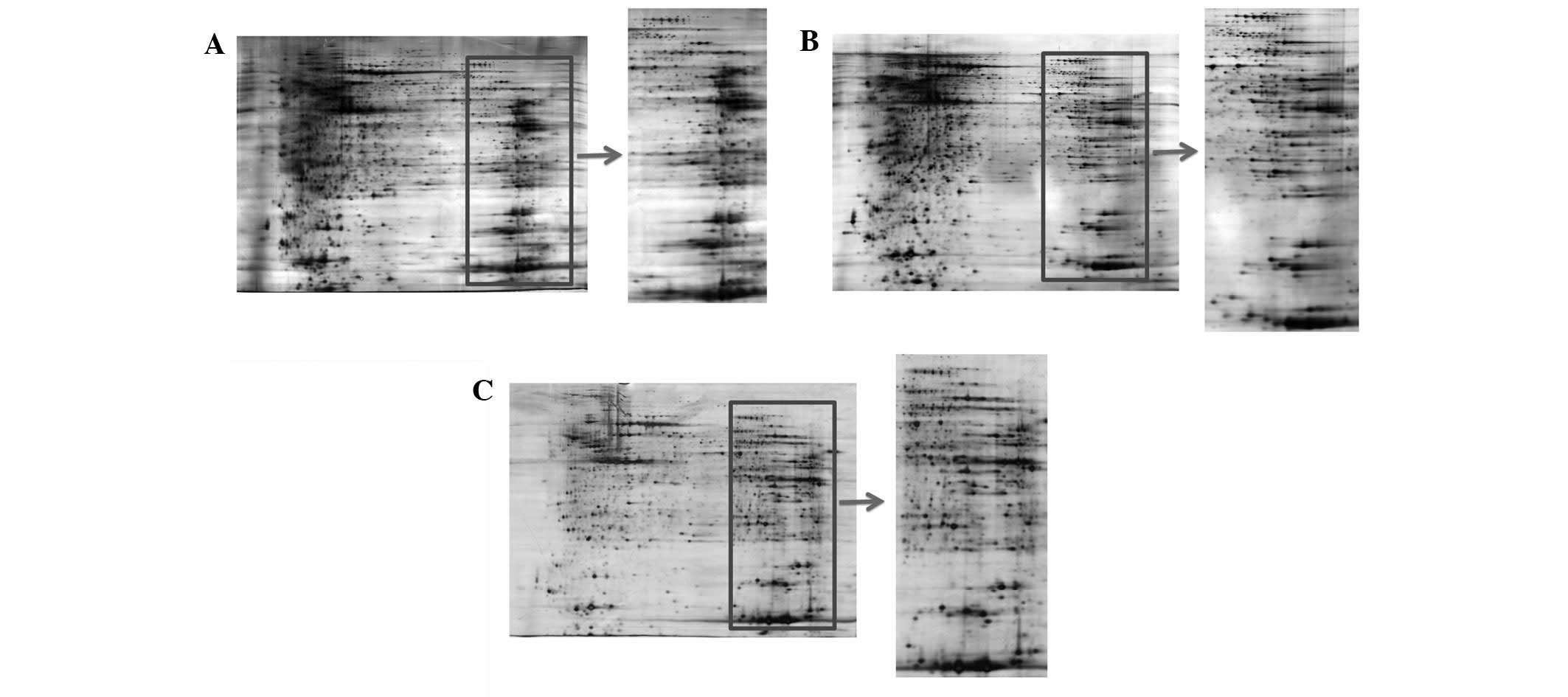
protein spots (9,10). In the present study, different
focusing times were compared (Fig.
3). When the sample was focused for 40,000 Vhr, incomplete
focusing and heavy horizontal streaking were observed in the 2-D
gel image (Fig. 3A), indicating
that the focusing duration is insufficient for most of the proteins
to achieve steady-state focusing. When the samples were focused for
60,000 and 70.000 Vhr, the image quality around neutral pH was
acceptable but heavy horizontal streaking was observed in the areas
near the cathode electrodes (Fig. 3C
and D), indicating overfocusing. By using 50,000 Vhr, optimal
gel resolution and minimized streaking were observed (Fig. 3B). Collectively, the results
indicated that 50,000 Vhr was optimal for the 2-D gel protein
analysis of tumor tissue samples.
Reduction and alkylation of unexpected
proteins prior to IEF
The reduction process is another pivotal factor in
protein separation and detection by 2-D gel analysis. DTT is a
strong reducing agent, which is often adopted for breaking
disulfide bonds and unfolding protein secondary and tertiary
structures. Depletion of DTT during IEF may induce horizontal
streaking due to protein re-oxidation, which is particularly common
in large gels (3). To achieve a
well-focused 1D separation, protein extracts in rehydration buffer
and 1 or 2% DTT were compared. Increasing the percentage of DTT in
the rehydration buffer reduced the horizontal streaking and
improved gel resolution at the cathode, although it also resulted
in distorted protein patterns (Fig. 4A
and B). In addition, a study has demonstrated that a shortage
of the reducing agent is avoidable by placing an extra paper soaked
with DTT near the cathode (11).
Nevertheless, each of these two methods alone was insufficient in
diminishing horizontal streaking, while improved resolution and
inhibition of re-oxidation of the sulfhydryl groups, produced by
the dynamic loss of DTT, was achieved with 1% DTT when an extra
paper soaked with 1% DTT was used near the cathode (Fig. 4C).
A previous study demonstrated that the reduction and
alkylation of proteins prior to IEF greatly reduces the streaking
caused by IEF (12). Following
reduction of the disulfide bonds by DTT, IAA was used to alkylate
the free thiol groups of cysteine residues to prevent their
re-oxidation (back reaction) during electrophoresis. The excess IAA
was neutralized by adding an equimolar amount of DTT to prevent
overalkylation. However, the protocol for pre-reduction and
alkylation has varied greatly in different studies and the
experimental conditions have not yet been clearly optimized
(12,13). Thus, all factors, in terms of the
time and temperature of the reaction, as well as the ratio of
DTT/IAA, that may influence 2-D gel analysis were systematically
examined in the present study (Table
I). The results indicate that reduction and alkylation with
DTT/IAA/DTT (10mM/40 mM/40 mM to 5 μg/μl protein) on ice for 1 h
for each step prior to IEF achieves higher resolution and improved
protein separation (Fig. 5).
Protein detection using two
electrophoresis conditions
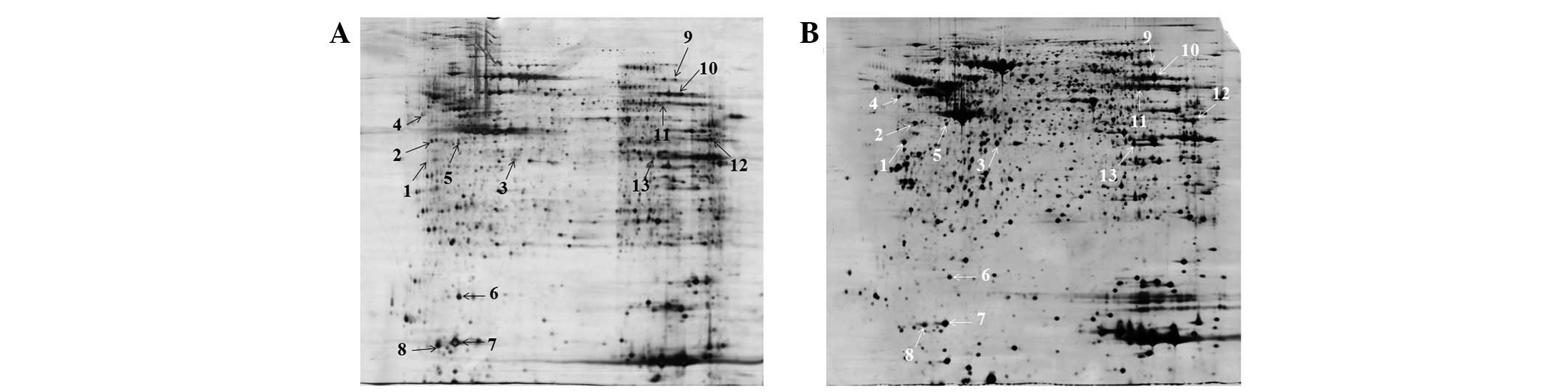
To investigate whether reduction and alkylation
prior to IEF affect the final protein identification, matched
protein spots from gels prepared with or without pre-reduction and
alkylation were subjected to gel-based protein identification. A
total of 13 matched spots were excised from different areas of the
2-D gels for analysis (Fig. 6).
Only eight spots (numbers 2, 3, 5, 6, 7, 8, 9 and 13) from the
existing procedure were successfully identified, while all spots
were identifiable following pre-reduction and alkylation (Table III). It was not possible to
identify confidently spot numbers 1, 4, 10, 11 and 12 from the
existing procedure as multiple overlapping peptides were recovered
due to horizontal streaking. The results indicate that the modified
conditions of the present study are more effective for finding
biomarkers and molecular targets.
| Table IIIComparison of the proteins identified
using two different electrophoresis conditions. |
Table III
Comparison of the proteins identified
using two different electrophoresis conditions.
| Identification |
|---|
|
|
|---|
| Spot no. | Previous
conditions | Modified
conditions |
|---|
| 1 | - | α-tropomyosin |
| 2 | 40S ribosomal protein
SA | 40S ribosomal protein
SA |
| 3 | Actin | Actin |
| 4 | - | p53-induced protein
phosphatase 1 |
| 5 | Galactokinase | Galactokinase |
| 6 | Vesicle protein
sorting 26B | Vesicle protein
sorting 26B |
| 7 | Cytochrome P450 | Cytochrome P450 |
| 8 | Thioredoxin OS | Thioredoxin OS |
| 9 | Transketolase | Transketolase |
| 10 | - | Pyruvate kinase
isozyme M2 |
| 11 | - | ATP synthase subunit
α |
| 12 | - | Vacuolar ATP synthase
subunit E |
| 13 | SID1 transmembrane
family member 2 precursor | SID1 transmembrane
family member 2 precursor |
Discussion
Overall, the present study has optimized the sample
preparation and 1D separation conditions for the 2-DE analysis of
tumor tissue samples. Compared with the previously reported
conditions, the present study demonstrated that tumor tissue
proteins extracted by a bead mill TissueLyser LT with urea/thiourea
lysis buffer, followed by 2-D Clean-Up kit purification produced
optimal throughput and protein yielding rates. In addition, the
reduction and alkylation of proteins with optimal focusing
conditions prior to IEF on a 24-cm strip was demonstrated to be
effective in preventing the horizontal streaking caused by
oxidation and in identifying biomarkers and molecular targets. In
conclusion, the modified conditions and protocols of the present
study allowed the 2-DE analysis of tumor tissue proteins with
enhanced reproducibility and sensitivity and higher 2-DE protein
recovery and resolution.
Acknowledgements
This study was supported by the Science and
Technology Development Fund, MSAR (Project code: 035/2011/A2).
References
|
1
|
Klose J: Protein mapping by combined
isoelectric focusing and electrophoresis of mouse tissues. A novel
approach to testing for induced point mutations in mammals.
Humangenetik. 26:231–243. 1975.
|
|
2
|
O’Farrell PH: High resolution
two-dimensional electrophoresis of proteins. J Biol Chem.
250:4007–4021. 1975.
|
|
3
|
Görg A, Obermaier C, Boguth G, et al: The
current state of two-dimensional electrophoresis with immobilized
pH gradients. Electrophoresis. 21:1037–1053. 2000.
|
|
4
|
Campostrini N, Areces LB, Rappsilber J, et
al: Spot overlapping in two-dimensional maps: a serious problem
ignored for much too long. Proteomics. 5:2385–2395. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Galvani M, Hamdan M and Righetti PG:
Investigating the reaction of a number of gel electrophoresis
cross-linkers with beta-lactoglobulin by matrix assisted laser
desorption/ionization-mass spectrometry. Electrophoresis.
21:3684–3692. 2000. View Article : Google Scholar
|
|
6
|
Shevchenko A, Tomas H, Havlis J, Olsen JV
and Mann M: In-gel digestion for mass spectrometric
characterization of proteins and proteomes. Nat Protoc.
1:2856–2860. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Herbert B, Righetti P, Citterio A and
Boschetti E: Sample preparation and prefractionation techniques for
electrophoresis-based proteomics. Proteome Research: Concepts,
Technology and Application. Wilkins MR, Appel RD, Williams KL and
Hochstrasser DF: 2nd edition. Springer; Heidelberg Berlin: pp.
15–40. 2007, View Article : Google Scholar
|
|
8
|
Rabilloud T: Solubilization of proteins in
2-D electrophoresis. An outline. Methods Mol Biol. 112:9–19.
1999.PubMed/NCBI
|
|
9
|
Görg A, Postel W and Günther S: The
current state of two-dimensional electrophoresis with immobilized
pH gradients. Electrophoresis. 9:531–546. 1988.
|
|
10
|
Görg A: Two-dimensional electrophoresis.
Nature. 349:545–546. 1991.
|
|
11
|
Görg A, Boguth G, Obermaier C, Posch A and
Weiss W: Two-dimensional polyacrylamide gel electrophoresis with
immobilized pH gradients in the first dimension (IPG-Dalt): the
state of the art and the controversy of vertical versus horizontal
systems. Electrophoresis. 16:1079–1086. 1995.PubMed/NCBI
|
|
12
|
Herbert B, Galvani M, Hamdan M, et al:
Reduction and alkylation of proteins in preparation of
two-dimensional map analysis: why, when, and how? Electrophoresis.
22:2046–2057. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Candiano G, Bruschi M, Musante L, et al:
Blue silver: a very sensitive colloidal Coomassie G-250 staining
for proteome analysis. Electrophoresis. 25:1327–1333. 2004.
View Article : Google Scholar : PubMed/NCBI
|