Introduction
Transient prosencephalic ischemia-reperfusion (I/R)
injury is able to cause neuronal death in the hippocampal CA1
region, thus resulting in damage to learning and memory, however,
its exact pathogenesis remains unclear (1). Factors that contribute to cerebral
I/R injury include oxidative stress caused by the production of
reactive oxygen species (ROS), disruption of Ca2+
homeostasis, activation of proteases and excitotoxicity of
glutamatergic neurons (1–4). Among these factors, increased ROS
production and cytosolic free Ca2+ overload are major
contributors to I/R-induced injury (2,3,5).
Increases in neuronal cytosolic Ca2+ levels have been
observed during ischemia and reperfusion (3,5). One
function of cytosolic Ca2+ in the pathogenesis of
I/R-induced cerebral injury has been hypothesized to be the
activation of the Ca2+-dependent protease calpain
(6). Calpain exists in myocytes in
two primary isoforms, namely, micro (calpain I) and milli (calpain
II), which are named based on the respective amounts of
Ca2+ required for their activation in vitro. When
whole or regional cerebral ischemia occurs, inner neuronal
Ca2+ is overloaded and calpain generation increases,
which in turn causes cell damage (6). The downstream factor of calpain,
cyclin-dependent kinase 5 (CDK5), is a multifaceted
serine/threonine kinase protein that has important functions in the
nervous system. Over the past decade, CDK5 activity has been
demonstrated to regulate numerous events during brain development,
including neuronal migration, as well as during axon and dendrite
development. Recent evidence also suggests that CDK5 is pivotal in
synaptic plasticity, behavior and cognition (7). Under pathological conditions, p35 may
be truncated into p25, which is able to markedly and consistently
activate CDK5, change the cellular localization of CDK5 and
ultimately cause neuronal death (8). CDK5 dysfunction has been implicated
in a number of neurological disorders and neurodegenerative
diseases, including Alzheimer’s disease (AD), amyotrophic lateral
sclerosis, Niemann-Pick type C disease and ischemia (7,8).
CDK5 hyperactivation, which is caused by the conversion of p35 to
p25 by calpain during neurotoxicity, also contributes to the
pathological state, however, its role in cognitive impairment
following I/R injury remains unclear.
ROS-mediated oxidative insults during cerebral I/R
injury may damage the vulnerable regions of the brain (9). Certain brain regions, including the
cortex and the hippocampus, are more vulnerable to ischemia than
other regions. Severe damage to the hippocampus results in
difficulties in forming new memories (10). Repeated cerebral ischemia may lead
to the accumulation of certain cellular active substances in the
body and thus demonstrate a neurotoxic effect. This phenomenon may
be an important factor in the occurrence of cognitive impairment
(11). Oxidative damage caused by
free radicals also involves mitochondrial damage, cell necrosis and
apoptotic processes. Superoxide dismutase (SOD) and malondialdehyde
(MDA) are important indicators in evaluating the extent of
oxidation and antioxidation (12).
Recent studies have linked I/R-induced ROS production to the
oxidative modification of Ca2+ handling proteins
(13). Therefore, the production
of ROS and the overexpression of the calpain/CDK5 pathway are
involved in the pathogenesis of I/R-induced cognitive
impairment.
Donepezil (±-2-(1-benzylpiperidin-4-yl)
methyl-5,6-dimethoxy-indan-1-one monohydrochloride) is a potent
acetylcholinesterase inhibitor that has been previously
demonstrated to be effective in improving cognition in patients
with AD (14). In addition,
pretreatment with donepezil hydrochloride (DH) prior to the
induction of focal cerebral ischemia has been demonstrated to
significantly attenuate cerebral infarction volume (15). DH may also protect rat cortical
neurons from glutamatergic neurotoxicity (16). These studies imply that DH may
provide benefits for the treatment of ischemia. Recent studies have
demonstrated that DH has a neuroprotective effect and this effect
does not involve anti-cholinesterase (17). DH is able to inhibit the fast
inflow of Na+ and Ca2+, reduce the release of
glutamate and therefore oppose the neurotoxicity induced by
depolarization (18). However, few
studies have investigated the effect of DH treatment on
prosencephalic ischemia and the involvement of the calpain I/CDK5
pathway in the ameliorating effect of DH on learning and
memory.
In the present study, the therapeutic effects of DH
on prosencephalic ischemia in mice, including its effect on spatial
memory function, are determined through histopathological
evaluation. Calpain I and CDK5/p25 expression, SOD activity and MDA
content are also analyzed in the ischemic mouse hippocampus.
Materials and methods
Animals and grouping
A total of 250 three-month old male mice were
provided by the Experimental Animal Center of Hebei Medical
University (Shijiazhuang, Hebei, China). The mice weighed 32.3±2.4
g and were bred in natural light. The circadian ratio was 12 h/12 h
at an ambient temperature of 20 to 25°C. The mice were provided
with food and water within the 1 week acclimatization period. The
mice were then randomly divided into three groups: the sham
operation group (SO, n=70), the model group (MG, n=90) and the
treatment group (TG, n=90). The present study was performed in
strict accordance with the recommendations in the Guide for the
Care and Use of Laboratory Animals of the National Institutes of
Health (Bethesda, MD, USA). The animal use protocol has been
reviewed and approved by the Institutional Animal Care and Use
Committee (IACUC) of the Hebei General Hospital (Shijiazhuang,
Hebei, China).
Animal model preparation
The following methods are based on literature with
slight modifications (7). The mice
were anesthetized with an intraperitoneal injection of 10% chloral
hydrate (0.035 ml/10 g). Next, the bilateral common carotid artery
was separated. The no. 4-0 silk thread was used to obstruct blood
flow for 30 min. Simultaneously, the tail was cut at ~1 cm from the
tail tip for ~0.3 ml of bloodletting. Once the blood flow was
restored for 10 min, the blood flow was obstructed again for 30
min. This operation was repeated three times. Following the third
blood flow reperfusion, careful observation was conducted for 30
min prior to the skin being sutured. The SO was only performed by
isolating the bilateral common carotid artery and by thread binding
without obstructing blood flow and tail bleeding. On the second day
postoperative, the TG was orally administered 3 mg/kg/day of DH
(Eisai Pharmaceutical Co., Ltd., Tokyo, Japan), whereas the SO and
the MG were administered the same volume of saline.
Achievement tests of mouse learning and
memory
The mouse water maze automatic recorder,
manufactured by the Institute of Materia Medica, Chinese Academy of
Medical Sciences (no. 3850; Beijing, China), is a brown Plexiglass
slot 1.1 m in length, 0.7 m in width and 0.4 m in height. The
ladder at one end of the slot is only able to be reached in one
way. The mouse may climb up the ladder and rest. The inner slot was
mazelike with multiple blind ends to prevent the mouse from seeing
the ladder. Numerous automatic sensing devices automatically
recorded the following data for 3 min: water depth, 10 cm and water
temperature, 22±1°C. Prior to the test, the mouse was trained to
swim through the maze. Swimming time was recorded as the duration
of time it took for the mouse to swim the entire distance; swimming
time >3 min was recorded as 3 min. On the 29th day (4th week),
43rd day (6th week) and 57th day (8th week) postoperative, the test
achievements were recorded as learning scores and the achievements
on the 30th day (4th week), 44th day (6th week) and 58th day (8th
week) postoperative were recorded as memory scores.
Morphological observation
Six mice from each group were subjected to
cardio-perfusion fixation. The section between the optic chiasm and
the mammillary bodies was obtained and a coronal slice with a
thickness of 6 μm was performed. The slice was stained with
hematoxylin for 5 min and 1% eosin for 4 min. Two slices from the
same parts of each mouse were selected and six non-overlapping
regions in the hippocampal CA1 region of each slice were selected
at a range of 0.1 mm for the regional normal neuron count and the
sum from all the regions. The round or oval nucleolus without cell
shrinkage and edema was counted as normal.
Immunohistochemical staining
Six mice from each group were subjected to
cardio-perfusion fixation. The brain tissue between the optic
chiasm and the mammillary bodies was obtained and subjected to
external fixation in 4% paraformaldehyde solution for 24 h.
Continuous coronal slices at a thickness of ~5 μm were then
performed. The operation strictly followed the manufacturer’s
instructions (purchased from Wuhan Boster Biological Technology,
Ltd., Wuhan, Hubei, China). Each slice was counted in 10 fields and
the amount of calpain I antigen was measured using the average
optical density (OD) value of the positive cell staining.
Western blot
Six mice from each group were anesthetized and
subsequently decapitated. The bilateral hippocampus was isolated on
ice and then lysed according to the manufacturer’s instructions.
Protein quantification was performed using the bicinchoninic acid
method. The products were then isolated by 15% SDS-polyacrylamide
gel electrophoresis, transferred onto a PVDF membrane and blocked
with the blocking solution at room temperature for 1 h. The
monoclonal primary antibody diluted with the blocking solution
(β-actin, 1:1,000) was then added, followed by the addition of
polyclonal antibody (CDK5, C8, 1:400 dilution; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA; p35/p25, C19, 1:100
dilution; Santa Cruz Biotechnology, Inc.) and overnight incubation
at 4°C. Secondary antibody horseradish peroxidase-labeled
anti-rabbit or mouse IgG (1:1,000 dilution; Proteintech Group,
Inc., Chicago, IL, USA) was added, the reaction was performed for 2
h and then developed with enhanced chemiluminiscence. β-actin was
used as an internal reference. The experiment was repeated three
times and gel image analysis was performed to determine the point
density.
Changes in SOD activity and MDA
content
Six mice from each group were anesthetized and
decapitated and, each mouse brain was placed on an ice tray.
Following rinsing with ice saline, the hippocampus was stripped and
homogenized with 10% specimen-weight saline. The homogenate was
then centrifuged at a low speed (2,000 × g) for 10 min. The
supernatant was placed inside a −80°C refrigerator. A xanthine
oxidase method was performed to test the SOD activity and a
thiobarbituric acid (TBA) assay was performed to test the MDA
content. SOD and MDA kits were purchased from the Nanjing Jiancheng
Bioengineering Institute (Nanjing, Jiangsu, China).
Statistical analysis
Data are expressed as the mean ± standard deviation
and processed using SPSS 13.0 software. The single-factor analysis
of variance was applied. An LSD test was used to compare the groups
and P<0.05 was considered to indicate a statistically
significant difference.
Results
General observation
The majority of the mice in the MG demonstrated dull
fur, depression and reduced activity. Mice in the TG also exhibited
similar symptoms after 1 week, however the symptoms gradually
disappeared after 1 month. These symptoms did not manifest in the
mice in the SO group. Mice in all groups had no significant limb
paralysis. The number of deaths in each treatment was as follows:
five mice in the SO (mortality rate, 7.14%), 30 mice in the MG
(mortality rate, 33.33%) and 28 mice in the TG (mortality rate,
31.1%).
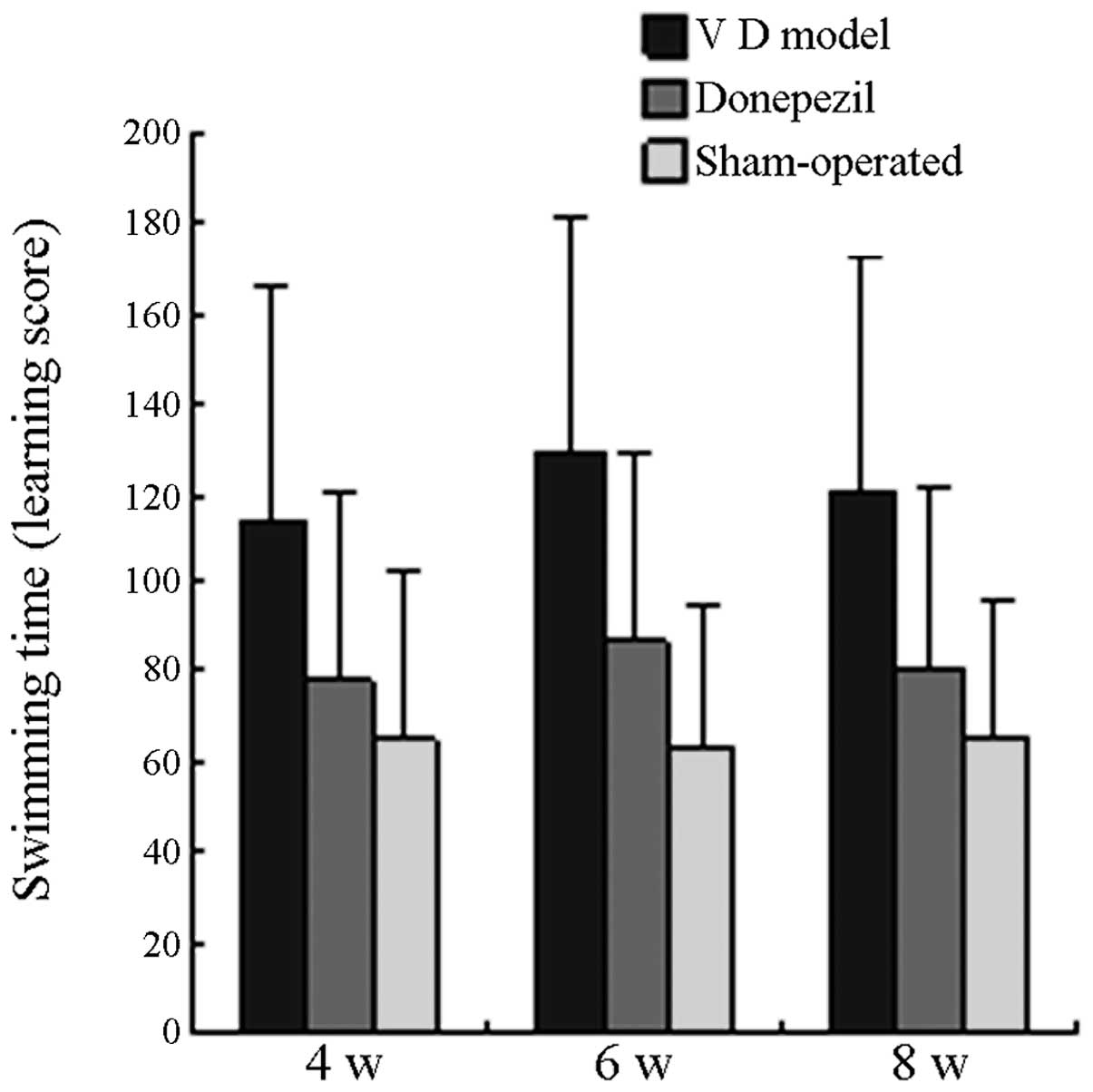
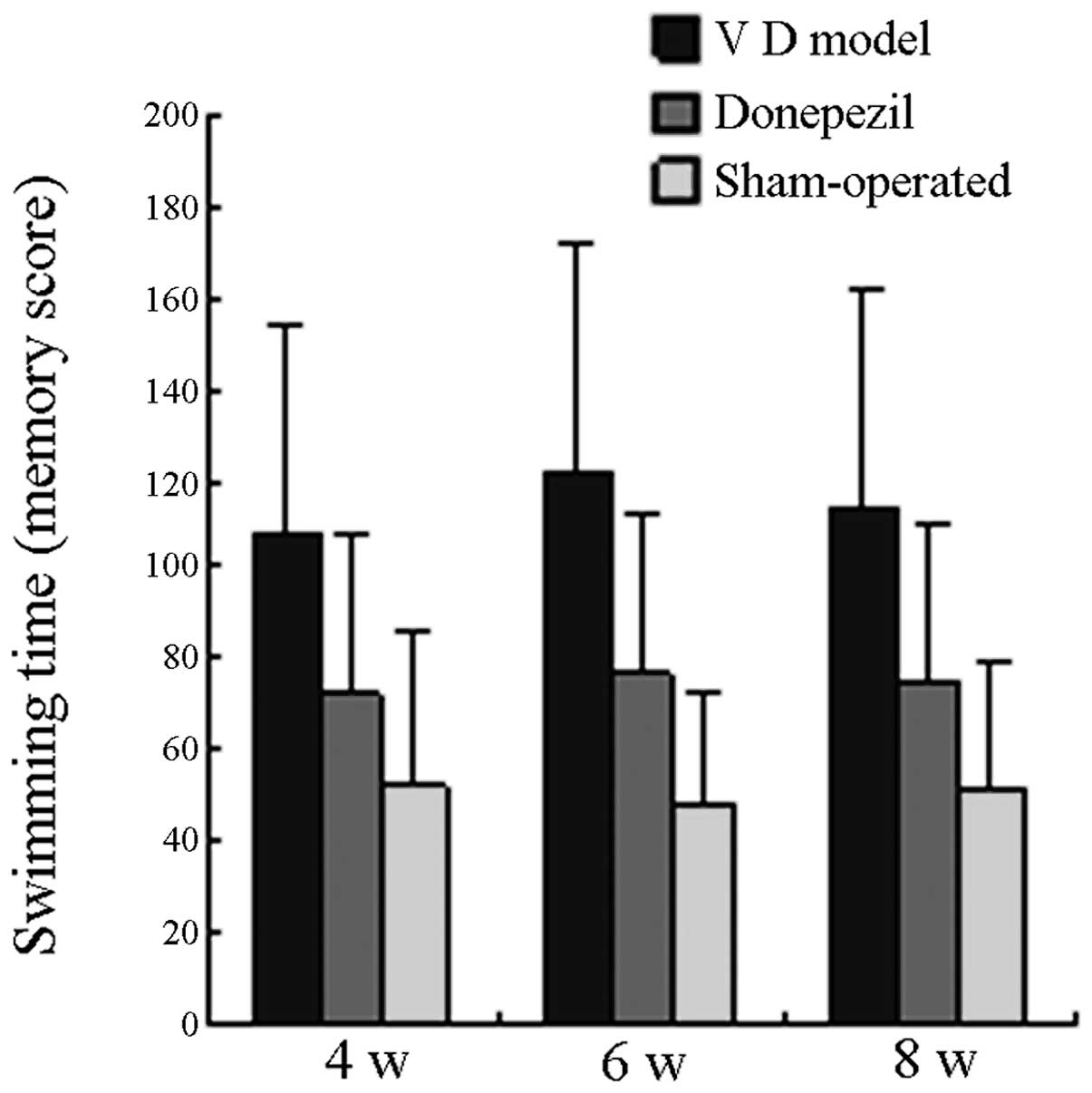
Comparison of learning and memory
scores
The swimming time of all the mice in the MG on the
29th, 43rd and 57th day postoperative was significantly longer than
that in the SO (P<0.05), whereas the swimming time of all the
mice in the TG was significantly shorter than that in the MG
(P<0.05). On the 30th, 44th and 58th day postoperative, the
swimming time of all mice in the MG was significantly longer than
that in the SO (P<0.05), whereas the swimming time of all the
mice in the TG was significantly shorter than that in the MG
(P<0.05). No significant difference was observed when the
swimming time in the TG was compared with that in the SO group
(P>0.05). The learning and memory scores of each group are
displayed in Fig. 1 and Fig. 2.
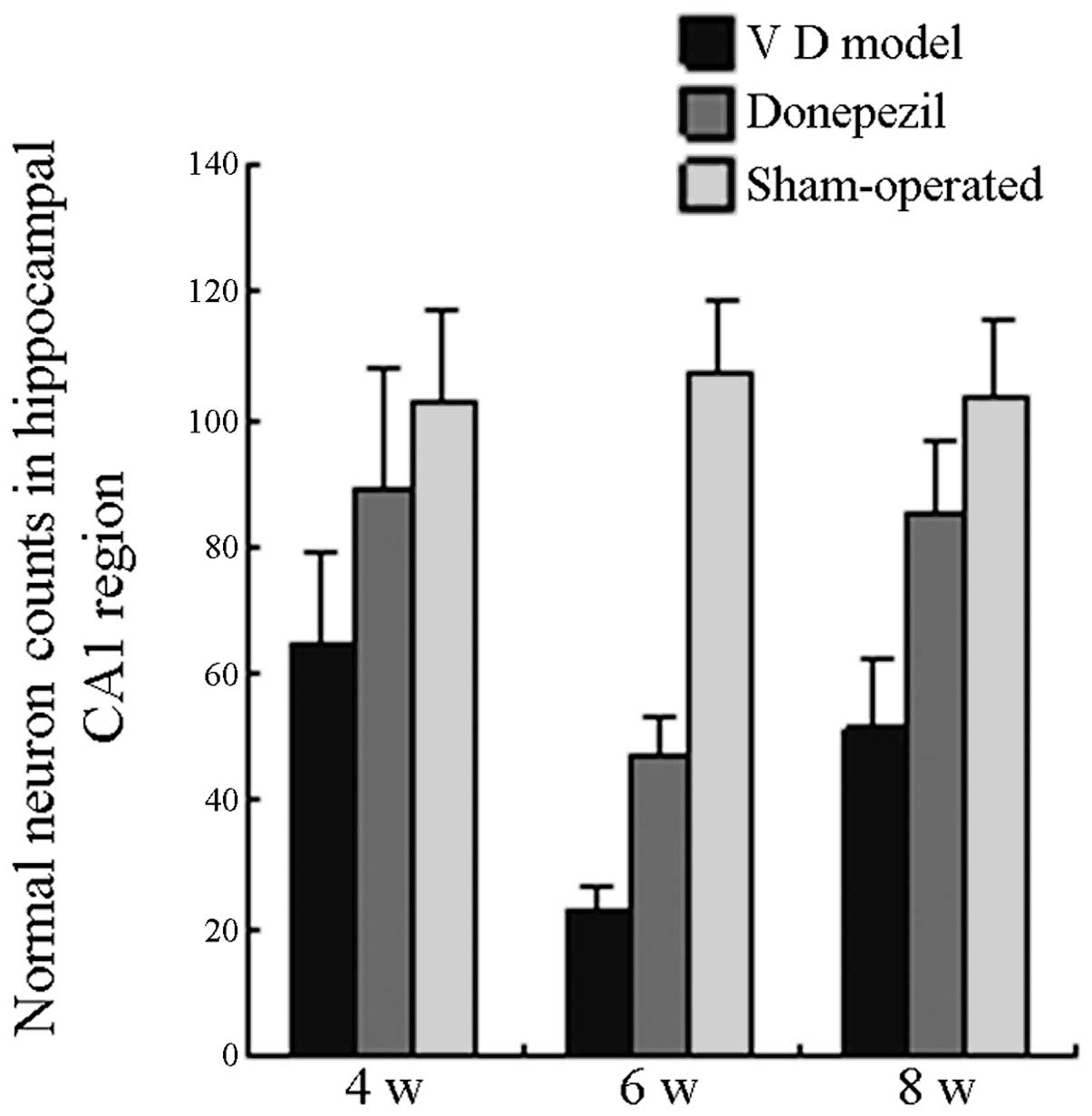
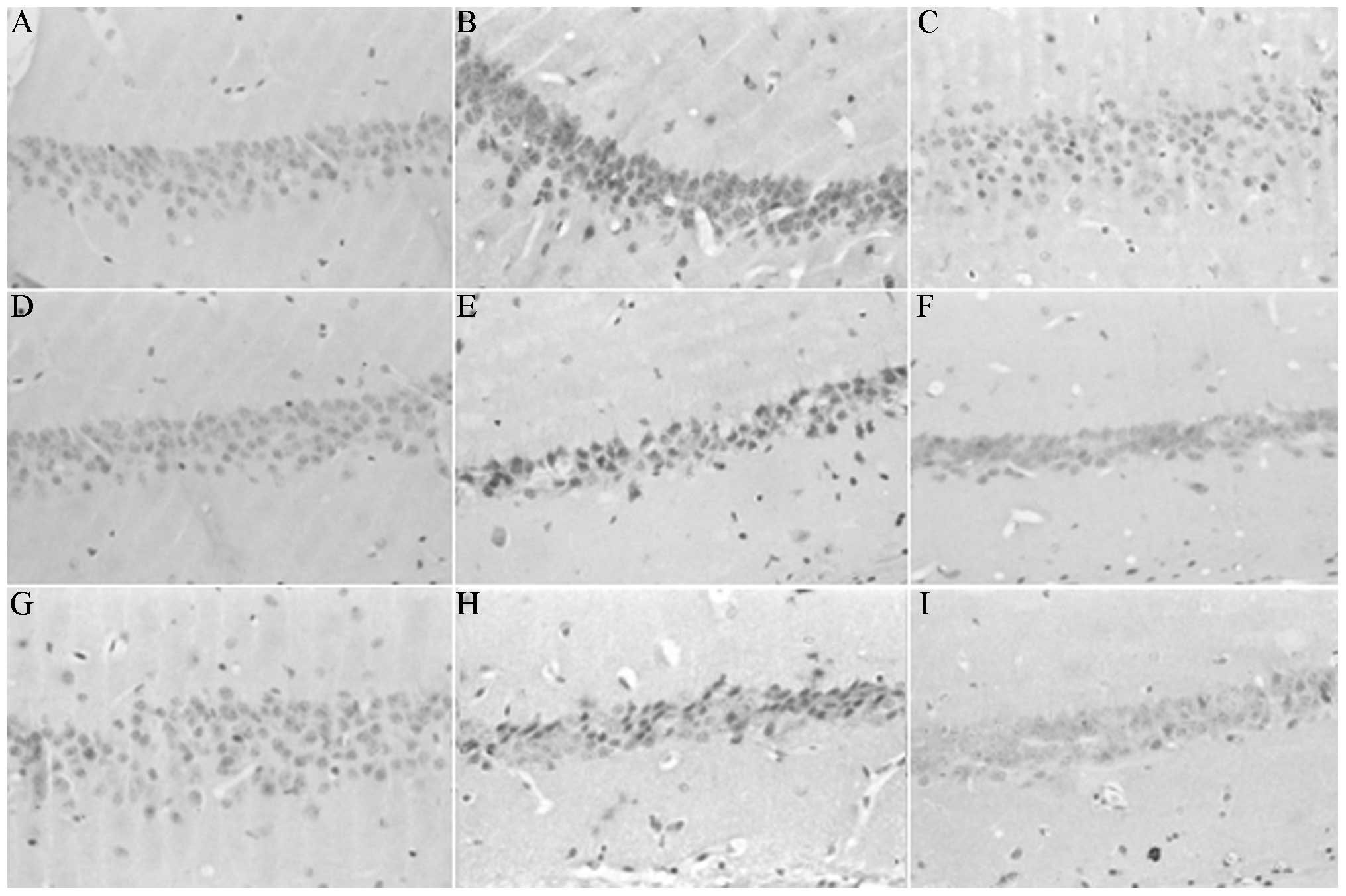
Pathological changes in the hippocampal
CA1 region
The pyramidal cells in the hippocampal CA1 region of
SO mice were tightly packed and demonstrated a clear outline; the
nucleus was large and round with a clear nucleolus. The hippocampal
pyramidal cell layers in the MG mice were few and loosely arranged.
The nucleus reduced in size and was deeply stained, and the normal
nerve cell count decreased. This decrease was most prominent
following six weeks of surgery. Only a small number of cells
exhibited a normal morphology. Compared with the normal neuron
count, the neuron count in the MG mice was significantly reduced
following 4 and 8 weeks postoperation (P<0.05). By contrast, the
pyramidal cells in the TG mice were arranged neatly. The layers
were clear and free from nuclear condensation. The count of normal
nerve cells was significantly higher than that in the MG mice
(P<0.05). The comparison between the TG and SO mice did not
demonstrate a significant difference on the 4 and 8 week
postoperation (P>0.05). As shown in Fig. 3, the neuron count in the TG mice
remained significantly lower than that in the SO mice 6 weeks
postoperation (P<0.05).
Calpain I expression in the hippocampal
CA1 region
In the SO, the cellular layers in the hippocampal
CA1 region on the 4, 6 and 8th week postoperative were clear, with
only a few traces of intracytoplasmically expressed calpain I. In
the MG, the neuron structure in the hippocampal CA1 region on the
4, 6 and 8th week postoperative was loose. The mean OD value of
calpain I in neuronic intracytoplasm significantly increased
(P<0.05) when compared with that in the SO. This increase was
more apparent in the 6th week postoperative. As displayed in
Fig. 4, the calpain I expression
in the TG at the three time points was significantly lower than
that in the MG (P<0.05) and close to that in the SO
(P>0.05).
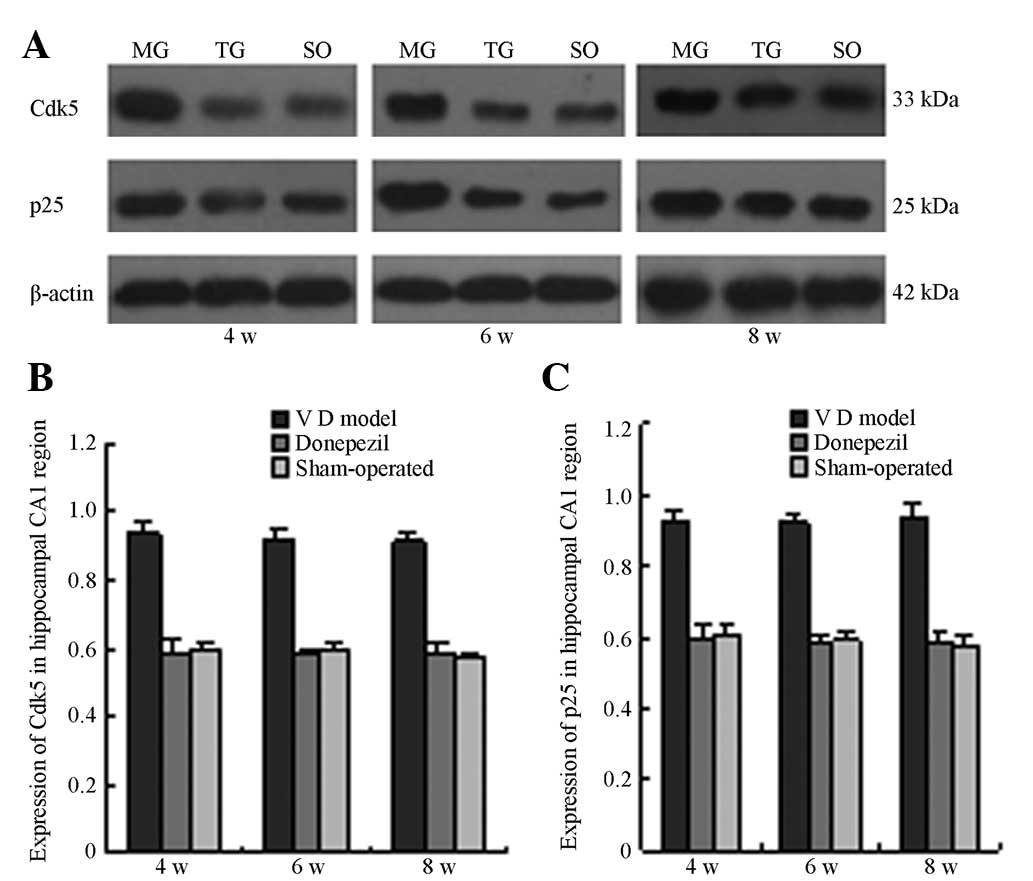
CDK5 and p25 protein expression
The CDK5 and p25 protein expression in the
hippocampal tissues of the MG mice on the 4, 6 and 8th week
postoperative of cerebral I/R were significantly higher than those
in the SO (P<0.05). This result suggests that CDK5/p25 protein
expression exhibited a sustained increase within eight weeks
following cerebral I/R. The CDK5 and p25 protein expression in the
TG at the three time points significantly decreased when compared
with those in the MG. The difference is statistically significant
at P<0.05 (Fig. 5 and Fig. 6).
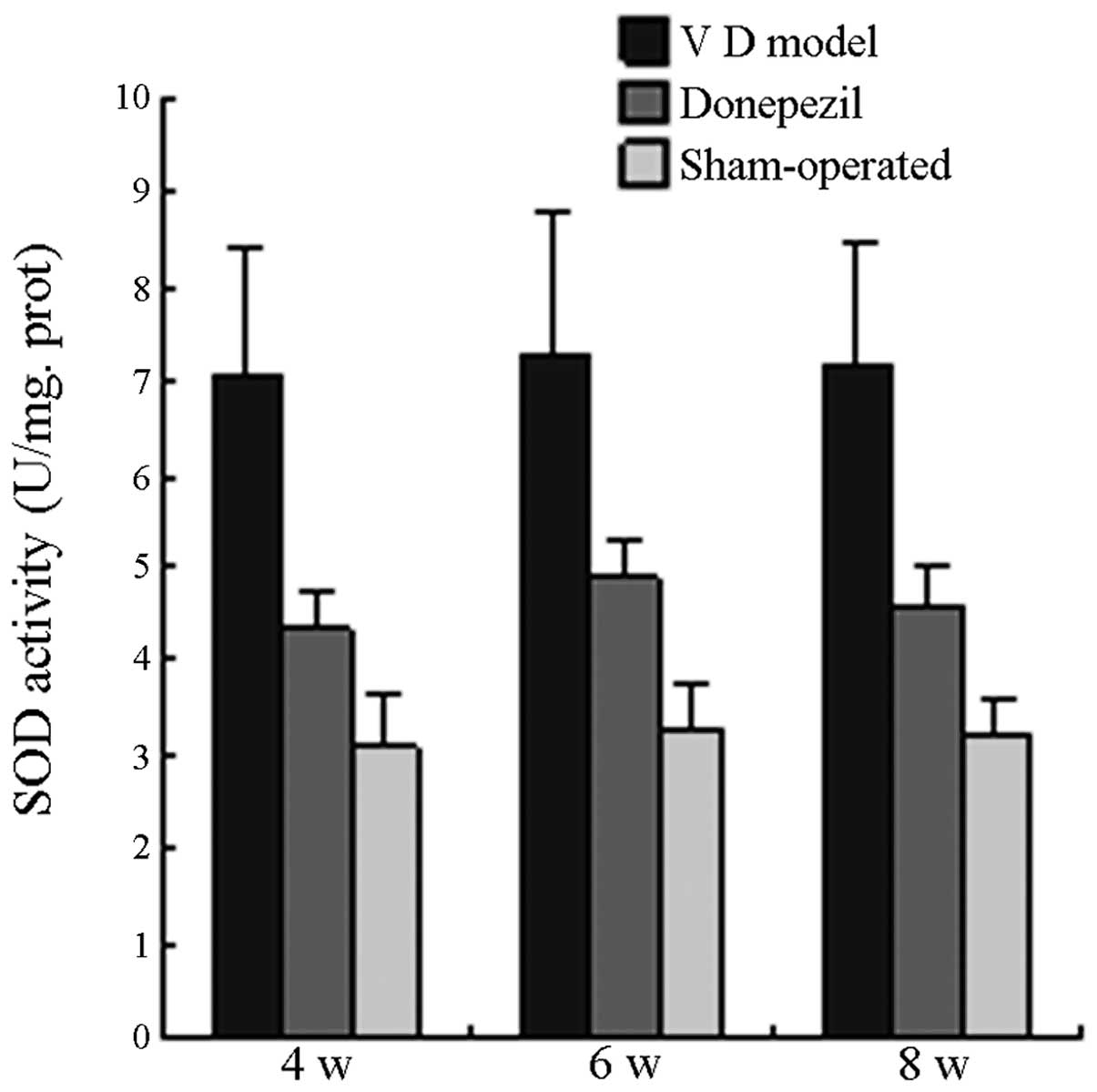
SOD activity and MDA content
On the 4th, 6th and 8th week postoperative, the SOD
activity in the MG was significantly higher than that in the SO
(P<0.05), whereas SOD activity in the TG was significantly lower
than that in the MG (P<0.05) and slightly higher than that in
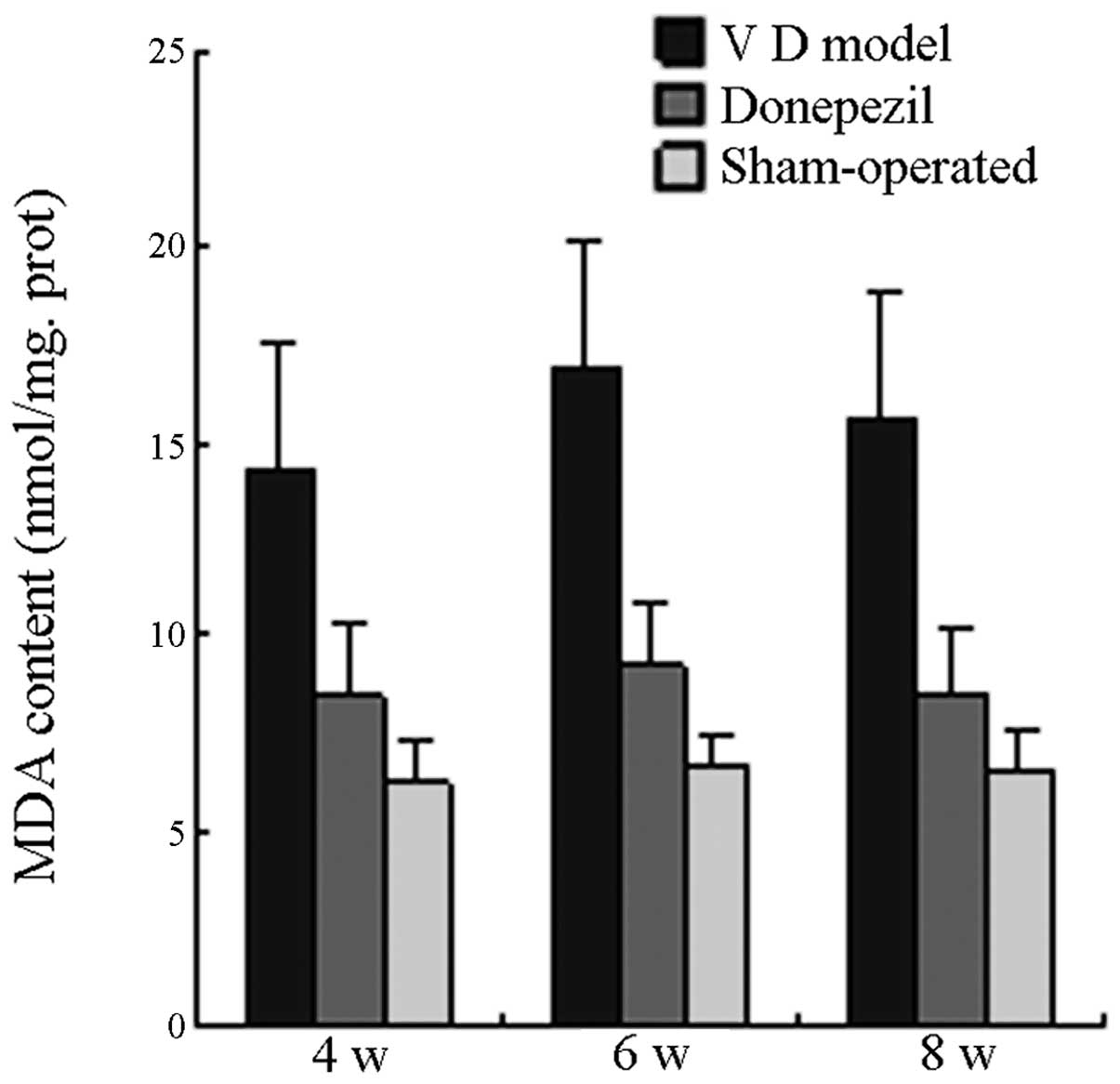
the SO with no significant difference (P>0.05). On the 4th, 6th
and 8th week postoperative, the MDA activity in the MG was
significantly higher than that in the SO group (P<0.05), whereas
MDA activity in the TG was significantly lower than that in the MG
(P<0.05). No significant difference was observed when MDA
activity in the TG was compared with that in the SO group
(P>0.05; Fig. 7).
Discussion
Previous studies demonstrated that ischemia is able
to lead to neuronal cell death in vulnerable regions of the brain,
particularly in the CA1 region of the hippocampus (19,20).
This finding is partly consistent with the results of the present
study. By observing the histological morphology, it was
demonstrated that the normal neuron count in the hippocampal CA1
region is significantly reduced within 8 weeks following cerebral
I/R, along with deficits in the water maze performance. The
findings of the present study support the theory that ischemic
damage in the hippocampus causes a serious impairment of spatial
memory tasks (21). The number of
normal neurons in the MG on the 6th week postoperative was
significantly lower than that in the SO and that in the MG on the 4
and 8th week postoperative. This result suggests that cerebral I/R
injury exhibited a progressively increasing trend within 6 weeks.
As time passed, the damage gradually reduced and the neuronal
function began to recover. Consistent with the present study is one
study that also highlights the inherent ability of the brain to
form new nerve cells following ischemic damage (22). Overall, ischemic animals treated
with DH performed better than those in the MG, suggesting that DH
has a neuroprotective effect.
To explore the biochemical mechanisms that may be
responsible for the observed histopathological and behavioral
changes, the expression of calpain I and CDK5/p25 was examined.
CDK5/p25 regulates numerous signaling cascades and is thought to be
an important mediator of learning and memory (23). A previous study revealed that the
inhibition of CDK5/p25 activity provides a promising therapeutic
avenue during or following stroke. This finding suggests that
increases in CDK5/p25 activity leads to neuronal death (24). These data support the results of
the present study, which demonstrated that calpain I and CDK5/p25
in the ischemic group were significantly increased in the
hippocampus, particularly during the 6th week (postoperative) of
ischemia. The increase in calpain I and CDK5/p25 in the ischemic
group was accompanied by neuronal impairment in the hippocampal CA1
region and a decrease in learning and memory ability. The possible
reasons were as follows: under the conditions of ischemia and
hypoxia, Ca2+ overload was observed in hippocampal
neurons, thereby activating calpain I (6) and consequently resulting in the
increase in p25 generation. Therefore, CDK5/p25 activity was
maladjusted, thus leading to the inability of CDK5 to perform
normal physiological functions toward synapse-related substances.
These conditions affect long term potentiation generation and cause
a decrease in learning and memory ability (7).
Oxidative stress is important in the pathogenesis of
I/R injury (2). The process is
accompanied by elevated free radicals. These radicals initiate a
radical chain reaction of signaling pathways that involve the
mitochondria and lead to cell death (25). The oxidation and antioxidation
status following cerebral ischemia in mice is determined by the
activities of lipid peroxides and SOD and by the levels of catalase
and glutathione peroxidase. MDA is used as a marker of lipid
peroxidation and its content may reflect the level of lipid
peroxide (26).
The present study explored SOD activity and MDA
content in brain tissue and demonstrated that MDA content was
significantly higher in the hippocampus following 8 weeks of
cerebral I/R; an increase in SOD activity was also observed. As SOD
may function as an antioxidant enzyme in ischemia-induced neuronal
damage, increased SOD activity found in this study may be explained
by a compensatory increase in antioxidant activity that targeted
the increase in free radical production associated with the brain
antioxidant mechanism, which was activated under serious oxidative
stress (27). The study also
confirmed that DH is able to reduce SOD activity and MDA content.
These findings suggest that DH has a protective function in
cerebral I/R-induced memory impairment.
Growing evidence suggests that increased ROS
production and cytosolic free Ca2+ overload, either
independently or co-operatively, are major contributors to
I/R-induced injury (2,3,5). Our
findings are consistent with this evidence. In the present study,
the expression of CDK5/p25 in the hippocampal CA1 region was
demonstrated to increase following cerebral I/R, along with the
increase in SOD activity and MDA content. Peroxiredoxin 2 (Prx2) is
an antioxidative enzyme with peroxidase activity (28). A previous study demonstrated that
Prx2 is a critical cytoplasmic target of Cdk5. In the cerebral
ischemia model, an abnormal increase in the expression of CDK5 in
the hippocampal CA1 region, accompanied by increasing Prx2
phosphorylation, was observed. This finding suggests that CDK5 may
affect ROS generation by affecting Prx2 (29). The involvement of this mechanism in
the pathogenesis of cerebral I/R requires further exploration.
In the mouse model of the present study, DH was
demonstrated to be capable of ameliorating learning and memory
function through the calpain I/CDK5 signaling pathway in the
hippocampus. DH is now in clinical trials for AD treatment, however
emerging information suggests a potential role of DH in the therapy
of ischemic disorders (20). The
pretreatment with DH may reduce the release of lactic
dehydrogenase, thus suggesting that anti-ischemic brain damage as a
consequence of DH is not associated with the inhibition of
cholinesterase (17). DH may
inhibit the fast inflow of Na+ and Ca2+,
reduce the release of glutamate and therefore oppose the
neurotoxicity induced by depolarization (30). The inhibition of Ca2+
influx by DH is likely to be the cause of the decreased expression
of calpain I and CDK5/p25, and improved memory function in the
present study. In addition, the neuroprotective effects of DH may
be caused by ROS reduction by SOD and MDA. These results suggest
that antioxidation may be one of the key mechanisms of
neuroprotection following DH treatment.
This study is not without its limitations. Firstly,
further study is needed to investigate which CDK5/p25 signaling
pathway regulates oxidative damage. Secondly, excitotoxicity,
inflammation and apoptosis are also involved in cerebral I/R injury
(31,32). Whether these mechanisms, except
Ca2+ overload and antioxidant effects, are responsible
for the neuroprotection of DH needs to be explored further.
In conclusion, the present study demonstrated that
DH has a significant protective effect on cerebral I/R-induced
brain injury. Inhibition of Ca2+ overload and
antioxidant ability appears to be the basic and important
mechanisms of the neuroprotective effect of DH. These results
suggest that DH may be a promising therapeutic drug for the
treatment of cognitive impairment following cerebral I/R
damage.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81241037).
References
|
1
|
Yan BC, Park JH, Ahn JH, Lee JC, Won MH
and Kang IJ: Postsynaptic density protein (PSD)-95 expression is
markedly decreased in the hippocampal CA1 region after experimental
ischemia-reperfusion injury. J Neurol Sci. 330:111–116. 2013.
View Article : Google Scholar
|
|
2
|
Tu Q, Wang R, Ding B, Zhong W and Cao H:
Protective and antioxidant effect of Danshen polysaccharides on
cerebral ischemia/reperfusion injury in rats. Int J Biol Macromol.
60:268–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang Q, Kalogeris TJ, Wang M, Jones AW and
Korthuis RJ: Antecedent ethanol attenuates cerebral
ischemia/reperfusion-induced leukocyte-endothelial adhesive
interactions and delayed neuronal death: role of large conductance,
Ca2+-activated K+ channels. Microcirculation.
17:427–438. 2010.
|
|
4
|
Zhang F, Guo A, Liu C, Comb M and Hu B:
Phosphorylation and assembly of glutamate receptors after brain
ischemia. Stroke. 44:170–176. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Racay P, Tatarkova Z, Chomova M, Hatok J,
Kaplan P and Dobrota D: Mitochondrial calcium transport and
mitochondrial dysfunction after global brain ischemia in rat
hippocampus. Neurochem Res. 34:1469–1478. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Peng S, Kuang Z, Zhang Y, Xu H and Cheng
Q: The protective effects and potential mechanism of Calpain
inhibitor Calpeptin against focal cerebral ischemia-reperfusion
injury in rats. Mol Biol Rep. 38:905–912. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Su SC and Tsai LH: Cyclin-dependent
kinases in brain development and disease. Annu Rev Cell Dev Biol.
27:465–491. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen J and Wang ZF: Roles of
cyclin-dependent kinase 5 in central nervous system development and
neurodegenerative diseases. Sheng Li Xue Bao. 62:295–308.
2010.PubMed/NCBI
|
|
9
|
Ghosh A, Sarkar S, Mandal AK and Das N:
Neuroprotective role of nanoencapsulated quercetin in combating
ischemia-reperfusion induced neuronal damage in young and aged
rats. PLoS One. 8:e577352013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Harry GJ and Lefebvre d’Hellencourt C:
Dentate gyrus: alterations that occur with hippocampal injury.
Neurotoxicology. 24:343–356. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Knapp LT and Klann E: Role of reactive
oxygen species in hippocampal long-term potentiation: contributory
or inhibitory? J Neurosci Res. 70:1–7. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kishida KT and Klann E: Sources and
targets of reactive oxygen species in synaptic plasticity and
memory. Antioxid Redox Signal. 9:233–244. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou L, Aon MA, Liu T and O’Rourke B:
Dynamic modulation of Ca2+ sparks by mitochondrial
oscillations in isolated guinea pig cardiomyocytes under oxidative
stress. J Mol Cell Cardiol. 51:632–639. 2011.
|
|
14
|
Winblad B, Engedal K, Soininen H, et al: A
1-year, randomized, placebo-controlled study of donepezil in
patients with mild to moderate AD. Neurology. 57:489–495.
2001.PubMed/NCBI
|
|
15
|
Fujiki M, Kobayashi H, Uchida S, Inoue R
and Ishii K: Neuroprotective effect of donepezil, a nicotinic
acetylcholine-receptor activator, on cerebral infarction in rats.
Brain Res. 1043:236–241. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takada Y, Yonezawa A, Kume T, et al:
Nicotinic acetylcholine receptor-mediated neuroprotection by
donepezil against glutamate neurotoxicity in rat cortical neurons.
J Pharmacol Exp Ther. 306:772–777. 2003. View Article : Google Scholar
|
|
17
|
Akasofu S, Kimura M, Kosasa T, Sawada K
and Ogura H: Study of neuroprotection of donepezil, a therapy for
Alzheimer’s disease. Chem Biol Interact. 175:222–226. 2008.
|
|
18
|
Akasofu S, Sawada K, Kosasa T, Hihara H,
Ogura H and Akaike A: Donepezil attenuates excitotoxic damage
induced by membrane depolarization of cortical neurons exposed to
veratridine. Eur J Pharmacol. 588:189–197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fang S, Yan B, Wang D, et al: Chronic
effects of venlafaxine on synaptophysin and neuronal cell adhesion
molecule in the hippocampus of cerebral ischemic mice. Biochem Cell
Biol. 88:655–663. 2010.PubMed/NCBI
|
|
20
|
Min D, Mao X, Wu K, et al: Donepezil
attenuates hippocampal neuronal damage and cognitive deficits after
global cerebral ischemia in gerbils. Neurosci Lett. 510:29–33.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Block F: Global ischemia and behavioural
deficits. Prog Neurobiol. 58:279–295. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
von Euler M, Bendel O, Bueters T, Sandin J
and von Euler G: Profound but transient deficits in learning and
memory after global ischemia using a novel water maze test. Behav
Brain Res. 166:204–210. 2006.PubMed/NCBI
|
|
23
|
Shukla V, Skuntz S and Pant HC:
Deregulated Cdk5 activity is involved in inducing Alzheimer’s
disease. Arch Med Res. 43:655–662. 2012.PubMed/NCBI
|
|
24
|
Mitsios N, Pennucci R, Krupinski J, et al:
Expression of cyclin-dependent kinase 5 mRNA and protein in the
human brain following acute ischemic stroke. Brain Pathol.
17:11–23. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Christophe M and Nicolas S: Mitochondria:
a target for neuroprotective interventions in cerebral
ischemia-reperfusion. Curr Pharm Des. 12:739–757. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guo C, Tong L, Xi M, Yang H, Dong H and
Wen A: Neuroprotective effect of calycosin on cerebral ischemia and
reperfusion injury in rats. J Ethnopharmacol. 144:768–774. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu C, Wu JL, Gu J, et al: Baicalein
improves cognitive deficits induced by chronic cerebral
hypoperfusion in rats. Pharmacol Biochem Behav. 86:423–430. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rhee SG, Yang KS, Kang SW, Woo HA and
Chang TS: Controlled elimination of intracellular H(2)O(2):
Regulation of peroxiredoxin, catalase, and glutathione peroxidase
via post-translational modification. Antioxid Redox Signal.
7:619–626. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rashidian J, Rousseaux MW, Venderova K, et
al: Essential role of cytoplasmic cdk5 and Prx2 in multiple
ischemic injury models, in vivo. J Neurosci. 29:12497–12505. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bendel O, Prunell G, Stenqvist A, et al:
Experimental subarachnoid hemorrhage induces changes in the levels
of hippocampal NMDA receptor subunit mRNA. Brain Res Mol Brain Res.
137:119–125. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Koh PO: Ischemic injury decreases
parvalbumin expression in a middle cerebral artery occlusion animal
model and glutamate-exposed HT22 cells. Neurosci Lett. 512:17–21.
2012. View Article : Google Scholar
|
|
32
|
Kliper E, Bashat DB, Bornstein NM, et al:
Cognitive decline after stroke: relation to inflammatory biomarkers
and hippocampal volume. Stroke. 44:1433–1435. 2013.PubMed/NCBI
|