Introduction
The ADP-ribosylation factor GTPase-activating
proteins (ArfGAPs) are a family of proteins that catalyze the
conversion of the active GTP-bound ADP-ribosylation factors (Arfs)
to the inactive GDP-bound Arfs. ArfGAPs are proposed to function in
fundamental biological processes, such as secretion, endocytosis,
phagocytosis, cytokinesis, cell adhesion, cell proliferation, cell
migration/invasion, membrane trafficking, cytoskeletal remodeling
and Ca2+ signaling (1–8).
Based on their protein domain structures and phylogeny, ArfGAPs
were classified into several subgroups, including ASAPs [ArfGAPs
containing Src-homology-3 (SH3), ankyrin (ANK) repeats and
plekstrin homology (PH) domain], ACAPs (ArfGAPs containing
coiled-coil, ANK repeats and PH domain) and ARAPs (ArfGAPs
containing Rho GAP domain).
Utilizing a genome-wide complementary DNA (cDNA)
microarray analysis, Okabe et al (9) identified a novel protein that was
highly overexpressed in human hepatocarcinoma tissues. Due to its
high amino acid sequence similarity with that of development and
differentiation enhancing factor-2, the newly identified protein
was termed development and differentiation enhancing factor-like-1
(DDEFL1), which was previously known as upregulated in liver
cancer-1 (UPLC1). In addition to liver cancer cells and tissues,
significant expression of DDEFL1 was identified in normal lung and
liver tissues and leukocytes in humans. Moreover, overexpression of
DDEFL1 in two cell lines that have low endogenous expression of
DDEFL1 (SNU423 and Alexander human hepatoma cells) appeared to
promote cell growth, but antisense RNA-mediated inhibition of
DDEFL1 suppressed the growth of hepatoma SNU475 cells
(9). Subsequently, Fang et
al (10) identified an ArfGAP
that utilizes Arf6 as the preferred substrate in HeLa cervical
cancer cells, using a proteomic approach. This Arf6-specific GAP
was found to contain two coiled coils, one PH domain, one GAP motif
and two ANK repeats, therefore, it was termed ACAP4. Notably, the
depletion of ACAP4 by small interfering RNA (siRNA), or inhibition
of Arf6 GTP hydrolysis by overexpressing GAP-deficient ACAP4,
suppressed Arf6-dependent cell migration in wound-healing assays,
demonstrating that ACAP4 is required for Arf6-mediated migratory
activity in HeLa cells. It was also demonstrated that ACAP4
effectively interacts with ezrin, an important
membrane-cytoskeletal linker protein. The formation of ACAP4-ezrin
was demonstrated to be important in acid secretion in gastric
parietal cells by orchestrating H,K-ATPase-containing
tubulovesicular trafficking to the apical plasma membrane (11). It was also shown that ACAP4
regulates integrin-β1 recycling in epidermal growth
factor-stimulated cell migration by interacting with growth factor
receptor-binding protein-2 (Grb2) via Tyr-733 phosphorylation
(12). Ha et al (13) analyzed the primary sequence and
phylogeny of the highly conserved Arf GAP domain of the reported
mammalian ArfGAPs and suggested that UPLC1/DDEFL1/ACAP4 is an
ASAP-type protein and should be named ASAP3; thus, in the present
study, we have decided to use this name. Enzyme characterizations
have indicated that ASAP3 is stimulated by phosphatidylinositol
4,5-bisphosphate and is capable of using Arf1, Arf5 and Arf6 as
substrates in vitro. ASAP3 has been demonstrated to be
associated with focal adhesions and circular dorsal ruffles in
MDA-MB-231 breast cancer and U118 glioma cells. ASAP3, however, did
not localize with invadopodia in MDA-MB-231 breast cancer cells or
podosomes in NIH-3T3 mouse fibroblasts. In MDA-MB-231 cells
transfected with plasmids overexpressing either the active or
inactive ASAP3, the distribution of vinculin or paxillin in focal
adhesions and invadopodia was not affected. However, in cells with
reduced ASAP3 protein, a significant reduction in actin-containing
stress fibers was observed, although the expression of β-actin was
not significantly affected. With the disruption of the stress fiber
network, the level of phosphomyosin was significantly decreased
(13). Phosphomyosin is able to
bind to the fibers in the network to stabilize them (14). Similarly, reduced ASAP3 levels have
been demonstrated to result in slowed migration and invasion in
mammary carcinoma cells (13).
Overall, the aforementioned studies suggest that
ASAP3 is essential in cytoskeletal remodeling and cancer cell
migration/invasion. However, the underlying mechanisms remain to be
further investigated. For instance, it is not clear how the stress
fibers are disrupted and what particular cytoskeletal actins are
affected by ASAP3. The present study reported that ASAP3 mediates
cancer cell migration and invasion at least in part by controlling
the expression of cytoskeletal γ-actin-1 (ACTG1) protein.
Materials and methods
Cell cultures, treatments and transient
transfections
H1299 human non-small cell lung cancer cells and
HepG2 hepatocellular carcinoma cells were purchased from the
American Type Culture Collection (Manassas, VA, USA). The cells
were normally maintained in Dulbecco’s modified Eagle’s medium
supplemented with 10% (v/v) fetal bovine serum, unless indicated
otherwise. For glucose or serum depletion treatments, four types
(serum free + 5 mM glucose, serum-free + 25 mM glucose, 10% serum +
5 mM glucose and 10% serum + 25 mM glucose) of reconstituted DMEM
media were prepared and used. Plasmid transfections were performed
with Lipofectamine® 2000 reagent (Invitrogen Life
Technologies, Carlsbad, CA, USA) according to the manufacturer’s
instructions.
Construction of recombinant adenoviral
vectors carrying shRNAs against ASAP3, and of the plasmid
overexpressing human ACTG1
Three gene cassettes containing short hairpin RNAs
(shRNAs) against ASAP3, as well as a gene cassette
containing a scrambled control sequence, were designed using an
siRNA Selection Program (http://jura.wi.mit.edu/siRNAext/home.php) and
chemically synthesized (Sangong Biotech, Co., Ltd, Shanghai,
China). The BamHI- (Cat. #FD0054)and HindIII [Cat.
#FD0504; Fermentas, Thermo Fisher Scientific (China) Co., Ltd.,
Beijing, China]-digested (gene cassettes were inserted into the
corresponding restriction sites on pSilencer 3.0-H1-neo (Ambion,
Austin, TX, USA), generating pSilencer-shRNA-1, -2 and -3 and its
control pSilencer-shRNA-CK. Subsequently, DNA fragments containing
the shRNA cassettes, as well as the H1 promoter, were polymerase
chain reaction (PCR)-amplified using the four plasmids and the
following primers: 5′-ACGGTACCTGATGACGGTGAAAACCTCT-3′ (forward) and
5′-GACCTCGAGGGCTTTACACTT TATGCTTCC-3′ (reverse). The four
KpnI/XhoI-digested PCR-amplified fragments were used
for subcloning into the Ad-Track cytomegalovirus (CMV) vector and
recombining into the Ad-Easy adenoviral vector using the AdEasy-XL
Adenoviral Vector system (Stratagene, Cedar Creek, TX, USA) as
instructed by the manufacturer (15), which generated recombinant
adenoviruses containing shRNAs against ASAP3 (Ad-shRNA-1, -2
and -3 respectively) and their control (Ad-shRNA-CK).
A 1,128-bp human ACTG1 (GenBank accession no.
NM-00119995) cDNA was PCR-amplified from cDNAs prepared from HepG2
human hepatoma cells using the following primers:
5′-CGCGAATTCAGAAGAAGAGATCGCCGC-3′ (forward) and
5′-CCCGGATC-CTTAGAAGC ATTTGCGGTG-3′ (reverse). The PCR-amplified
DNA fragment was digested with EcoRI and HindIII, and
subcloned into the pFlag-CMV2 vector (Addgene, Cambridge, MA, USA)
to generate a human ACTG1-overexpressing plasmid, pFlag-ACTG1.
Analysis of mRNA expression
Total RNA was extracted from HepG2 or H1299 cells
with TRIzol reagent (Invitrogen Life Technologies) according to the
manufacturer’s instructions. For the analysis of ASAP3 mRNA,
semi-quantitative analyses of mRNA expression by reverse
transcription (RT)-PCR was conducted according to previously
described methods (16), using the
following primer pair: 5′-AGCTGAGACATTTGTTCTCTTG-3′ (forward) and
5′-TATAAACCAGCTGAGTCCAGAG-3′ (reverse). GAPDH served as a control
[5′-ACAACAGCCTCAAGATCATCAG-3′ (forward) and
5′-GGTCCACCACTGACACGTTG-3′ (reverse)] (9).
Western blot analysis of proteins
Lysates from cultured cells were mixed with the
radioimmunoprecipitation assay buffer [25 mM Tris-HCl, pH 7.6, 150
mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl
sulfate, 1 ml protease inhibitor cocktails (Cat. #KGP603; Keygene,
Nanjing, China) and 5 ml phosphatase inhibitor cocktail (Cat.
#KGP602; Keygene) were added to 1 liter RIPA buffer immediately
before use], boiled for 5 min and sodium dodecyl
sulfate-polyacrylamide gel electrophoresis was applied. The protein
in the gel was electronically transferred to a nitrocellulose
membrane. Following blocking by 5% skimmed milk solution diluted in
Tris-buffered saline Tween 20 (TBST) for 1 h at room temperature,
the membrane was incubated with the primary antibody (diluted
1:1,000 with coating buffer) solutions overnight at 4°C and washed
with TBST three times. The washed membrane was further incubated
with secondary antibody in TBST for 1 h at room temperature. The
primary antibodies used were rabbit polyclonal anti-Flag antibody
(Octa-probe antibody, Cat. #sc-807, 1:1,000 dilution; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), rabbit polyclonal
anti-ASAP3/DDEFL1/UPLC1 antibody (Cat. #200-401-990, 1:1,000
dilution; Rockland Immunochemicals Inc., Gilbertsville, PA, USA),
mouse monoclonal anti-ACTG1 (1–24)
antibody (Cat. #sc-65635, 1:1,000 dilution; Santa Cruz
Biotechnology, Inc.), mouse monclonal anti-β-actin (C4) antibody
(Cat. #sc47778, 1:1,000 dilution; Santa Cruz Biotechnology, Inc.)
and mouse monoclonal anti-α-tubulin antiody (Cat. #5286, 1:1,000
dilution; Santa Cruz Biotechnology, Inc.). Detection was achieved
using the Immobilon Western Chemiluminescent Horseradish Peroxidase
Substrate kit (Millipore, Billerica, MA, USA). The western blot
images were captured by the Biosense SC8108 Gel Documentation
system with GeneScope V1.73 software (Shanghai BioTech, Shanghai,
China). The digital density values were acquired by Image-Pro Plus
6.0 software (MediaCybernetics, Rockville, MD, USA).
Cell proliferation, wound-healing and
transwell assays
For the cell proliferation assays, a total density
of 4×103 cells were seeded in 96-well plates and
infected with the recombinant adenoviruses at a dosage of ~400
viral particles/cell. After 48 h, 20 μl of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT;
5 mg/ml) was applied to each well and incubated for a further 4 h.
Cells were lysed with the addition of acidic isopropyl alcohol
(containing 0.04 M HCl) or dimethylsulfoxide for 15 min. The
absorbance was measured at 570 nm using a POLARstar Omega
microplate reader (BMG Labtech, Ortenberg, Germany).
For the cell migration assays, recombinant
adenovirus-infected cells or plasmid-transfected cells were marred
with a linear scratch by a sterile pipette tip. Wound healing was
recorded by photographing at specified time points following the
scratch. In addition, cell migration was analyzed by the transwell
assays (BD Biosciences, Sparks, MD, USA) according to the
manufacturer’s instructions. The comparison of the rates of cell
migration was achieved by direct visualization of the recorded
images.
Proteomic analyses of differentially
expressed proteins by two-dimensional gel electrophoresis and mass
spectrometry
H1299 cells were cultured and infected by
recombinant adenovirus containing either pAd-shRNA-3 or
pAd-shRNA-CK for 72 h. The cell lysates were collected for
proteomic analyses by two-dimensional gel electrophoresis and mass
spectrometry analyses (4800 Plus MALDI-TOF/TOF™ Analyzer; Applied
Biosystems, Foster City, CA, USA), that were performed by the
Beijing Proteome Research Center (Beijing, China).
Statistical analyses
Numerical data were analyzed by Graphpad Prism 5.0
(GraphPad Software, La Jolla, CA, USA) and expressed as the means ±
standard error of the mean. One-way analysis of variance with
Bonferroni’s post hoc test was used for multiple comparisons and
Student’s t-test (two-tailed) was used for pair-wise
comparisons.
Results
Adenovirus-mediated ASAP3 knockdown
inhibits cancer cell migration but does not affect cell
proliferation or the cell cycle
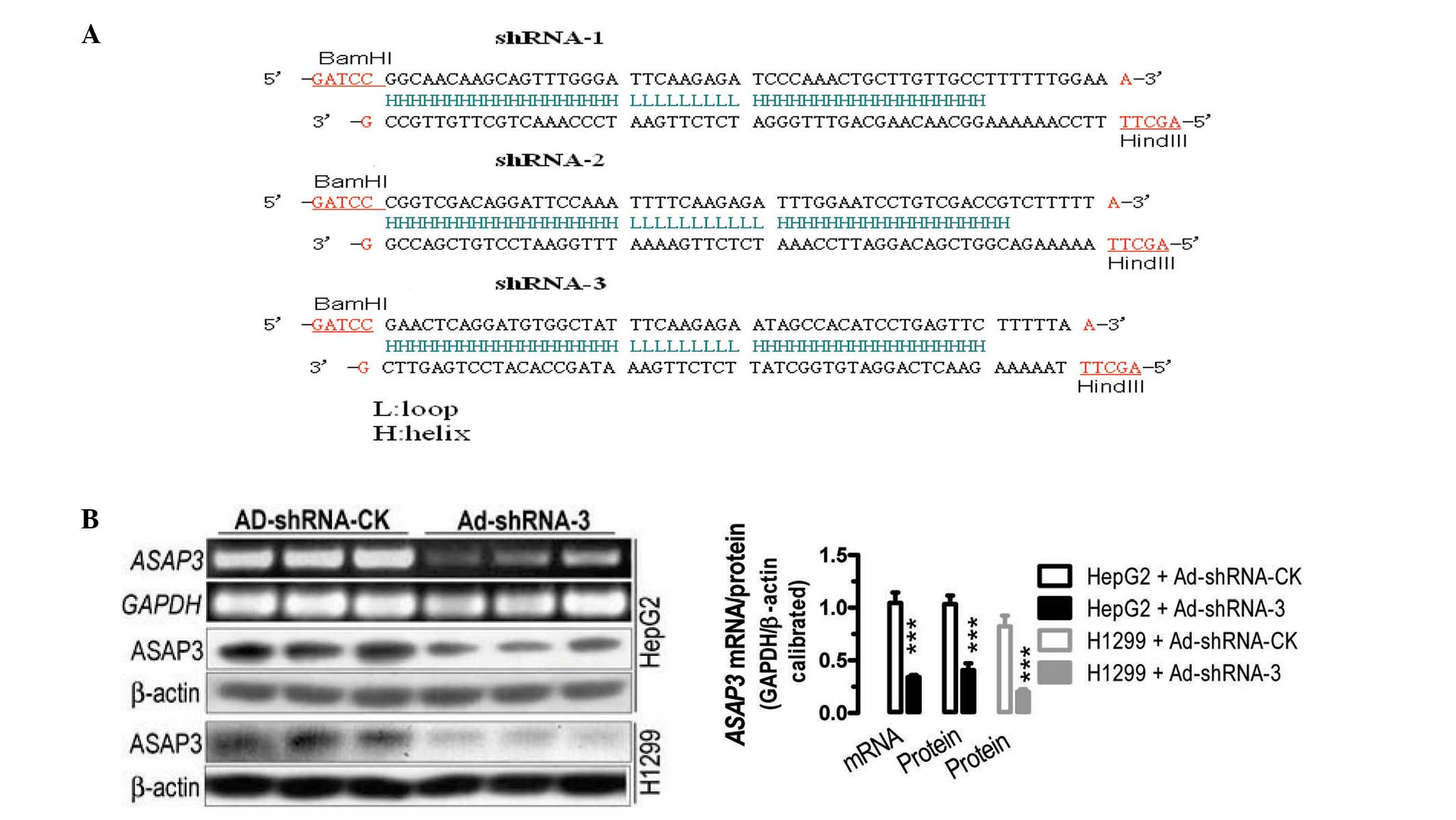
Based on bioinformatics analyses, four gene
cassettes were designed, with three cassettes carrying shRNAs
against human ASAP3 (GenBank accession no. NM-017707)
(Fig. 1A) and one cassette
carrying a scrambled control sequence. The four chemically
synthesized gene cassettes were used to create four recombinant
adenoviruses carrying the corresponding shRNA (Ad-shRNA-1, -2 and
-3) and their control (Ad-shRNA-CK). The infection studies
demonstrated that infection with Ad-shRNA-1, -2 (data not shown)
and -3 significantly reduced the mRNA and protein levels of
ASAP3 in cultured H1299 and HepG2 cancer cells, compared
with those in cells infected with Ad-shRANA-CK (Fig. 1B). For instance, in comparison with
cells infected with Ad-shRNA-CK, infection with Ad-shRNA-3 caused a
significant reduction of ~64% in ASAP3 mRNA expression in
the HepG2 cells (P<0.001), as determined by semi-quantitative
RT-PCR analyses. Simililarly, infection with Ad-shRNA-3 led to 60
and 80% reductions in ASAP3 protein expression in HepG2 and H1299
cells, respectively (P<0.001). The results demonstrated the
successful creation of recombinant viruses carrying shRNAs against
ASAP3.
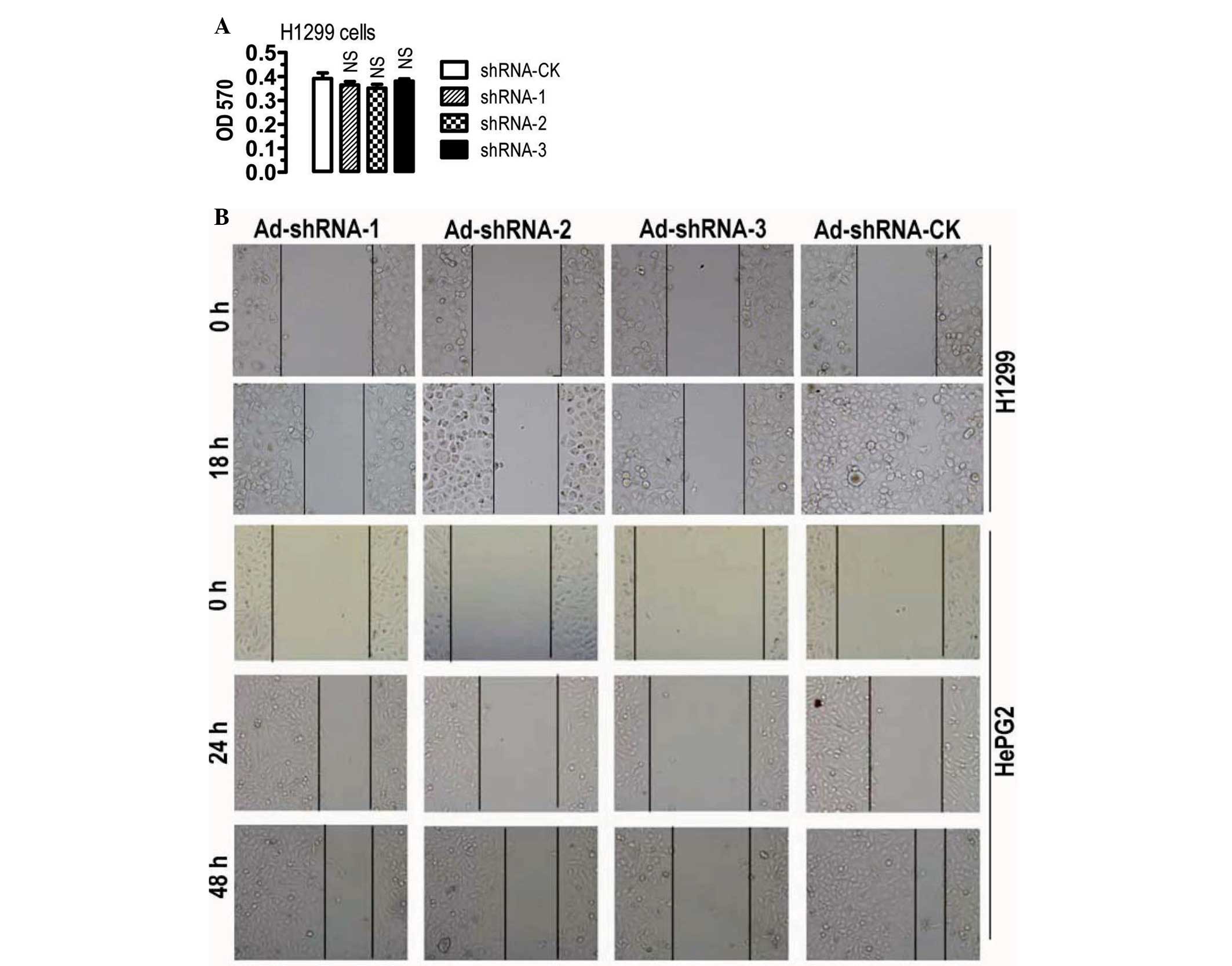
To determine the possible effects of
adenovirus-mediated ASAP3 knockdown on cancer cells, cell
proliferation, the cell cycle and cell migration in H1299 and HepG2
cells were analyzed. ASAP3 knockdown in H1299 cells did not
significantly affect the rate of cell proliferation (Fig. 2A) and neither did the cell cycle
appear to be affected (data not shown). By contrast, ASAP3
knockdown appeared to significantly reduce the likelihood of cell
migration in H1299 and HepG2 cells (Fig. 2B). The results, therefore, are
consistent with the notion that ASAP3 is an important regulator of
cell migration.
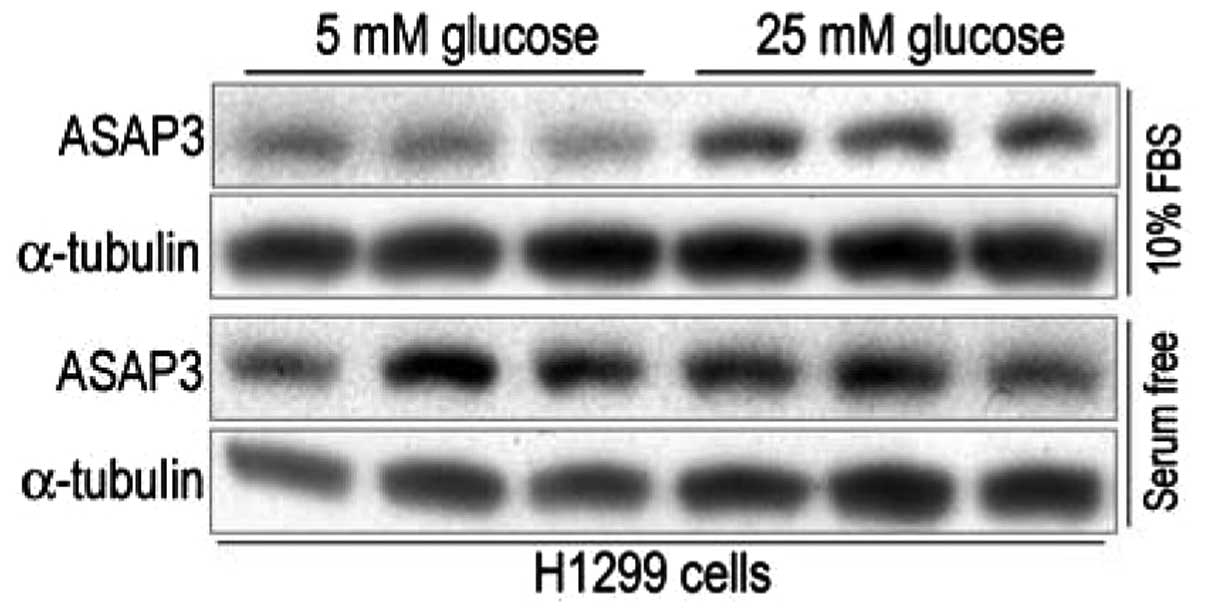
Effects of glucose and serum
concentration on protein expression of ASAP3
Okabe et al (9) demonstrated that ASAP3 may have
growth-promoting ability, particularly when under stressed
conditions. Therefore the present study aimed to determine whether
low glucose or serum deprivation affect ASAP3 expression. As shown
in Fig. 3, among the four
treatment groups, H1299 cells grown in media containing 10% serum
and 5 mM glucose had the lowest level of ASAP3 protein expression.
In comparison with this, ASAP3 expression appeared to be
significantly increased in cells grown in media containing 10%
serum and 25 mM glucose, while H1299 cells grown in media that were
serum free also had high levels of ASAP3 protein expression,
compared with that of H1299 cells grown in media containing 10%
serum and 5 mM glucose. These results suggest that high glucose and
serum deprivation are both capable of inducing ASAP3 protein
expression in H1299 cells.
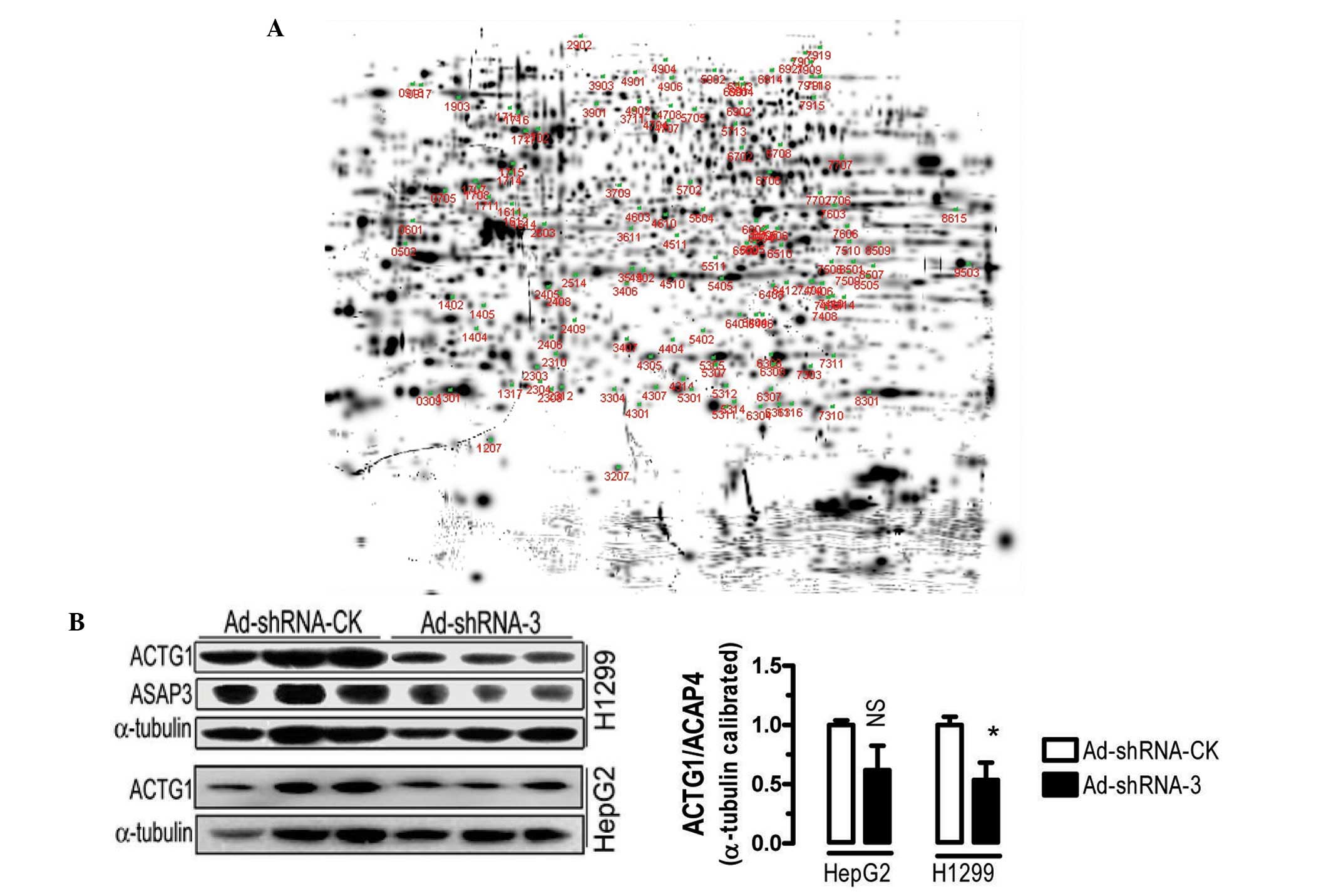
ASAP3 may regulate cell migration through
controlling the stability of cytoskeletal protein ACTG1
The demonstration that ASAP3 controlled cell
migration in H1299 and HepG2 cells is largely consistent with
previous findings in other cancer cell lines, including the HeLa
cell line (10,13). However, the underlying mechanisms
were not clear. Utilizing two-dimensional gel electrophoresis and
mass spectrometry analyses, proteomic analyses were performed with
H1299 cells infected with Ad-shRNA-3 and Ad-shRNA-CK. The
two-dimensional gel analyses identified 124 protein spots that were
differentially expressed by at least two-fold between the
Ad-shRNA-3- and Ad-shRNA-CK-transfected cells. Protein spot no.
1207 was significantly downregulated (Fig. 4A) and spot no. 8507 was
significantly upregulated compared with the control (with the
expression of Ad-shRNA-CK transfected cells being 1, the expression
of Ad-shRNA-3 transfected cells for no. 1207 corresponding spot was
0.11 and no. 8507 corresponding spot was 10.34). Mass spectrometry
analyses indicated that the two proteins were non-muscle
cytoskeletal proteins, ACTG1 and GAPDH, respectively.
To verify the regulation of ACTG1 by ASAP3,
protein expression analyses were performed in H2199 and HepG2
cells. Adenovirus-mediated knockdown of ASAP3 caused downregulation
of ACTG1 protein expression in the two cell lines compared with the
control, although the difference was only statistically significant
in the H1299 cells (Fig. 4B).
These results suggested that ASAP3 may be able to regulate the
stability or expression of ACTG1.
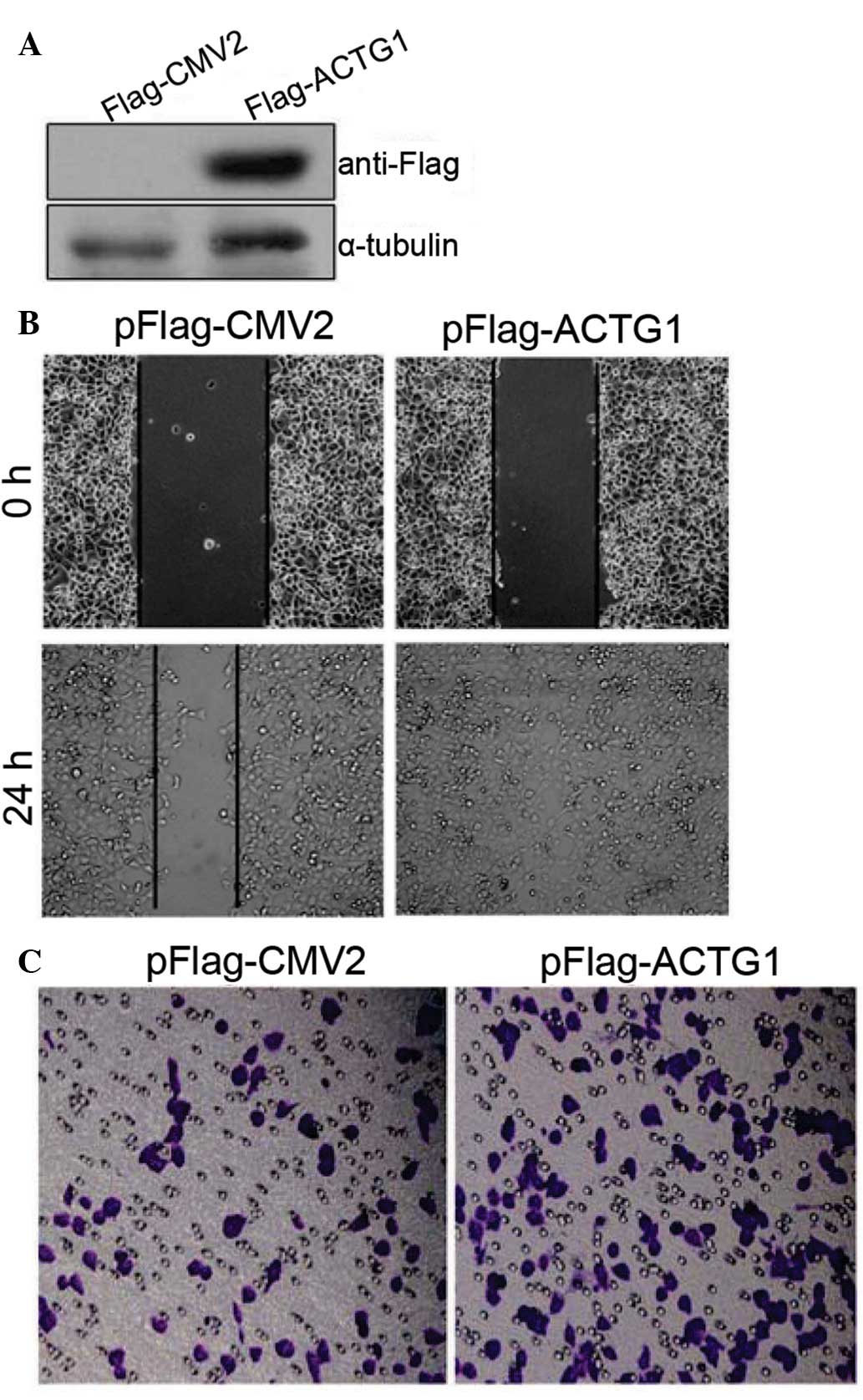
Overexpression of ACTG1 enhances cancer
cell migration
To determine the potential effects of ACTG1 on
cancer cells, wound-healing and transwell assays were performed.
Notably, overexpression of ACTG1 (Fig.
5A) significantly enhanced the likelihood of cell migration as
assayed by wound-healing (Fig. 5B)
and transwell (Fig. 5C) assays in
the H1299 cells. Similar results were observed for HepG2 cells
(data not shown). Together these results suggested that ASAP3 may
regulate cell migration at least in part by downregulating the
expression ACTG1, thereby disrupting the cytoskeletal fiber
networks.
Discussion
Previous studies have suggested that ASAP3 is
pivotal in cell migration (10,12,13).
However, the mechanisms through which ASAP3 mediates cell migration
were not clearly elucidated. Although, Yu et al (12) demonstrated that ASAP3 may cooperate
with Grb2 to regulate integrin β1 recycling in cancer cell
migration. Grb2 is an adaptor protein important for intracellular
signal transduction, cell proliferation, actin-based cell motility
and endocytic trafficking, whereas integrins are transmembrane
receptors that are important for cell signaling and the regulation
of the cell cycle, shape and motility. Despite this, there may be
other mechanisms involved. In breast cancer MDA-MB-231 cells, Ha
et al (13) demonstrated
that overexpression of wild-type ASAP3 or of a GAP-inactive mutant
ASAP3, or ASAP3 knockdown did not affect the distribution of
vinculin or paxillin, suggesting that ASAP3 does not affect focal
adhesions. ASAP3 knockdown, however, was demonstrated to
significantly reduce actin stress fibers, although it is not clear
which particular actins may be affected. Notably, using proteomic
and other approaches in the present study, cytoskeletal ACTG1 was
identified as one of the actins directly targeted by ASAP3 in H1299
lung cancer cells and appeared to contribute significantly to
ASAP3 knockdown-induced disruption of the actin stress
fibers.
Actins are highly conserved proteins that are
involved in cell motility and maintenance of the cytoskeleton,
among other functions (17). In
vertebrates, three main groups of actin isoforms, namely α, β and
γ, have been identified. The α-actins are found in muscle tissues
and are a major constituent of the contractile apparatus.
Conversely the β- and γ-actins, co-exist in the majority of cell
types as components of the cytoskeleton, and as mediators of
internal cell motility. A previous study associated alterations in
the expression of ACTG1 with diseases, such as hearing loss and
cancer (18). In auditory cells,
multiple mutations in ACTG1 were demonstrated to be
associated with dominant progressive deafness (19–26).
Certain studies have demonstrated that ACTG1 is important for
cytoskeletal maintenance but not for development (27,28).
However, a more recent study suggested that a de novo
mutation in ACTG1 causes a form of brain malformation in humans
(Baraitser-Winter syndrome) (29).
Moreover, a study in ACTG1 null mice suggested that ACTG1
deficiency led to growth impairment and reduced cell viability
(30). Using oligonucleotide
microarrays, Sun et al (31) demonstrated that a group of
microtubule proteins and intermediate filament proteins, including
ACTG1, were significantly upregulated in the liver tissues of
transgenic mice overexpressing the oncogenic hepatitis B virus X
protein (HBx), suggesting that the dysregulation of ACTG1 and/or
other cytoskeletal proteins may contribute significantly to
HBx-induced hepatocarcinogenesis.
Notably, Shum et al (32) demonstrated that in SH-EP
neuroblastoma cells, ACTG1 knockdown resulted in a
significant decrease in wound healing and transwell migration. In
contrast to ASAP3 knockdown in MDA-MD-231 cells, however,
there was a significant increase in the size and number of
paxillin-containing focal adhesions, coupled with a significant
decrease in phosphorylated paxillin in ACTG1 knockdown
neuroblastoma cells. It is not clear whether this effect of
ACTG1 knockdown is cell type-specific. In addition,
inhibition of the Rho-associated kinase (ROCK) with the inhibitor
Y-27632 restored the ability of ACTG1-knockdown cells to
migrate, suggesting that ACTG1 is an upstream regulator of
ROCK-mediated cell migration. More recently, the same research
group further demonstrated that ACTG1 can modulate microtubule
dynamics (33). Largely consistent
with the two aforementioned studies, the present study demonstrated
that overexpression of ACTG1 enhanced cancer cell migration in the
H1299 and HepG2 cells (Fig. 5).
Concordant with a previous suggestion that ArfGAPs may function as
upstream regulators of Rho family proteins (1), the results of the present study
indicate that in cancer cells, ASAP3 may positively regulate ACTG1
to promote cytoskeletal remodeling and cell migration, particularly
ROCK signaling pathway-mediated cell migration (34). At present, however, whether ASAP3
regulates ACTG1 directly or indirectly and the underlying
mechanisms are not clear and awaits further investigation. Our
demonstration that ASAP3 controls cell migration at least in part
by destabilizing cytoskeletal protein ACTG1, may aid future
development of drugs aiming at cancer cell migration, invasion and
metastasis.
Acknowledgements
This study was supported in part by grants from the
973 Program, China (grant no. 2009CB941601), the National Natural
Science Foundation of China (grant no. 31271239), the Fujian
Provincial Department of Science and Technology (grant no.
2010L0002) and the Open Research Fund of State Key Laboratory of
Cellular Stress Biology, Xiamen University (grant no.
SKLCSB2012KF005).
References
|
1
|
Randazzo PA, Inoue H and Bharti S: Arf
GAPs as regulators of the actin cytoskeleton. Biol Cell.
99:583–600. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Spang A, Shiba Y and Randazzo PA: Arf
GAPs: gatekeepers of vesicle generation. FEBS Lett. 584:2646–2651.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Randazzo PA and Hirsch DS: Arf GAPs:
multifunctional proteins that regulate membrane traffic and actin
remodelling. Cell Signal. 16:401–413. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
D’Souza-Schorey C and Chavrier P: ARF
proteins: roles in membrane traffic and beyond. Nat Rev Mol Cell
Biol. 7:347–358. 2006.
|
|
5
|
Bharti S, Inoue H, Bharti K, Hirsch DS,
Nie Z, Yoon HY, Artym V, Yamada KM, Mueller SC, Barr VA and
Randazzo PA: Src-dependent phosphorylation of ASAP1 regulates
podosomes. Mol Cell Biol. 27:8271–8283. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Randazzo PA, Andrade J, Miura K, Brown MT,
Long YQ, Stauffer S, Roller P and Cooper JA: The Arf
GTPase-activating protein ASAP1 regulates the actin cytoskeleton.
Proc Natl Acad Sci USA. 97:4011–4016. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
de Curtis I: Cell migration: GAPs between
membrane traffic and the cytoskeleton. EMBO Rep. 2:277–281.
2001.PubMed/NCBI
|
|
8
|
Ismail SA, Vetter IR, Sot B and
Wittinghofer A: The structure of an Arf-ArfGAP complex reveals a
Ca2+ regulatory mechanism. Cell. 141:812–821. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Okabe H, Furukawa Y, Kato T, Hasegawa S,
Yamaoka Y and Nakamura Y: Isolation of development and
differentiation enhancing factor-like 1 (DDEFL1) as a drug target
for hepatocellular carcinomas. Int J Oncol. 24:43–48.
2004.PubMed/NCBI
|
|
10
|
Fang Z, Miao Y, Ding X, Deng H, Liu S,
Wang F, Zhou R, Watson C, Fu C, Hu Q, et al: Proteomic
identification and functional characterization of a novel ARF6
GTPase-activating protein, ACAP4. Mol Cell Proteomics. 5:1437–1449.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ding X, Deng H, Wang D, Zhou J, Huang Y,
Zhao X, Yu X, Wang M, Wang F, Ward T, et al: Phospho-regulated
ACAP4-Ezrin interaction is essential for histamine-stimulated
parietal cell secretion. J Biol Chem. 285:18769–18780. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu X, Wang F, Liu H, Adams G, Aikhionbare
F, Liu D, Cao X, Fan L, Hu G, Chen Y, et al: ACAP4 protein
cooperates with Grb2 protein to orchestrate epidermal growth
factor-stimulated integrin β1 recycling in cell migration. J Biol
Chem. 286:43735–43747. 2011.PubMed/NCBI
|
|
13
|
Ha VL, Bharti S, Inoue H, Vass WC, Campa
F, Nie Z, de Gramont A, Ward Y and Randazzo PA: ASAP3 is a focal
adhesion-associated Arf GAP that functions in cell migration and
invasion. J Biol Chem. 283:14915–14926. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pellegrin S and Mellor H: Actin stress
fibres. J Cell Sci. 120:3491–3499. 2007. View Article : Google Scholar
|
|
15
|
He TC, Zhou S, da Costa LT, Yu J, Kinzler
KW and Vogelstein B: A simplified system for generating recombinant
adenoviruses. Proc Natl Acad Sci USA. 95:2509–2514. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qiu L, Wu X, Chau JF, Szeto IY, Tam WY,
Guo Z, Chung SK, Oates PJ, Chung SS and Yang JY: Aldose reductase
regulates hepatic peroxisome proliferator-activated receptor α
phosphorylation and activity to impact lipid homeostasis. J Biol
Chem. 283:17175–17183. 2008.PubMed/NCBI
|
|
17
|
Herman IM: Actin isoforms. Curr Opin Cell
Biol. 5:48–55. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chou CC, Davis RC, Fuller ML, Slovin JP,
Wong A, Wright J, Kania S, Shaked R, Gatti RA and Salser WA:
Gamma-actin: unusual mRNA 3′-untranslated sequence conservation and
amino acid substitutions that may be cancer related. Proc Natl Acad
Sci USA. 84:2575–2579. 1987.
|
|
19
|
Zhu M, Yang T, Wei S, DeWan AT, Morell RJ,
Elfenbein JL, Fisher RA, Leal SM, Smith RJ and Friderici KH:
Mutations in the γ-actin gene (ACTG1) are associated with dominant
progressive deafness (DFNA20/26). Am J Hum Genet. 73:1082–1091.
2003.
|
|
20
|
van Wijk E, Krieger E, Kemperman MH, De
Leenheer EM, Huygen PL, Cremers CW, Cremers FP and Kremer H: A
mutation in the gamma actin 1 (ACTG1) gene causes autosomal
dominant hearing loss (DFNA20/26). J Med Genet. 40:879–884.
2003.PubMed/NCBI
|
|
21
|
de Heer AM, Huygen PL, Collin RW, Oostrik
J, Kremer H and Cremers CW: Audiometric and vestibular features in
a second Dutch DFNA20/26 family with a novel mutation in ACTG1. Ann
Otol Rhinol Laryngol. 118:382–390. 2009.PubMed/NCBI
|
|
22
|
Liu P, Li H, Ren X, Mao H, Zhu Q, Zhu Z,
Yang R, Yuan W, Liu J, Wang Q and Liu M: Novel ACTG1 mutation
causing autosomal dominant non-syndromic hearing impairment in a
Chinese family. J Genet Genomics. 35:553–558. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morin M, Bryan KE, Mayo-Merino F, Goodyear
R, Mencia A, Modamio-Hoybjor S, del Castillo I, Cabalka JM,
Richardson G, Moreno F, et al: In vivo and in vitro effects of two
novel gamma-actin (ACTG1) mutations that cause DFNA20/26 hearing
impairment. Hum Mol Genet. 18:3075–3089. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rendtorff ND, Zhu M, Fagerheim T, Antal
TL, Jones M, Teslovich TM, Gillanders EM, Barmada M, Teig E, Trent
JM, et al: A novel missense mutation in ACTG1 causes dominant
deafness in a Norwegian DFNA20/26 family, but ACTG1 mutations are
not frequent among families with hereditary hearing impairment. Eur
J Hum Genet. 14:1097–1105. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Perrin BJ, Sonnemann KJ and Ervasti JM:
β-actin and γ-actin are each dispensable for auditory hair cell
development but required for Stereocilia maintenance. PLoS Genet.
6:e10011582010.
|
|
26
|
Kruth KA and Rubenstein PA: Two
deafness-causing (DFNA20/26) actin mutations affect
Arp2/3-dependent actin regulation. J Biol Chem. 287:27217–27226.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Belyantseva IA, Perrin BJ, Sonnemann KJ,
Zhu M, Stepanyan R, McGee J, Frolenkov GI, Walsh EJ, Friderici KH,
Friedman TB and Ervasti JM: γ-actin is required for cytoskeletal
maintenance but not development. Proc Natl Acad Sci USA.
106:9703–9708. 2009.
|
|
28
|
Sonnemann KJ, Fitzsimons DP, Patel JR, Liu
Y, Schneider MF, Moss RL and Ervasti JM: Cytoplasmic γ-actin is not
required for skeletal muscle development but its absence leads to a
progressive myopathy. Dev Cell. 11:387–397. 2006.
|
|
29
|
Riviere JB, van Bon BW, Hoischen A,
Kholmanskikh SS, O’Roak BJ, Gilissen C, Gijsen S, Sullivan CT,
Christian SL, Abdul-Rahman OA, et al: De novo mutations in the
actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat
Genet. 44:440–444. S1–S2. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bunnell TM and Ervasti JM: Delayed
embryonic development and impaired cell growth and survival in
Actg1 null mice. Cytoskeleton (Hoboken). 67:564–572. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun Q, Wang Y, Zhang Y, Liu F, Cheng X,
Hou N, Zhao X and Yang X: Expression profiling reveals
dysregulation of cellular cytoskeletal genes in HBx-induced
hepatocarcinogenesis. Cancer Biol Ther. 6:668–674. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shum MS, Pasquier E, Po’uha ST, O’Neill
GM, Chaponnier C, Gunning PW and Kavallaris M: γ-Actin regulates
cell migration and modulates the ROCK signaling pathway. FASEB J.
25:4423–4433. 2011.
|
|
33
|
Po’uha ST, Honore S, Braguer D and
Kavallaris M: Partial depletion of gamma-actin suppresses
microtubule dynamics. Cytoskeleton (Hoboken). 70:148–160.
2013.PubMed/NCBI
|
|
34
|
Patel RA, Liu Y, Wang B, Li R and Sebti
SM: Identification of novel ROCK inhibitors with anti-migratory and
anti-invasive activities. Oncogene. Feb 11–2013.(Epub ahead of
print).
|