Introduction
Osteoporosis is a systemic skeletal disease
characterized by low bone mineral density and micro-architectural
deterioration of bone, leading to increased bone fragility and
susceptibility to fractures (1).
The clinical complications of osteoporosis include pain, fractures
and disability. Recently, osteoporosis has become a predominant
public health problem in elderly individuals, particularly in
postmenopausal females (2).
Reactive oxygen species (ROS) such as superoxide anions, hydroxyl
radicals and hydrogen peroxide (H2O2) lead to
severe damage to DNA, protein and lipids. Therefore, ROS is a
predominant cause of cell damage and death in numerous pathological
conditions (3). Furthermore, it
has been demonstrated that ROS-induced bone cell cytotoxicity is
critical in the development of osteoporosis (4). Under normal physiological and
pathological conditions, the mitochondria are the predominant
producers of ROS, such as hydrogen peroxide, hydroxyl radicals,
superoxide radicals and singlet oxygen (5). Additionally, the mitochondria are the
key targets of ROS-mediated damage (6). It has been shown that mitochondrial
dysfunction is involved in ROS-induced bone cell cytotoxicity
(2,7). Therefore, it is plausible that
therapeutic strategies which aim to prevent or delay ROS-induced
bone cell cytotoxicity by maintaining mitochondrial function may be
suitable for the prevention or treatment of bone loss-related
disorders.
Melatonin is an indoleamine primarily secreted by
the pineal gland and is also synthesized in other organs, such as
the skin (8), gastrointestinal
tract (9), thymus (10), retina (11) and bone marrow (12). Melatonin is involved in the
regulation of several physiological processes such as sleep-wake
rhythms (13,14), regulation of the circadian cycle
(15,16), seasonal control of reproductive
processes (17) and immune
processes (18). In addition to
these physiological actions, melatonin is a potent scavenger of
ROS, such as the carbonate radical (19), which is presumed to be involved in
mitochondrial damage (20).
Furthermore, melatonin easily enters the mitochondria and exerts a
direct beneficial effect on the maintenance of mitochondrial
homeostasis (21). It has also
been demonstrated that melatonin stabilizes the mitochondrial inner
membrane, improves electron transport chain activity, increases ATP
synthesis and protects mitochondrial DNA from oxidative damage
(22–25). Based on the mitochondrial
dysfunction observed during ROS-induced bone cell cytotoxicity and
the beneficial effects of melatonin on the mitochondria, melatonin
may be a useful molecule for preventing or treating diseases
related to bone loss.
The aim of the present study was to investigate the
potential efficacy of melatonin in the protection against
ROS-induced bone cell cytotoxicity. To address this issue, the MG63
osteoblastic cell line was exposed to different concentrations of
H2O2. As expected, melatonin pretreatment
successfully attenuated H2O2-induced
cytotoxicity in MG63 cells. The protective effects of melatonin
were correlated with its ability to reduce oxidative damage,
increase ATP production, maintain the mitochondrial membrane
potential and preserve mitochondrial DNA content in
H2O2-treated cells. This study suggests that
melatonin effectively protects against
H2O2-induced cytotoxicity in MG63
osteoblastic cells by maintaining mitochondrial function.
Materials and methods
Chemicals
H2O2 was purchased from
Sigma-Aldrich (St. Louis, MO, USA). Melatonin was obtained from
Sangon Biotech Co., Ltd. (Shanghai, China). The cell lysis buffer
and bicinchoninic acid (BCA) protein assay kit were purchased from
Beyotime (Shanghai, China).
Cell culture and treatments
MG63 osteoblast-like cells (American Type Culture
Collection, Manassas, VA, USA) were cultured in Dulbecco’s modified
Eagle’s medium (DMEM; Hyclone, Logan, UT, USA) supplemented with
10% heat-inactivated fetal bovine serum (FBS; HyClone) and 80 μg/ml
penicillin/streptomycin (Sigma-Aldrich) in a 5%
CO2-humidified atmosphere at 37°C. To estimate the
toxicity of H2O2, MG63 cells were seeded at a
density of 1×104 cells/well in 96-well plates, cultured
overnight and then exposed to various concentrations of
H2O2 (0, 200, 400 or 800 μM) for 8 h, or
exposed to 200 or 400 μM H2O2 for different
periods of time (0.5, 4, 8 and 12 h). In other assays unless
otherwise stated, the cells were seeded at a density of
2×105/well in 6-well plates, cultured overnight,
pretreated with melatonin (1 mM) for 2 h and then exposed to
H2O2 (200 or 400 μM) for 8 h.
Measurement of cell viability and lactate
dehydrogenase (LDH) release
The viability of MG63 cells was assessed using the
Cell Counting kit-8 (CCK-8; Dojindo, Kumamoto, Japan) according to
the manufacturer’s instructions. Following treatment with
H2O2, the cells were cultured in 100 μl fresh
growth medium supplemented with 10 μl CCK-8-solution for 2 h at
37°C and the absorption values were measured at 450 nm using a
microplate reader. Cell viability was expressed as a percentage of
untreated control cells (set at 100%). All the experiments were
performed in triplicate and were repeated three times.
LDH release was measured using the Cytotoxicity
Detection kit (Roche, Mannheim, Germany) according to the
manufacturer’s instructions. The cells were pretreated with
melatonin and exposed to H2O2 in a low-serum
(1% FBS) medium, in order to minimize the effects of LDH in the
serum. Following treatment, cell-free culture supernatants were
collected and incubated with LDH assay solution at 25°C for 30 min.
The optical density values were analyzed at 490 nm by subtracting
the reference value at 620 nm. The experiment was repeated three
times and the results were expressed as a percentage of the maximum
LDH release, obtained by lysing the cells in 1% Triton X-100.
Measurement of ROS and MDA levels
Intracellular ROS generation was detected by flow
cytometry (BD FACSCanto II; BD Biosciences, Mississauga, ON,
Canada) using 2′,7′-dichlorofluoresceindiacetate (DCFH-DA;
Beyotime, Shanghai, China). Subsequent to the indicated treatments,
the cells were trypsinized, centrifuged and washed with fresh
preheated DMEM to remove any H2O2 remaining
at the end of the exposure time. The cells were then resuspended in
preheated serum-free medium supplemented with DCFH-DA and incubated
at 37°C for 20 min. Following three washes with PBS, the
fluorescence intensity of the cells in each group was measured by
flow cytometry. The level of ROS is expressed as the mean of the
DCF fluorescence intensity in each group. The experiment was
performed in triplicate and repeated three times.
Intracellular malondialdehyde (MDA) levels were
measured using a Lipid Peroxidation MDA assay kit (Beyotime)
according to the manufacturer’s instructions. Following
H2O2 treatment, the cells were collected,
lysed with cell lysis buffer and centrifuged. The supernatants were
reacted with thiobarbituric acid (TBA) and the absorption values of
the reaction products were measured with a microplate reader
(Varioskan™ Flash 3001; Thermo Scientific, Waltham, MA, USA) at 535
nm. The protein concentration was measured by the Bradford Protein
assay. The experiment was repeated three times and the MDA levels
were expressed as nmol/mg protein.
Measurement of ATP content
The ATP determination kit (Beyotime, Nanjing, China)
was used to determine the ATP content. Following the termination of
the treatment period, the cells were incubated with cell lysis
buffer and centrifuged. The supernatants (10 μl) were mixed with
reaction buffer (100 μl) and measured using a luminometer (Turner
Designs Inc., Sunnyvale, CA, USA) and the experiments were repeated
four times. The cellular ATP content was determined from an ATP
standard curve and the results were expressed as a percentage,
assuming that the ATP content of untreated control cells was
100%.
Measurement of mitochondrial membrane
potential
The mitochondrial membrane potential (ΔΨm) of MG63
cells was determined by the fluorescent, lipophilic and cationic
probe, JC-1, according to the manufacturer’s instructions. Briefly,
the cells were seeded at a density of 1×104 cells/well
in 96-well plates. Subsequent to the indicated treatments, the
cells were incubated with 1X JC-1 in growth medium at 37°C for 20
min and then rinsed twice with JC-1 washing buffer. The green
fluorescence intensities from the JC-1 monomer (with a 530-nm
excitation) and the red fluorescence intensities from the
aggregated form of JC-1 (with a 590-nm emission) in the cells were
measured by spectrofluorometry (FACSCalibur; Becton-Dickinson,
Franklin Lakes, NJ, USA). The ΔΨm of MG63 cells in each group was
calculated as the fluorescence ratio of red to green. The
experiment was repeated at least three times.
Measurement of mitochondrial DNA (mtDNA)
copy number
qPCR was used to determine the mtDNA copy number,
and was conducted using the Mx3000p Real-Time PCR detection system
(Stratagen Inc., La Jolla, CA, USA) with the SYBR-Green I detection
method. Total DNA was extracted from the treated cells using a DNA
extract kit (Omega Bio-Tek, Norcross, GA, USA). The mtDNA copy
number was expressed relative to the nuclear DNA copy number. Two
different segments of mtDNA were amplified: cytochrome coxidase
subunit I (COX I), encoded by the heavy chain of mtDNA, and
NADH dehydrogenase subunit 6 (ND6), encoded by the light
chain of mtDNA. The nuclear amplicon was generated by amplification
of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which
was chosen as an internal standard. The primers for mtDNA were:
Sense: 5′-CATCGGGGTAGTCCGAGTAA-3′ and antisense:
5′-ACGTTGTAGCCCACTTCCAC-3′ for COXI; sense:
5′-TGATTGTTAGCGGTGTGGTC-3′ and antisense:
5′-CCACAGCACCAATCCTACCT-3′ for ND6; and sense:
5′-TCAGTGGTGGACCTGACCTG-3′ and antisense:
5′-TGCTGTAGCCAAATTCGTTG-3′ for GAPDH. Each measurement was
performed in triplicate, repeated at least three times and
normalized against control cells.
Statistical analysis
Experimental data are presented as the mean ± SD.
Each experiment was repeated at least three times. Differences
between two groups were analyzed using the Student’s t-test.
Multiple comparisons were analyzed by analysis of variance and
Tukey’s post hoc tests. P<0.05 was considered to indicate a
statistically significant difference. Images are representative of
three or more experiments.
Results
Melatonin reduces the cytotoxicity of
H2O2 in MG63 cells
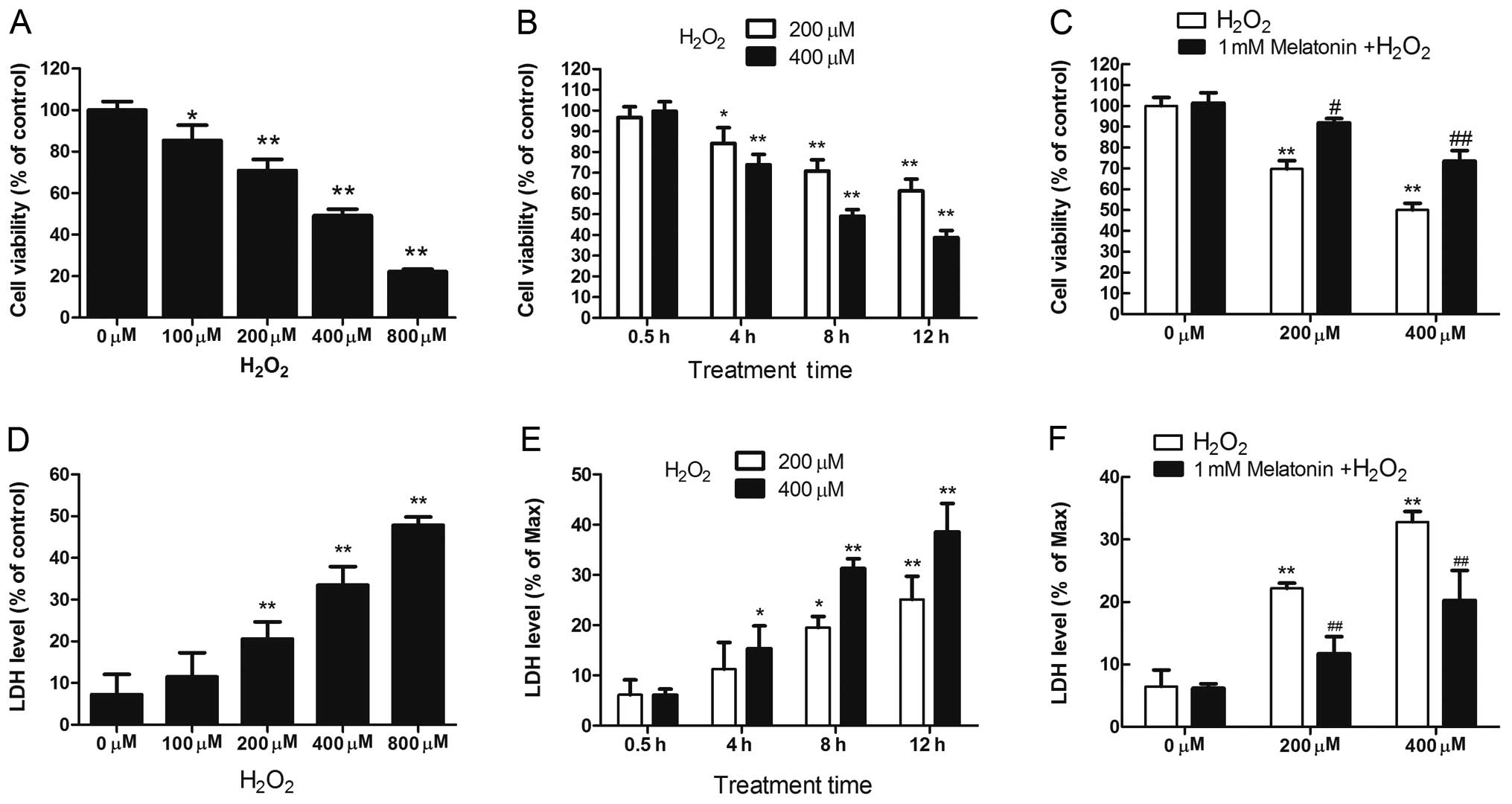
The cytotoxicity of H2O2 in
MG63 cells was analyzed by assaying cell viability and measuring
LDH release. The viability of MG63 cells exposed to
H2O2 was reduced significantly in a dose- and
time-dependent manner (Fig. 1A and
B). Additionally, H2O2 treatment resulted
in a dose- and time-dependent increase in LDH release by MG63 cells
(Fig. 1D and E).
H2O2 (200 and 400 μM) induced significant
toxicity in MG63 cells after 8 h of treatment; therefore, these
treatment conditions were used for subsequent experiments.
Melatonin pretreatment (1 mM) significantly prevented the loss of
cell viability and reduced LDH release in
H2O2-treated MG63 cells (Fig. 1C and F), demonstrating that
melatonin protected against the cytotoxic effects of
H2O2 in MG63 cells.
Melatonin ameliorates
H2O2-mediated oxidative stress in MG63
cells
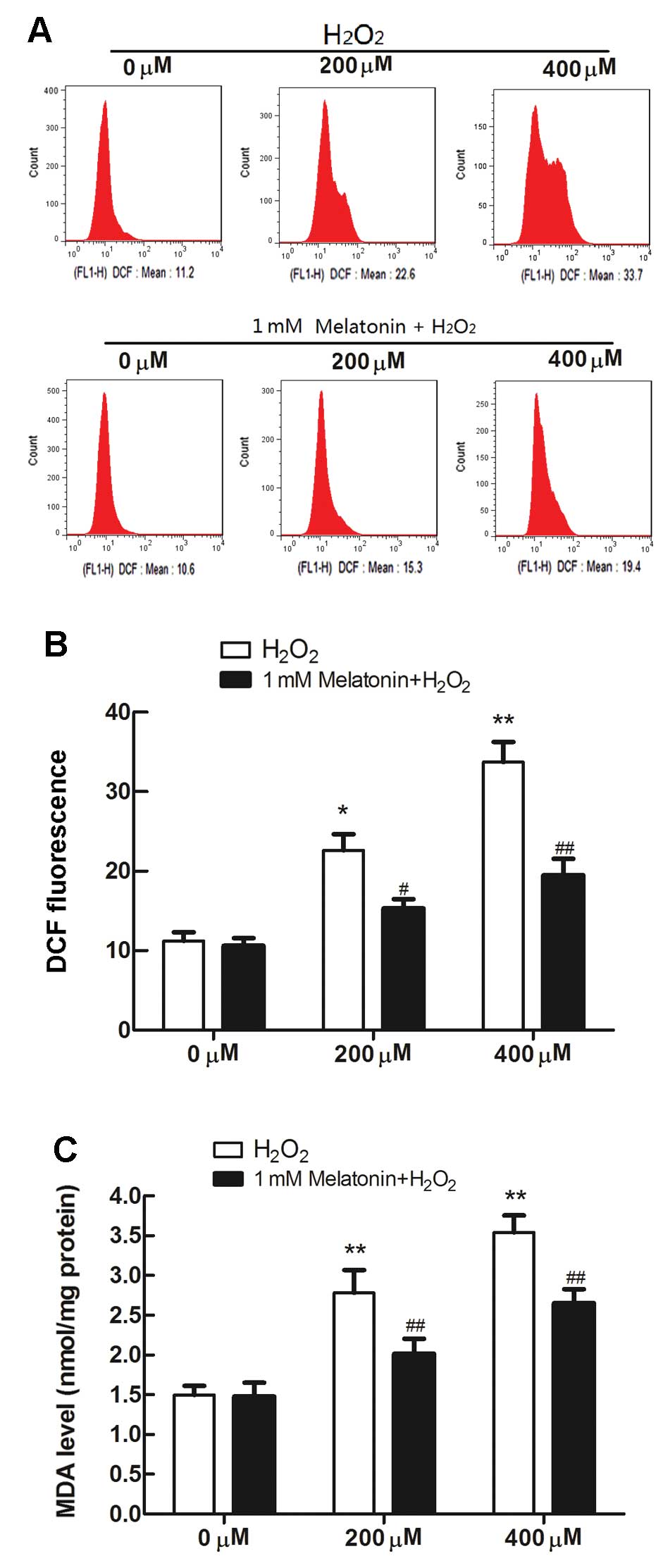
The levels of intracellular ROS in MG63 cells
following H2O2 treatment were measured by
flow cytometry using the fluorescent probe DCFH-DA. ROS generation
increased significantly in MG63 cells exposed to
H2O2 (Fig. 2A
and B). Furthermore, to investigate the effects of
H2O2 on oxidative damage, the intracellular
levels of MDA (a marker of lipid peroxidation) were measured. The
concentration of MDA increased from 1.49 nmol/mg protein in
untreated control cells to 2.78 and 3.54 nmol/mg protein in 200 and
400 μM H2O2-treated cells, respectively
(Fig. 2C). However, melatonin
pretreatment (1 mM) successfully attenuated the
H2O2-induced increases in ROS release and
intracellular MDA levels in MG63 cells (Fig. 2).
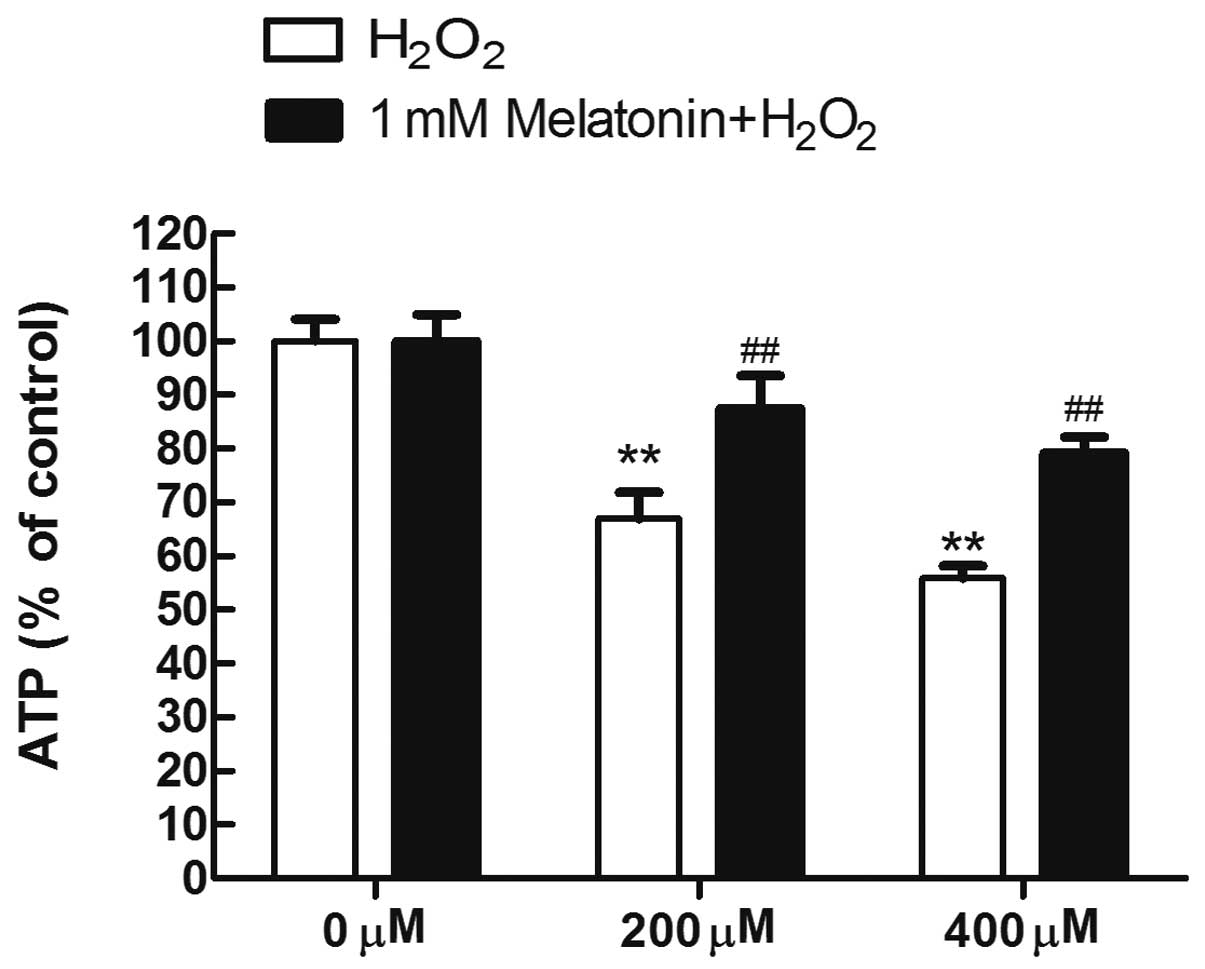
Melatonin maintains the ATP content in
MG63 cells exposed to H2O2
The protective effects of melatonin on respiratory
function were evaluated by measuring the ATP concentrations in MG63
cells exposed to H2O2 following pretreatment
with melatonin. Compared with the untreated control group, the ATP
concentrations were significantly reduced in the
H2O2-treated groups (all P<0.05). However,
pretreatment with melatonin maintained the ATP content in MG63
cells exposed to H2O2 (Fig. 3).
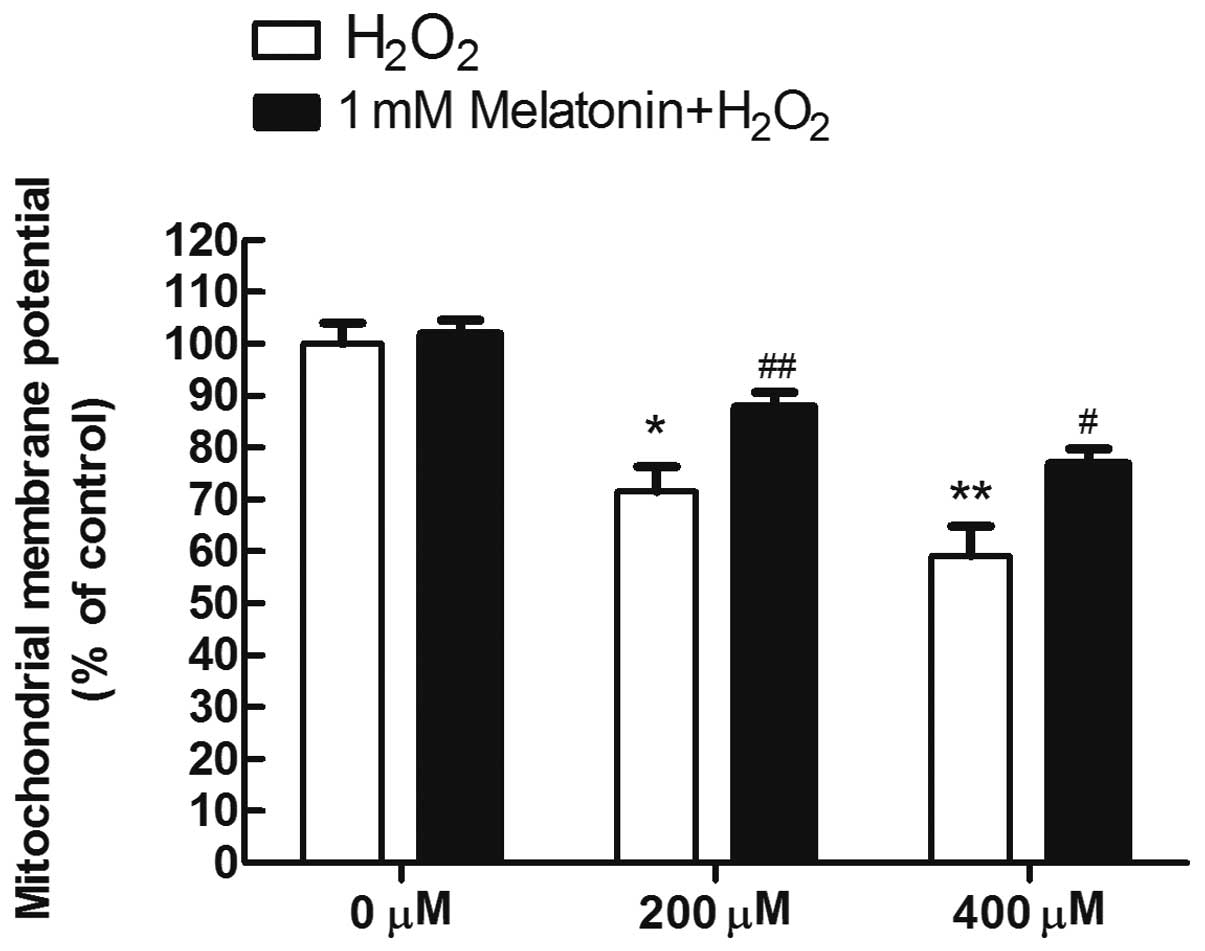
Melatonin prevents the loss of
mitochondrial membrane potential in MG63 cells exposed to
H2O2
To analyze whether the inhibition of mitochondrial
disruption is a mechanism underlying the protective effects of
melatonin, the fluorescence probe JC-1 was used to estimate the
mitochondrial membrane potential (ΔΨm), which is a sensitive
indicator of the mitochondrial integrity and bioenergetic function.
As a result, it was demonstrated that exposure to 200 and 400 μM
H2O2 for 8 h, respectively, yielded marked
decreases of the red/green fluorescence ratio, to 71 and 59% of the
control in the H2O2-treated MG63 cells,
indicating that H2O2 induced depolarization
of ΔΨm. By contrast, pretreatment of melatonin largely reversed the
depolarization of ΔΨm induced by H2O2
(Fig. 4).
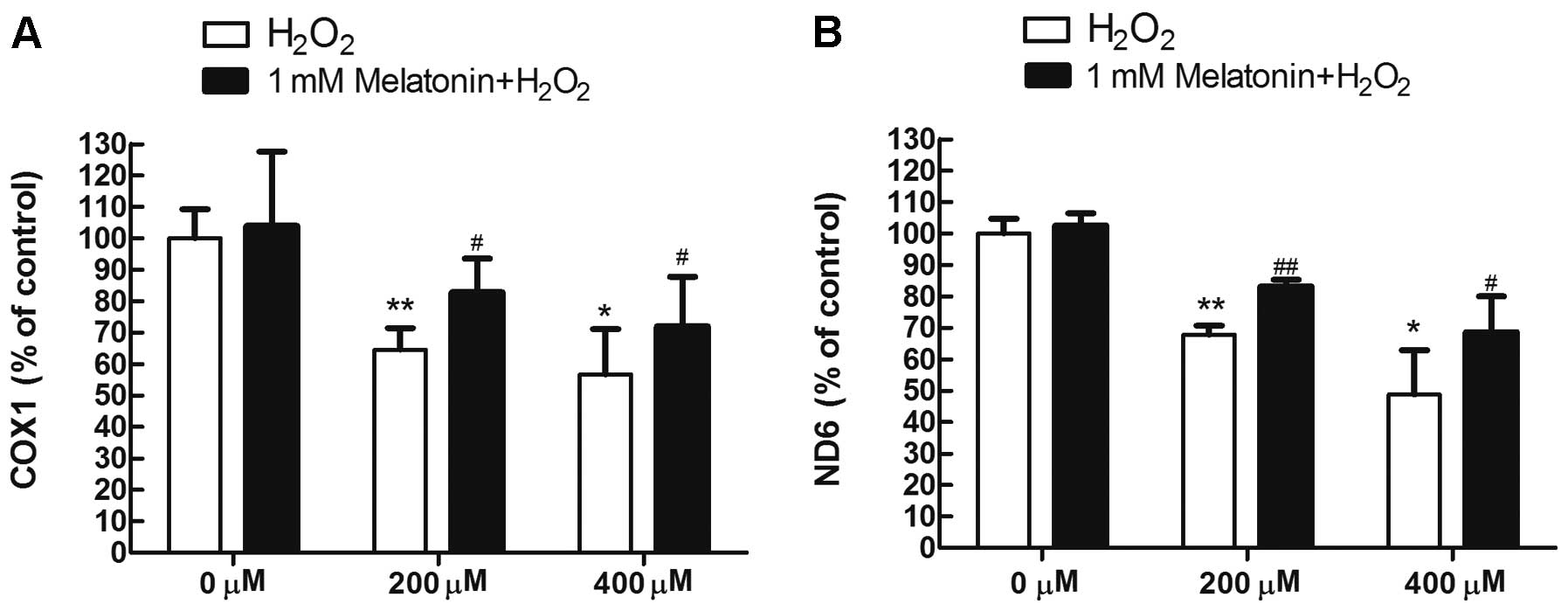
Melatonin attenuates the reduction in
mtDNA copy number in MG63 cells exposed to
H2O2
To determine whether melatonin attenuates the
reduction in mtDNA copy number observed in MG63 cells exposed to
H2O2, two mitochondrial gene fragments, the
COX I gene (representative of the mitochondrial heavy chain)
and ND6 gene (representative of the mitochondrial light
chain) were analyzed by qPCR. As shown in Fig. 5, H2O2
treatment significantly reduced the mtDNA copy number in MG63
cells; however, this reduction in mtDNA copy number was efficiently
attenuated by pretreatment with melatonin.
Discussion
Osteoporosis is a bone disease that leads to an
increased risk of fractures (1)
and which has been recognized as a major threat to public health
(2). It has been demonstrated that
ROS-induced bone cell cytotoxicity is critical in the development
of osteoporosis (4).
Identification of an agent that protects bone cells from
ROS-induced cytotoxicity may be a beneficial strategy in the
prevention of osteoporosis. In the present study, it was
demonstrated that H2O2 was cytotoxic to MG63
cells, as H2O2 reduced cell viability and
increased LDH release in a dose- and time-dependent manner, within
the range of concentrations used. We investigated whether 1 mM
melatonin prevented H2O2-induced toxicity, as
melatonin exerts a protective effect against oxidative damage
(26–28). As expected, melatonin pretreatment
effectively prevented H2O2-induced
cytotoxicity. Melatonin has been proposed to exert protective
benefits by improving mitochondrial energetics and function
(22–25). Therefore, melatonin may protect
against H2O2-induced cytotoxicity by
maintaining mitochondrial function.
ROS accumulation due to improper electron transport
in the mitochondrial respiratory chain results in oxidative damage
to biomacromolecules, including mitochondrial proteins and DNA,
leading to lipid peroxidation, oxidation of amino acid residues and
formation of protein-protein cross-links (29). These processes are important in the
etiology of pathological conditions including osteoporosis
(3). The ability of melatonin to
protect against H2O2-induced intracellular
oxygen species generation and oxidative damage in MG63 cells was
also investigated. Following H2O2 treatment,
the levels of intracellular ROS and lipid peroxidation
(malondialdehyde, MDA) increased in MG63 osteoblastic cells.
However, pretreatment with melatonin reduced
H2O2-induced ROS generation and oxidative
damage in MG63 cells. Melatonin directly increases the activity of
complex I and IV of the electron transfer chain (30), which prevents ROS production and
secondary oxidative damage. Additionally, melatonin induces the
expression of genes encoded by mtDNA, which is essential for
maintaining the activity of the respiratory chain (31). These results indicate that
melatonin may act on respiratory chain complexes to promote
mitochondrial homeostasis in H2O2-treated
MG63 cells.
The beneficial effects of melatonin on mitochondrial
function in H2O2-treated cells were
demonstrated by the following results. Melatonin pretreatment
prevented the reduction in ATP content induced by
H2O2. Melatonin also directly increased the
activity of complex I and IV of the electron transport chain.
Therefore, melatonin may efficiently decrease
H2O2-induced oxidative stress by improving
the function of the electron transport chain and increasing ATP
generation (21,30). Melatonin also maintained ΔΨm during
exposure to H2O2 in MG63 cells. Melatonin
interacts with the lipid bilayer and stabilizes the inner
mitochondrial membrane, which is essential for maintaining ΔΨm
(21,30). This ability of melatonin to
maintain ΔΨm may be beneficial for improving mitochondrial
oxidative phosphorylation and ATP generation under
H2O2-induced oxidative stress (21,32).
In addition, melatonin prevented a reduction in mtDNA copy number
in H2O2-treated MG63 cells. A reduction in
mtDNA content amplifies oxidative stress by reducing the
replication and transcription of mtDNA-encoded genes that are
required for the respiratory chain (33). The ability of melatonin to
stimulate the expression of mtDNA-encoded genes may protect the
activity of the respiratory chain (31) and attenuate
H2O2-induced cytotoxicity.
Due to its amphiphilic properties, melatonin freely
accesses all compartments of the cell, and is able to become highly
concentrated in the mitochondria (34), where it may exert direct beneficial
effects. Furthermore, melatonin is administered in pharmacological
doses without causing significant side-effects. Doses of >1,200
mg/kg melatonin have been administered to humans without signs of
toxicity and a median lethal dose (LD50) has not been
identified for melatonin (35).
Therefore, the results of the present study and the validity and
safety of melatonin in clinical applications suggests that
melatonin is a potential therapeutic agent for preventing
ROS-induced bone loss in diseases, such as osteoporosis.
In conclusion, mitochondrial dysfunction may
underlie the cytotoxic effects of H2O2 in
osteoblastic cells and melatonin may provide protective benefits
through maintaining mitochondrial function in cells exposed to ROS.
The beneficial effects of melatonin in the mitochondria may provide
a novel strategy for protecting against ROS-induced bone cell
cytotoxicity by improving mitochondrial function.
Acknowledgements
This study was partially supported by the National
Natural Science Foundation of China (grant no. 81000803).
References
|
1
|
Sandhu SK and Hampson G: The pathogenesis,
diagnosis, investigation and management of osteoporosis. J Clin
Pathol. 64:1042–1050. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Choi EM: Magnolol protects osteoblastic
MC3T3-E1 cells against antimycin A-induced cytotoxicity through
activation of mitochondrial function. Inflammation. 35:1204–1212.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu ZS, Wang XY, Xiao DM, et al: Hydrogen
sulfide protects MC3T3-E1 osteoblastic cells against
H2O2-induced oxidative damage-implications
for the treatment of osteoporosis. Free Radic Biol Med.
50:1314–1323. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park BG, Yoo CI, Kim HT, Kwon CH and Kim
YK: Role of mitogen-activated protein kinases in hydrogen
peroxide-induced cell death in osteoblastic cells. Toxicology.
215:115–125. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee HC and Wei YH: Oxidative stress,
mitochondrial DNA mutation, and apoptosis in aging. Exp Biol Med
(Maywood). 232:592–606. 2007.PubMed/NCBI
|
|
6
|
Raha S and Robinson BH: Mitochondria,
oxygen free radicals, disease and ageing. Trends Biochem Sci.
25:502–508. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Choi EM: Deoxyactein isolated from
Cimicifuga racemosa protects osteoblastic MC3T3-E1 cells
against antimycin A-induced cytotoxicity. J Appl Toxicol.
33:488–494. 2013.
|
|
8
|
Slominski A, Fischer TW, Zmijewski MA, et
al: On the role of melatonin in skin physiology and pathology.
Endocrine. 27:137–148. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bubenik GA: Gastrointestinal melatonin:
localization, function, and clinical relevance. Dig Dis Sci.
47:2336–2348. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jimenez-Jorge S, Jimenez-Caliani AJ,
Guerrero JM, et al: Melatonin synthesis and melatonin-membrane
receptor (MT1) expression during rat thymus development: role of
the pineal gland. J Pineal Res. 39:77–83. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rosenstein RE, Pandi-Perumal SR,
Srinivasan V, Spence DW, Brown GM and Cardinali DP: Melatonin as a
therapeutic tool in ophthalmology: implications for glaucoma and
uveitis. J Pineal Res. 49:1–13. 2010.PubMed/NCBI
|
|
12
|
Conti A, Conconi S, Hertens E,
Skwarlo-Sonta K, Markowska M and Maestroni JM: Evidence for
melatonin synthesis in mouse and human bone marrow cells. J Pineal
Res. 28:193–202. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rajaratnam SM, Middleton B, Stone BM,
Arendt J and Dijk DJ: Melatonin advances the circadian timing of
EEG sleep and directly facilitates sleep without altering its
duration in extended sleep opportunities in humans. J Physiol.
561:339–351. 2004. View Article : Google Scholar
|
|
14
|
Arendt J and Skene DJ: Melatonin as a
chronobiotic. Sleep Med Rev. 9:25–39. 2005. View Article : Google Scholar
|
|
15
|
Armstrong SM: Melatonin and circadian
control in mammals. Experientia. 45:932–938. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Deacon S and Arendt J: Melatonin-induced
temperature suppression and its acute phase-shifting effects
correlate in a dose-dependent manner in humans. Brain Res.
688:77–85. 1995. View Article : Google Scholar
|
|
17
|
Srinivasan V, Spence WD, Pandi-Perumal SR,
Zakharia R, Bhatnagar KP and Brzezinski A: Melatonin and human
reproduction: shedding light on the darkness hormone. Gynecol
Endocrinol. 25:779–785. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guerrero JM and Reiter RJ:
Melatonin-immune system relationships. Curr Top Med Chem.
2:167–179. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hardeland R, Poeggeler B, Niebergall R and
Zelosko V: Oxidation of melatonin by carbonate radicals and
chemiluminescence emitted during pyrrole ring cleavage. J Pineal
Res. 34:17–25. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Coto-Montes A and Hardeland R: New vistas
on oxidative damage and aging. Open Biol J. 3:39–52. 2010.
View Article : Google Scholar
|
|
21
|
León J, Acuña-Castroviejo D, Escames G,
Tan DX and Reiter RJ: Melatonin mitigates mitochondrial
malfunction. J Pineal Res. 38:1–9. 2005.PubMed/NCBI
|
|
22
|
Klongpanichapak S, Phansuwan-Pujito P,
Ebadi M and Govitrapong P: Melatonin protects SK-N-SH neuroblastoma
cells from amphetamine-induced neurotoxicity. J Pineal Res.
43:65–73. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Duan Q, Wang Z, Lu T, Chen J and Wang X:
Comparison of 6-hydroxylmelatonin or melatonin in protecting
neurons against ischemia/reperfusion-mediated injury. J Pineal Res.
41:351–357. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Herrera F, Martin V, Garcia-Santos G,
Rodriguez-Blanco J, Antolin I and Rodriguez C: Melatonin prevents
glutamate-induced oxytosis in the HT22 mouse hippocampal cell line
through an antioxidant effect specifically targeting mitochondria.
J Neurochem. 100:736–746. 2007. View Article : Google Scholar
|
|
25
|
Chen LJ, Gao YQ, Li XJ, Shen DH and Sun
FY: Melatonin protects against MPTP/MPP+-induced
mitochondrial DNA oxidative damage in vivo and in vitro. J Pineal
Res. 39:34–42. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jou MJ, Peng TI, Yu PZ, et al: Melatonin
protects against common deletion of mitochondrial DNA-augmented
mitochondrial oxidative stress and apoptosis. J Pineal Res.
43:389–403. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tan DX, Manchester LC, Terron MP, Flores
LJ and Reiter RJ: One molecule, many derivatives: a never-ending
interaction of melatonin with reactive oxygen and nitrogen species?
J Pineal Res. 42:28–42. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Reiter RJ, Tan DX, Jou MJ, Korkmaz A,
Manchester LC and Paredes SD: Biogenic amines in the reduction of
oxidative stress: melatonin and its metabolites. Neuro Endocrinol
Lett. 29:391–398. 2008.PubMed/NCBI
|
|
29
|
Choi EM, Kim GH and Lee YS: Diazoxide
protects against hydrogen peroxide-induced toxicity in the
osteoblastic MC3T3-E1 cells. Eur J Pharmacol. 624:45–50. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
López A, García JA, Escames G, et al:
Melatonin protects the mitochondria from oxidative damage reducing
oxygen consumption, membrane potential, and superoxide anion
production. J Pineal Res. 46:188–198. 2009.PubMed/NCBI
|
|
31
|
Xu S, Zhong M, Zhang L, et al:
Overexpression of Tfam protects mitochondria against
beta-amyloid-induced oxidative damage in SH-SY5Y cells. FEBS J.
276:3800–3809. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Acuña Castroviejo D, Escames G, Carazo A,
León J, Khaldy H and Reiter RJ: Melatonin, mitochondrial
homeostasis and mitochondrial-related diseases. Curr Top Med Chem.
2:133–151. 2002.PubMed/NCBI
|
|
33
|
Xu S, Zhou Z, Zhang L, et al: Exposure to
1800 MHz radiofrequency radiation induces oxidative damage to
mitochondrial DNA in primary cultured neurons. Brain Res.
1311:189–196. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Escames G, López A, García JA, et al: The
role of mitochondria in brain aging and the effects of melatonin.
Curr Neuropharmacol. 8:182–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Vairetti M, Ferrigno A, Bertone R, et al:
Exogenous melatonin enhances bile flow and ATP levels after cold
storage and reperfusion in rat liver: implications for liver
transplantation. J Pineal Res. 38:223–230. 2005. View Article : Google Scholar : PubMed/NCBI
|