Introduction
Chromosomal translocations are frequent events that
occur in leukemia. The translocation t(8;21)(q22;q22) is one of the
most frequent chromosomal translocations in leukemia and accounts
for 12–15% of acute myeloid leukemia (AML) and ~40–50% of M2 AML
(French-American-British classification) (1). This translocation involves the
AML1 gene at q22 on chromosome 21 and the ETO gene at
q22 on chromosome 8, resulting in an AML1/ETO fusion gene.
This fusion gene encodes a chimeric protein, AML1/ETO. The chimeric
protein silences target gene transcription by recruiting histone
deacetylases (HDACs), which remove acetyl groups from histone
lysine residues. The abnormal recruitment of HDAC due to
chromosomal rearrangements often occurs in the development of
malignant tumors and contributes to their pathogenesis (2). Several studies have demonstrated that
the abnormal AML1/ETO protein and the silencing of hematopoietic
genes contribute to the hematopoietic developmental abnormalities
of AML with t(8;21) (3–6). Inhibition of HDAC activity has been
reported to restore the abnormal histone acetylation in tumors,
thus resulting in the growth arrest, differentiation and/or
apoptotic cell death of tumor cells (7). Therefore, HDAC inhibitors represent a
promising treatment for patients with AML with t(8;21), as they may
enhance histone acetylation via inhibition of HDAC activities, thus
restoring the disrupted gene transcripts in AML (8).
Angiogenesis is critical for tumor growth and
metastasis. Vascular endothelial growth factor (VEGF), VEGF
receptors (VEGFRs) and basic fibroblast growth factors (bFGFs) are
the most potent pro-angiogenic factors and are critical in tumor
angiogenesis (9,10). Anti-angiogenic approaches are a
novel strategy to treat AML. It has been reported that the HDAC
inhibitor, FK228, inhibits the expression of angiogenic factors,
including VEGF and bFGF, in PC-3 xenografts implanted in nude mice,
indicating that the antitumor effects of FK228 are mediated through
the inhibition of angiogenesis (11). Valproic acid (VPA), which is widely
used clinically for the treatment of epilepsy, has been
demonstrated to be a strong HDAC inhibitor (12). Our previous in vitro studies
revealed that VPA exerted antitumor effects on Kasumi-1 cells,
human acute myeloid leukemia cells with an 8;21 chromosome
translocation, via downregulation of VEGF and VEGFR (13,14).
The purpose of the present study was to investigate the effect of
VPA on tumor growth and the expression of angiogenic factors in
mice transplanted with Kasumi-1 cells, and also to analyze the
histone acetylation on VEGF promoters in these cells.
Materials and methods
Tumor cells and animals
The Kasumi-1 cell line was a gift from Dr Jianxiang
Wang at the Institute of Hematology, Chinese Academy of Medical
Science (Tianjin, China). The cells were maintained in culture with
RPMI-1640 medium supplemented with 20% fetal bovine serum in a 37°C
incubator with 5% CO2 and 95% humidity. Female BALB/c
nude mice (SPF grade; 10–15 g; 4–6 weeks old) were purchased from
Beijing Vital River Lab Animal Technology Co., Ltd. (Beijing,
China). The study was approved by the Chengde Medical College
Animal Research Ethics Committee.
Tumor generation and VPA treatment
Splenectomies were performed on the BALB/c nude
mice. One week after the splenectomies, the mice received whole
body irradiation with 137Cs at a dose of 4 Gy. At 48–72
h post-irradiation, the mice were subcutaneously implanted with
Kasumi-1 cells (2×107 cells/mouse with 0.15–0.2 ml) in
the right axillary region. The mice were randomly assigned to two
groups, the VPA (n=6) and control (n=6) groups. When the tumors
were ~200 mm3 in size at ~10 days post-implantation, 0.2
ml VPA (500 mg/kg body weight) or 0.2 ml saline was injected
intraperitoneally every day. VPA (Sigma-Aldrich, St. Louis, MO,
USA) was dissolved in saline at a concentration of 25 mg/ml. The
longest diameter (a) and the shortest diameter (b) of the tumor
were measured every three days, and the tumor volume (TV) was
calculated according to the following formula: TV = 1/2 × a ×
b2. The relative TV (RTV) was calculated as the ratio
between the TV on day N and the TV on the day of injection (day 0)
according to the following formula: RTV = (TV on day N) / (TV on
day 0). The tumor growth inhibition rate (IR) was calculated from
the following formula: IR (%) = [1 − RTV(VPA group) /
RTV(control)) × 100, where RTV(VPA group) is
the RTV in the VPA-treated group and RTV(control) is the
RTV in the control group. Following two weeks of injections, the
mice were sacrificed by cervical dislocation and the tumor masses
were removed for the following experiments.
Immunohistochemistry
The tumor masses were fixed with 10% formalin and
embedded with paraffin. Tissue sections (5-μm thick) were obtained
from paraffin-embedded tissue blocks. The tissue sections were
immunohistochemically stained for CD34, VEGF, VEGFR2 and bFGF.
Briefly, the sections were washed in xylene to remove the paraffin,
rehydrated with serial dilutions of alcohol and then washed in
phosphate-buffered saline. The samples were then incubated in
primary antibodies against CD34, VEGF, VEGFR2 and bFGF overnight at
4°C. All primary antibodies were rat anti-human antibodies (Santa
Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Subsequent to the
primary antibody being washed off, the biotinylated goat anti-rat
IgG secondary antibody (Santa Cruz Biotechnology, Inc.) was applied
and then reacted with horseradish peroxidase-conjugated
streptavidin. The sections were stained with diaminobenzidine
solution and counterstained with hematoxylin.
Tumor microvessel density (MVD) analysis
performed following staining with the anti-CD34 antibody
Microvessels with brownish staining in the cytoplasm
of the endothelium were included. A single endothelial cell was
counted as a single vessel. An endothelial cell cluster with a
branching structure, which was clearly separated from the adjacent
endothelial cells, was also counted as a single vessel. At a
low-power field (x40), the tissue sections with the most intense
vascular density were selected. At a high-power field (x200),
microvessels in five fields were counted in the areas with the most
intense vascular density. The mean microvessel count of the five
most vascular areas was used as the MVD.
Semi-quantitative reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from the tumor masses using
TRIzol reagent (Invitrogen, Life Technologies, Carlsbad, CA, USA)
according to the manufacturer’s instructions. The final RNA
concentration was adjusted to 1 μg/μl. RNA was reverse transcribed
into complementary DNA using the reverse transcriptase of Moloney
murine leukemia virus (Bio Basic Canada Inc., Markham, ON, Canada).
PCR was performed using Taq DNA polymerase [Takara Biotechnology,
Co., Ltd., Dalian, China]. The primers were as follows: Sense:
5′-GAAGTGGTGAAGTTCATGGATGTC-3′ and antisense:
5′-CGATCGTTCTGTATCAGTCTTTCC-3′ for VEGF (size, 260 bp); sense:
5′-AGAGCGACCCTCACATCAAG-3′ and antisense:
5′-TCGTTTCAGTGCCACATACC-3′ for bFGF (size, 224 bp); and sense:
5′-GGGGATTGACTTCAACTGG-3′ and antisense: 5′-GACCCTGACAAATGTGCTG-3′
for VEGFR2 (size, 211 bp). β-actin (size, 453 bp) was used as an
internal control. The reaction conditions consisted of 38 cycles of
95°C for 45 sec, 61°C for 45 sec and 72°C for 60 sec. The PCR
products were analyzed by electrophoresis on a 1.8% agarose gel
containing ethidium bromide, and the gel was visualized with a
digital imaging system (Fujifilm; Fuji, Tokyo, Japan). The relative
mRNA expression of VEGF, bFGF and VEGFR2 was normalized to the
β-actin concentration.
Western blotting
For western blotting, the tumor masses were
homogenized on ice in RIPA lysis buffer [50 mM Tris-HCl, 150 mM
NaCl and 1% NP-40 (pH 7.4)]. Nuclear proteins were extracted using
the Nucleoprotein Extraction kit (Sangon Biotech, Co., Ltd.,
Shanghai, China) according to the manufacturer’s instructions. The
protein concentrations were determined with the bicinchoninic acid
method. Equal quantities of proteins were loaded and separated by
electrophoresis in 120 g/l SDS-PAGE and then transferred onto
polyvinylidene fluoride membranes. The membranes were incubated
with primary antibodies against HDAC1 (rabbit anti-human polyclonal
antibodies; Proteintech Group, Chicago, IL, USA) or acetylated
histone H3 (Ac-H3; rabbit anti-human monoclonal antibodies;
Epitomics, Burlingham, CA, USA) at 4°C overnight. Blots were
stained with horseradish peroxidase-linked goat anti-rabbit Ig
secondary antibodies and developed with a chemiluminescence
detection system (Fujifilm). The nuclear protein, lamin B served as
a loading control.
Chromatin immunoprecipitation (ChIP)
ChIP was performed using the ChIP assay kit (Upstate
Biotechnology, Billerica, MA, USA) according to the manufacturer’s
instructions. Briefly, tumor masses were cut into small sections
(Xinzhi Co. Ltd., Ningbo, China) and treated with 1% formalin for
10 min at room temperature. The tissues were then homogenized with
lysis buffer and sonicated five times at 80 W for 10 sec/time, at
60 sec intervals. Following centrifugation (5,000 × g, 1 min), the
supernatant was removed, and 20 μl supernatant was saved for the
controls (DNA input). Rabbit anti-human monoclonal antibodies
against Ac-H3 (3 μg) were added into the supernatant and incubated
overnight at 4°C with gentle rotation. Antibodies against RNA
polymerase were used as a positive control and mouse IgG was used
as a negative control. Protein G agarose beads were added into the
solution, and the samples were incubated at 4°C for 1 h. The beads
were washed five times with buffers according to the the ChIP assay
kit protocol. The protein-DNA complex was eluted with an elution
buffer containing 20% SDS and 1M NaHCO3. The
immunoprecipitated chromatin was dissolved in 0.9% saline and then
incubated at 65°C for 4 h to reverse the cross-linking and separate
the protein-DNA complex. The DNA was extracted, and RT-PCR was
performed with primers specific for the VEGF promoter. The primers
were as follows: Sense: 5′-CTT CGA GAG TGA GGA CGT GTG T-3′ and
antisense: 5′-GGA GCA GGA AAG TGA GGT TAC G-3′ for P1; sense:
5′-CCA GAC TCC ACA GTG CAT ACG T-3′ and antisense: 5′-TGGGAC TGG
AGT TGC TTC ATG-3 for P2; sense: 5′-TGC TGC ATT CCC ATT CTC AGT-3′
and antisense: 5′-ATC TTC CCTAAG TGC TCC CAA AG-3′ for P3; sense:
5′-CAG GGA AAG GAT GAT CAC TGT CA-3′ and antisense: 5′-TGC CTT TCA
CCA GGA CAA AGT-3′ for I1; and sense: 5′-ATG GAT GTC TAT CAG CGC
AGCT-3′ and antisense: 5′-TGG TGA TGT TGG ACT CCTCAG T-3′ for E3.
The reaction conditions were 94°C for 3 min, followed by 32 cycles
of 94°C for 20 sec, 57°C for 30 sec and 72°C for 30 sec and a final
extension of 72°C for 2 min.
Statistical analysis
Statistical analyses were performed using SPSS 17.0
(SPSS, Inc., Chicago, IL, USA). All the data presented are based on
an average of at least three independent experiments. The values
are presented as the mean ± standard deviation. Student’s t-test’s
were used to compare the differences between groups. P<0.05 was
used to indicate a statistically significant difference.
Results
VPA inhibits tumor growth in mice
transplanted with Kasumi-1 cells
The effect of VPA on tumor growth was investigated
in mice transplanted with Kasumi-1 cells. None of the mice died
prior to sacrifice at the end of the experiments. The size of the
tumor was measured every three days following daily injection of
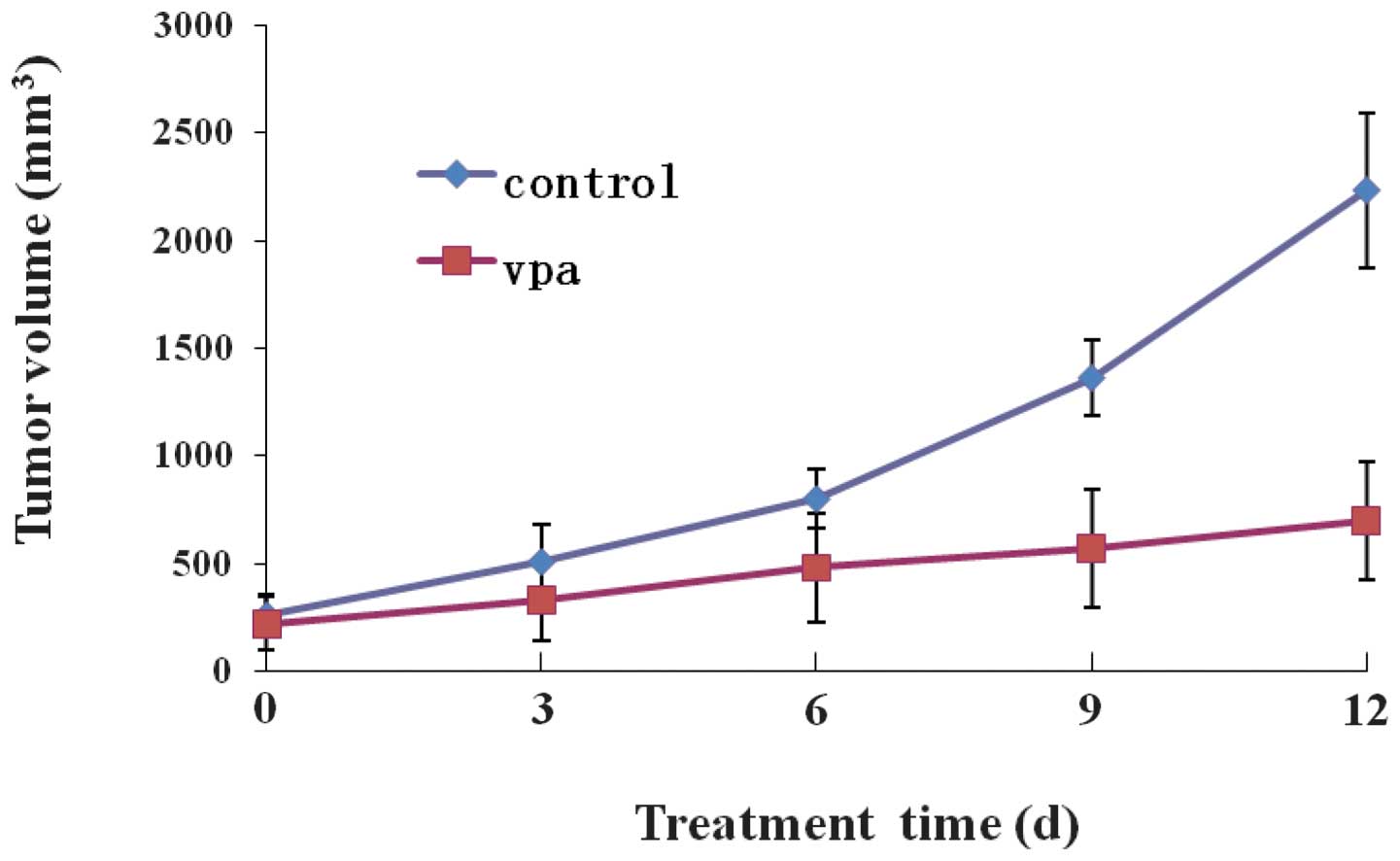
VPA for two weeks. Fig. 1 shows
the time-course of tumor growth in the control and VPA-treated
groups. The TV in the VPA group increased with time more slowly
than that in the control group. The final TV in the VPA group was
699.4±271.01 mm3, which was significantly less than the
TV of 2235.0±360.21 mm3 observed in the control group
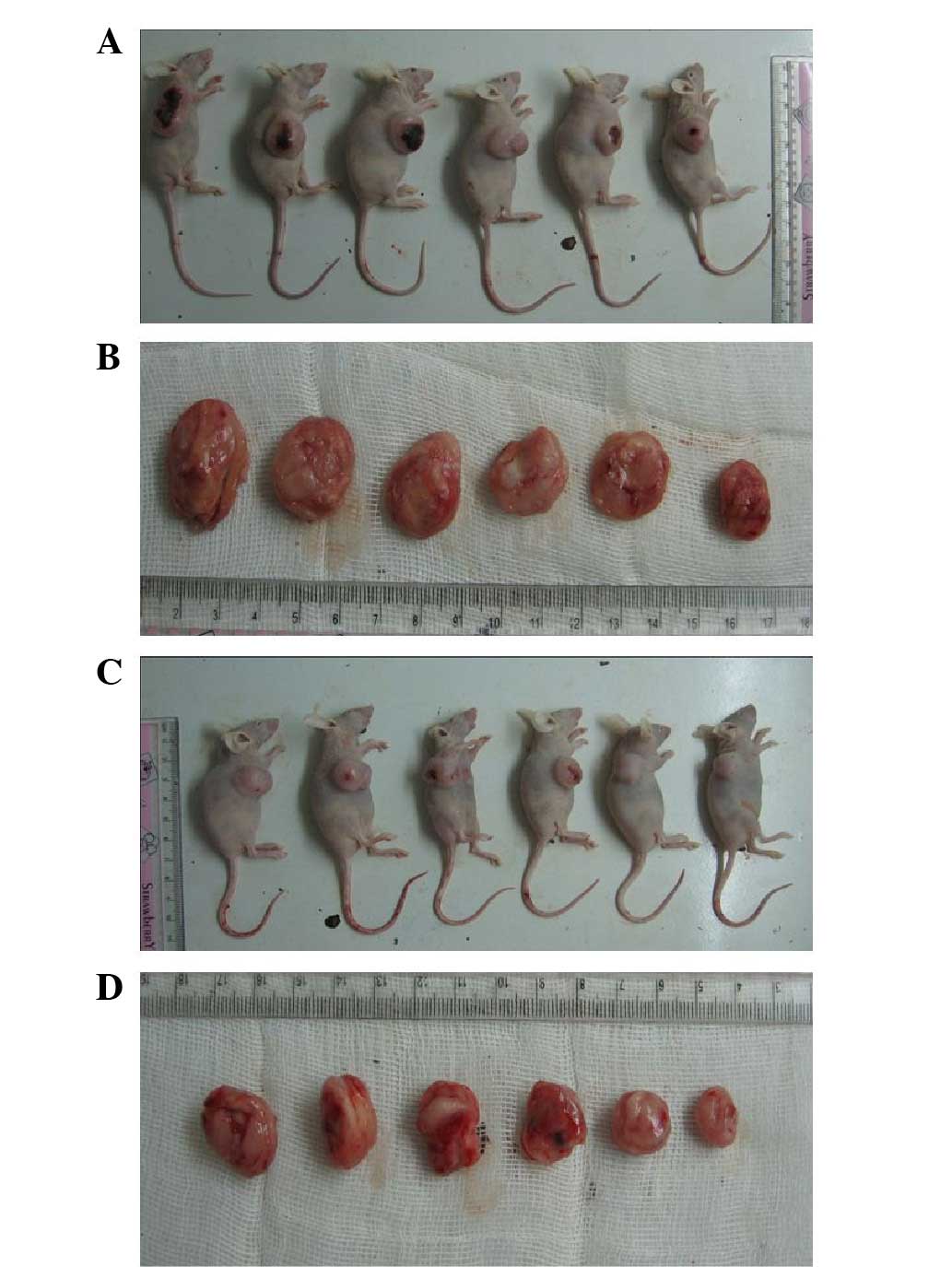
(P<0.05; Table I). Following
sacrificing the mice, the size, weight and RVT of the tumors in the
VPA group were significantly less than those in the control group
(P<0.05; Table I; Figs. 1 and 2), indicating that VPA inhibited the
tumor growth in the mice transplanted with Kasumi-1 cells. The IR
rate in the VPA group was 57.25% at the end of the experiment.
 | Table IVolume and weight of tumors following
sacrifice of the mice. |
Table I
Volume and weight of tumors following
sacrifice of the mice.
| Group | n | TV,
mm3 | Tumor weight, g | RTV | IR, % |
|---|
| Control | 6 | 2235.0±360.21 | 1.57±0.25 | 9.50±2.13 | |
| VPA | 6 | 699.4±271.01a | 0.46±0.17a | 4.06±1.05a | 57.25 |
VPA inhibits tumor angiogenesis in mice
transplanted with Kasumi-1 cells
The effect of VPA on tumor angiogenesis was tested
in mice transplanted with Kasumi-1 cells by measuring MVD using
CD34 immunostaining, which has been used in previous studies
(15,16). Specific staining of capillary-like
vessels by anti-CD34 was observed in the control and VPA groups
(Fig. 3). The mean MVD in the VPA
group (12.23±4.11; number of microvessels) was significantly lower
than that in the control group (32.59±5.76) (P<0.05), indicating
that VPA inhibited angiogenesis in the mice transplanted with
Kasumi-1 cells.
VPA inhibits the mRNA and protein
expression of VEGF, VEGFR2 and bFGF
The mechanisms underlying the VPA-induced inhibition
of angiogenesis were studied further in the mice transplanted with
Kasumi-1 cells. The mRNA and protein expression levels of VEGF,
VEGFR2 and bFGF, which have been reported to be involved in tumor
angiogenesis, were examined (9,10).
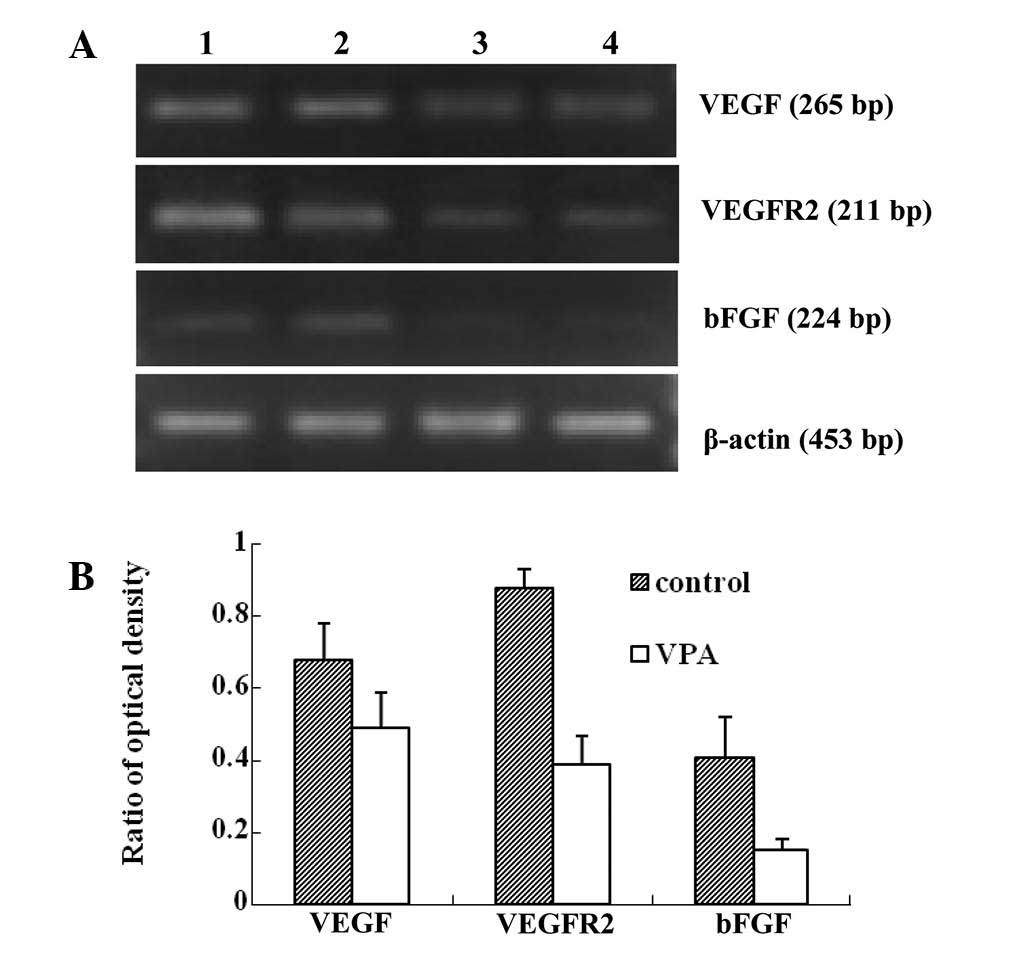
RT-PCR demonstrated that the mRNA levels of VEGF,
VEFGR2 and bFGF were significantly downregulated in
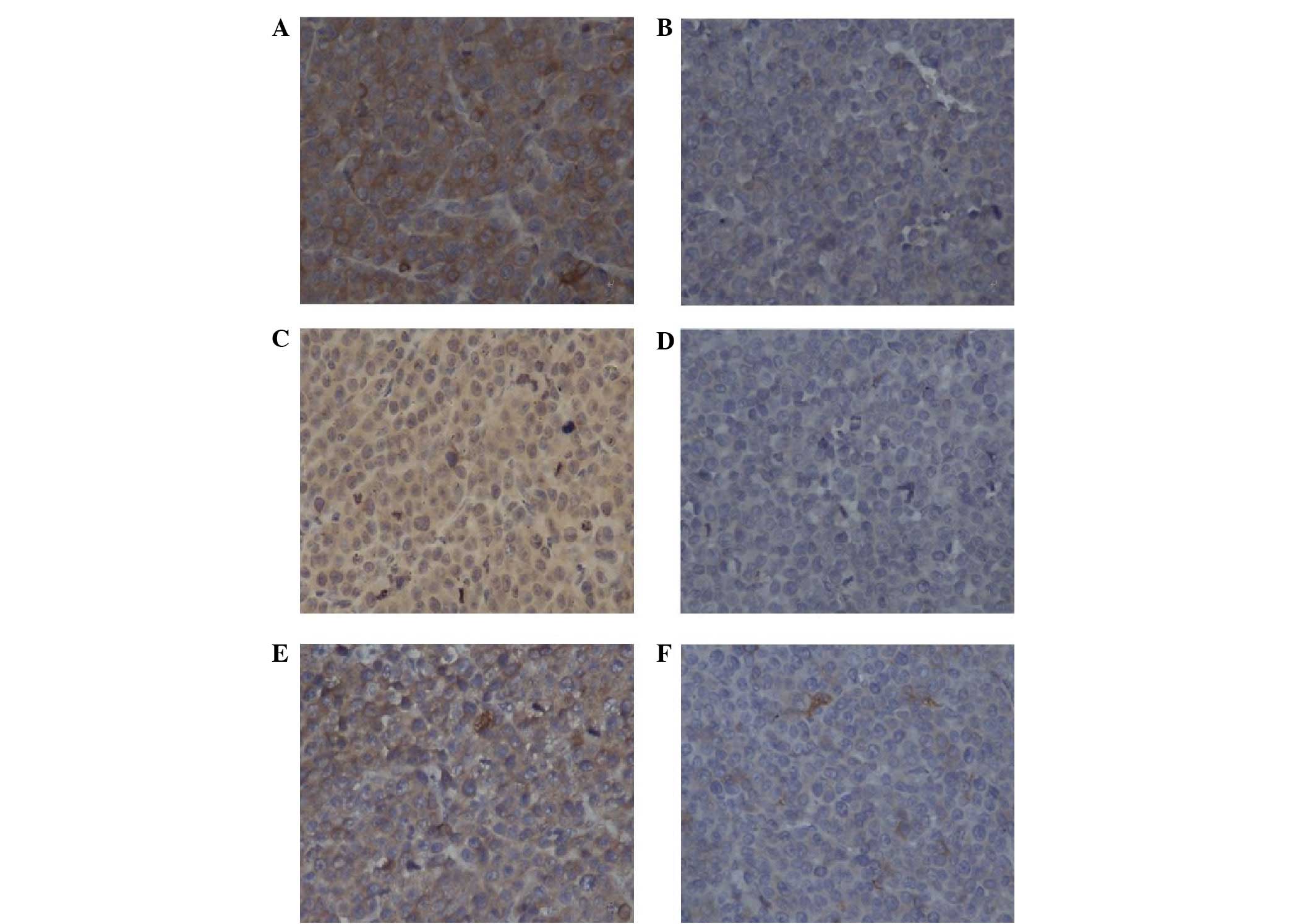
the VPA group compared with those in the control group (Fig. 4). The protein expression of VEGF,
VEFGR2 and bFGF was detected using immunohistochemistry (Fig. 5). Consistent with the mRNA levels,
the protein expression of VEGF, VEFGR2 and bFGF was suppressed in
the VPA group compared with the control group. These results
indicated that VPA inhibited tumor angiogenesis most likely through
its inhibition of VEGF, VEFGR2 and bFGF.
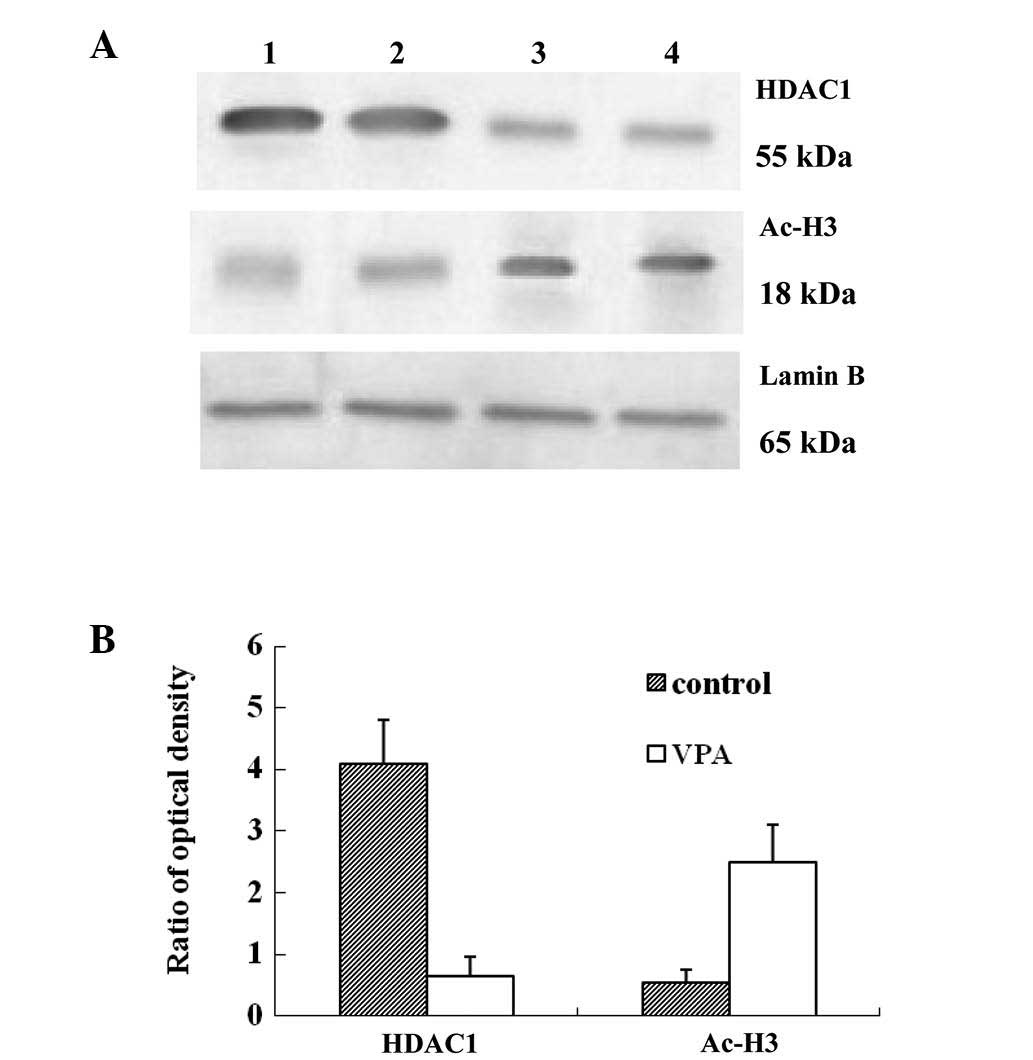
VPA inhibits HDAC activity and increases
acetylation of histone H3
As it has already been shown that the histone
acetylation of the VEGF promoters regulates VEGF protein expression
(11), the present study
investigated the effects of VPA on HDAC activity by detecting the
nuclear expression of HDAC1 and the acetylation of histone H3
(Fig. 6). Western blotting
revealed that the expression of HDAC1 was downregulated, while
histone H3 acetylation was increased in the VPA group compared with
the control group, indicating that VPA increased the acetylation of
histone H3 via the inhibition of HDAC.
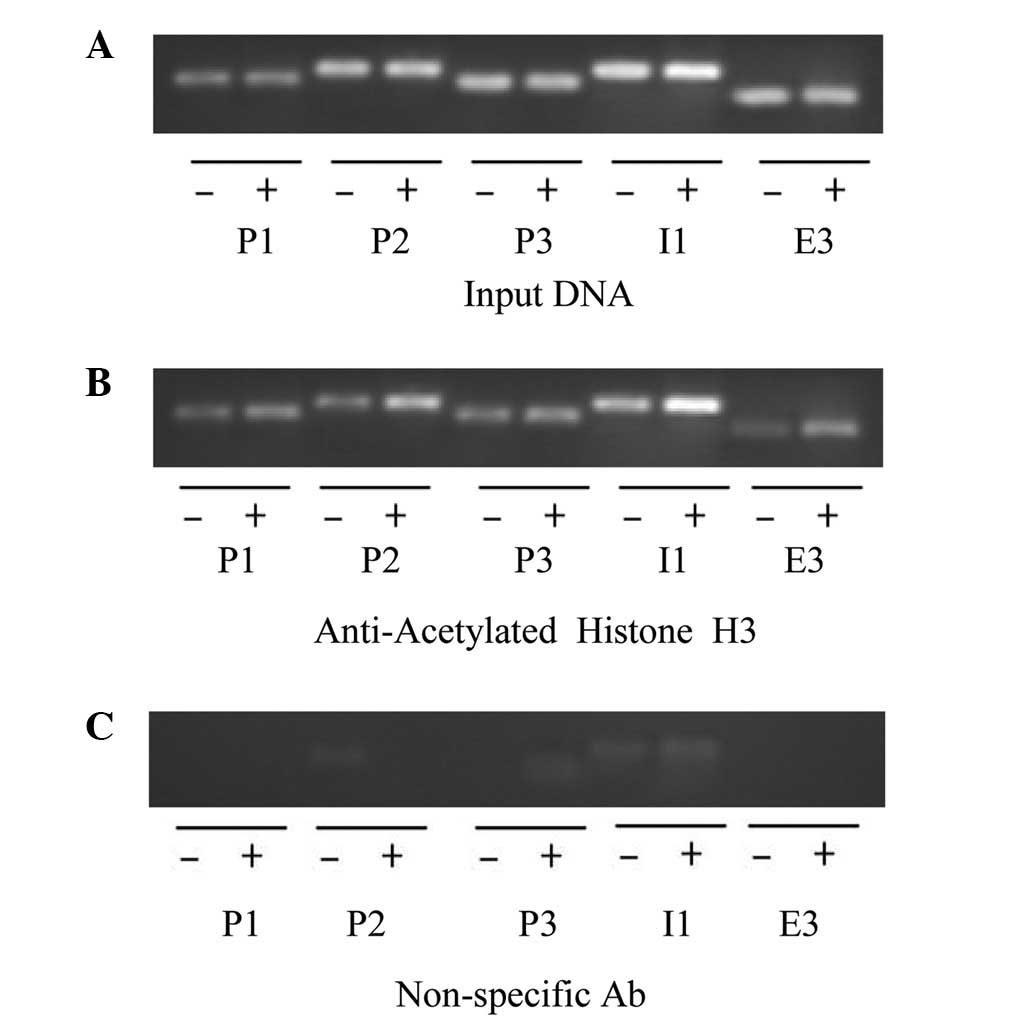
VPA enhances the accumulation of
hyperacetylated histone H3 on VEGF promoters
To investigate whether VPA induced the
hyperacetylation of histone H3 at the VEGF promoter, a ChIP
assay was performed using anti-Ac-H3 antibodies to precipitate
chromatin from Kasumi-1 cell-induced tumors (Fig. 7). Anti-Ace-H3 antibodies enriched
more VEGF promoter DNA fragments in the VPA group than in
the control group. By contrast, non-specific IgG antibodies did not
precipitate VEGF promoter DNA in either the VPA group or the
control group. Anti-polymerase II antibodies, which served as a
positive control, precipitated similar quantities of VEGF
promoter DNA in the VPA and control groups.
Discussion
In the present study, a mouse model of leukemia was
established using Kasumi-1 cells, which are human acute myeloid
leukemia cells with an 8;21 chromosome translocation. Kasumi-1
cells express the AML-1/ETO fusion protein and are ideal for the
study of AML with t(8;21) (17).
The in vitro study by Liu et al (18) in Kasumu-1 cells revealed that VPA
had pronounced antileukemic effects that were associated with an
inhibition of the protein expression of HDAC in the nuclei, a
disruption of the physical interaction between AML1/ETO and HDAC1,
an increase in their dissociation from the promoters of AML1/ETO
target genes and a translocation of these nuclear proteins to the
perinuclear region. In agreement with these reported in
vitro antileukemic effects of VPA, the present in vivo
study demonstrated that intraperitoneal injections of VPA reduced
tumor growth in mice implanted with Kasumi-1 cells, and the tumor
weights and volumes were significantly smaller in the VPA group
compared with those in the control group.
It has been reported that AML with t(8;21) and
Kasumi-1 cell lines demonstrate enhanced expression of VEGF and
VEGFR2, which is associated with the growth of Kasumi-1 cells and
the angiogenesis of AML (19,20).
Blockade of the VEGFR2 signaling pathway inhibits the growth of
Kasumi-1 cells (20). Furthermore,
several in vivo and in vitro studies have revealed
that HDAC inhibitors, including NaB, SAHA, TSA and FK228, inhibit
tumor angiogenesis via downregulation of the mRNA and protein
expression of VEGF (21–24). In addition, our previous study
revealed that VPA exerted antitumor effects on Kasumi-1 cells, most
likely through the downregulation of the mRNA and protein
expression of VEGF, VEGFR2 and bFGF (14). In the present study, the effects of
VPA were directly tested on tumor angiogenesis in mice transplanted
with Kasumi-1 cells by measuring the MVD with CD34 immunostaining.
VPA was observed to decrease the MVD and downregulate the VEGF,
VEGFR2 and bFGF mRNA and protein levels compared with the control.
Thus, the present study provided direct evidence that VPA inhibited
tumor angiogenesis in mice transplanted with Kasumi-1 cells.
Several mechanisms have been reported to be involved
in HDAC inhibitor-induced anti-angiogenesis, including the
inhibition of HDAC activity, the overexpression of HIF-1α, the
downregulation of tumor suppressor genes, including VHL and p53,
and the downregulation of VEGF expression (25–27).
In the present study, VPA, a HDAC inhibitor, was detected to
downregulate HDAC protein expression and increase the acetylation
of histone H3 in Kasumi-1 cell-induced tumors. In addition, VPA
enhanced the accumulation of hyperacetylated histone H3 on the
VEGF promoters. However, it remains unclear how histone
acetylation inhibits gene transcription. There are two possible
mechanisms underlying the regulation of gene transcription by
histone acetylation: i) Histone has several acetylation sites, and
the acetylation state of each lysine is different in each site. It
has been reported that the patterns of histone acetylation, which
are termed the ‘histone code’ reveal the sign of transcription
(28); and ii) histone acetylation
by HDAC inhibitors induces alterations in the chromatin structure,
which may lead to the disruption of the binding of transcription
factors to gene promoters. It has been reported that VPA may alter
the chromatin structure by regulating chromatin modulation proteins
(29). Although the mechanism
underlying the alteration of the chromatin structure by histone
acetylation has not been clarified, it may be important to explain
how HDAC inhibitors regulate gene expression.
In conclusion, VPA inhibits tumor growth and tumor
angiogenesis in mice implanted with Kasumi-1 cells. This antitumor
effect of VPA is possibly due to the inhibition of VPA on the
expression of angiogenic factors. In addition, VPA may increase the
accumulation of acetylated histones on the promoters of the genes,
which may contribute to the regulation of the expression of
angiogenic factors.
Abbreviations:
|
HDAC
|
histone deacetylase
|
|
VPA
|
valproic acid
|
|
MVD
|
microvessel density
|
|
AML
|
acute myeloid leukemia
|
|
VEGF
|
vascular endothelial growth factor
|
References
|
1
|
Wichmann C, Chen L, Heinrich M, et al:
Targeting the oligomerization domain of ETO interferes with
RUNX1/ETO oncogenic activity in t(8;21)-positive leukemic cells.
Cancer Res. 67:2280–2289. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Minucci S, Nervi C, Lo Coco F and Pelicci
PG: Histone deacetylases: a common molecular target for
differentiation treatment of acute myeloid leukemias? Oncogene.
20:3110–3115. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gelmetti V, Zhang J, Fanelli M, Minucci S,
Pelicci PG and Lazar MA: Aberrant recruitment of the nuclear
receptor corepressor-histone deacetylase complex by the acute
myeloid leukemia fusion partner ETO. Mol Cell Biol. 18:7185–7191.
1998.PubMed/NCBI
|
|
4
|
Hug BA and Lazar MA: ETO interacting
proteins. Oncogene. 23:4270–4274. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Le XF, Claxton D, Kornblau S, Fan YH, Mu
ZM and Chang KS: Characterization of the ETO and AML1-ETO proteins
involved in 8;21 translocation in acute myelogenous leukemia. Eur J
Haematol. 60:217–225. 1998.PubMed/NCBI
|
|
6
|
Lutterbach B, Westendorf JJ, Linggi B, et
al: ETO, a target of t(8;21) in acute leukemia, interacts with the
N-CoR and mSin3 corepressors. Mol Cell Biol. 18:7176–7184.
1998.PubMed/NCBI
|
|
7
|
Marks PA, Richon VM and Rifkind RA:
Histone deacetylase inhibitors: inducers of differentiation or
apoptosis of transformed cells. J Natl Cancer Inst. 92:1210–1216.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Redner RL, Wang J and Liu JM: Chromatin
remodeling and leukemia: new therapeutic paradigms. Blood.
94:417–428. 1999.PubMed/NCBI
|
|
9
|
Hoeben A, Landuyt B, Highley MS, Wildiers
H, Van Oosterom AT and De Bruijn EA: Vascular endothelial growth
factor and angiogenesis. Pharmacol Rev. 56:549–580. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nussenbaum F and Herman IM: Tumor
angiogenesis: insights and innovations. J Oncol. 2010:1326412010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sasakawa Y, Naoe Y, Noto T, et al:
Antitumor efficacy of FK228, a novel histone deacetylase inhibitor,
depends on the effect on expression of angiogenesis factors.
Biochem Pharmacol. 66:897–906. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Göttlicher M, Minucci S, Zhu P, et al:
Valproic acid defines a novel class of HDAC inhibitors inducing
differentiation of transformed cells. EMBO J. 20:6969–6978.
2001.
|
|
13
|
Zhao L, Zhang ZH and Zhu CM: Inhibitory
effect of valproic acid on cell cycle of Kasumi-1 cell line and its
mechanism. Zhonghua Xue Ye Xue Za Zhi. 29:802–805. 2008.(In
Chinese).
|
|
14
|
Zhu CM, Zhang ZH, Jiang FY, et al: Effects
of histone deacetylase inhibitor on the expression of angiogenesis
related factors in Kasumi-1 leukemic cell line. Zhonghua Xue Ye Xue
Za Zhi. 31:466–469. 2010.(In Chinese).
|
|
15
|
Poon RT, Ng IO, Lau C, et al: Tumor
microvessel density as a predictor of recurrence after resection of
hepatocellular carcinoma: a prospective study. J Clin Oncol.
20:1775–1785. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Weidner N: Intratumor microvessel density
as a prognostic factor in cancer. Am J Pathol. 147:9–19.
1995.PubMed/NCBI
|
|
17
|
Larizza L, Magnani I and Beghini A: The
Kasumi-1 cell line: a t(8;21)-kit mutant model for acute myeloid
leukemia. Leuk Lymphoma. 46:247–255. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu S, Klisovic RB, Vukosavljevic T, et
al: Targeting AML1/ETO-histone deacetylase repressor complex: a
novel mechanism for valproic acid-mediated gene expression and
cellular differentiation in AML1/ETO-positive acute myeloid
leukemia cells. J Pharmacol Exp Ther. 321:953–960. 2007. View Article : Google Scholar
|
|
19
|
Hiramatsu A, Miwa H, Shikami M, et al:
Disease-specific expression of VEGF and its receptors in AML cells:
possible autocrine pathway of VEGF/type1 receptor of VEGF in
t(15;17) AML and VEGF/type2 receptor of VEGF in t(8;21) AML. Leuk
Lymphoma. 47:89–95. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Imai N, Shikami M, Miwa H, et al: t(8;21)
acute myeloid leukaemia cells are dependent on vascular endothelial
growth factor (VEGF)/VEGF receptor type2 pathway and
phosphorylation of Akt. Br J Haematol. 135:673–682. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mühlethaler-Mottet A, Meier R, Flahaut M,
et al: Complex molecular mechanisms cooperate to mediate histone
deacetylase inhibitors anti-tumour activity in neuroblastoma cells.
Mol Cancer. 7:552008.
|
|
22
|
Yang QC, Zeng BF, Shi ZM, et al:
Inhibition of hypoxia-induced angiogenesis by trichostatin A via
suppression of HIF-1a activity in human osteosarcoma. J Exp Clin
Cancer Res. 25:593–599. 2006.PubMed/NCBI
|
|
23
|
Mie Lee Y, Kim SH, Kim HS, et al:
Inhibition of hypoxia-induced angiogenesis by FK228, a specific
histone deacetylase inhibitor, via suppression of HIF-1alpha
activity. Biochem Biophys Res Commun. 300:241–246. 2003.PubMed/NCBI
|
|
24
|
Williams RJ: Trichostatin A, an inhibitor
of histone deacetylase, inhibits hypoxia-induced angiogenesis.
Expert Opin Investig Drugs. 10:1571–1573. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim MS, Kwon HJ, Lee YM, et al: Histone
deacetylases induce angiogenesis by negative regulation of tumor
suppressor genes. Nat Med. 7:437–443. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tovar-Castillo LE, Cancino-Díaz JC,
García-Vázquez F, et al: Under-expression of VHL and
over-expression of HDAC-1, HIF-1alpha, LL-37, and IAP-2 in affected
skin biopsies of patients with psoriasis. Int J Dermatol.
46:239–246. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ellis L, Hammers H and Pili R: Targeting
tumor angiogenesis with histone deacetylase inhibitors. Cancer
Lett. 280:145–153. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Strahl BD and Allis CD: The language of
covalent histone modifications. Nature. 403:41–45. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Grunstein M: Histone acetylation in
chromatin structure and transcription. Nature. 389:349–352. 1997.
View Article : Google Scholar : PubMed/NCBI
|