Introduction
Macrosomia is characterized by a high birth weight
of ≥4,000 g (1) and has previously
been observed in ~10% of newborns in certain regions of China
(2). The incidence of macrosomia
is increasing (3). Macrosomia
increases the risk of fetal asphyxia, shoulder dystocia, birth
trauma and neonatal hypoglycemia (4,5).
Furthermore, macrosomia is associated with long-term health
problems (6). The developmental
origin hypothesis (7) indicates
that nutrition and other environmental stimuli affect prenatal and
postnatal development, causing permanent changes in the metabolism
and increasing susceptibility to chronic diseases. Birth weight is
considered to be an indicator of risk for developing future
metabolic disorders. Numerous studies have documented associations
between birth weight and the increased incidence of metabolic
diseases (8–10), including insulin resistance
(11), obesity (12) and cancer (13).
The placenta has important functions in controlling
fetal growth and development. In particular, the placenta functions
as a gatekeeper of nutrient and waste exchange between the mother
and the developing fetus, and as a regulator of the intrauterine
environment (14). An adverse
intrauterine environment may affect fetal birth weight and
long-term health outcomes (15).
Epigenetic mechanisms regulate gene expression and contribute to
adverse intrauterine growth and fetal development. Thus, by
investigating epigenetic alterations in the placenta we may gain an
improved understanding of the molecular mechanisms behind a number
of developmental outcomes (16),
including macrosomia, which may be affected by intrauterine
conditions.
Leptin (LEP), a 16-kDa protein hormone, was
initially identified in adipose tissue and is also known to be
expressed in placental and fetal tissues. LEP is considered to be a
significant fetal growth factor that maintains energy and metabolic
balance during pregnancy (17,18).
Studies involving rats and humans have shown that LEP is regulated
in part by epigenetic mechanisms, specifically DNA methylation. The
CpG islands of the LEP promoter region may be subject to dynamic
methylation, which may affect LEP gene expression. The dynamic
methylation process may be affected by environmental or endogenous
factors. A study by Milagro et al reported that a high-fat
diet altered the methylation pattern of the LEP promoter in rats,
and that the methylation of at least one of the analyzed CpG sites
was significant in the regulation of leptin transcription in
adipose tissue (19). Melzner
et al (20) also provided
evidence that LEP promoter demethylation induces gene transcription
in human adipocytes. Additionally, LEP methylation levels in the
placenta were associated with maternal glycemia during pregnancy in
individuals with gestational impaired glucose tolerance (IGT; 2 h
post-oral glucose tolerance test, glycemia of >7.8 mmol/l)
(21). However, the placental LEP
methylation pattern in macrosomia remains unclear. In the present
study, differences between placental LEP promoter methylation in
infants with macrosomia and infants with normal birth weights who
were born to non-diabetic and/or non-hypertensive mothers were
examined. Furthermore, the contribution of placental LEP to
macrosomia was investigated.
Materials and methods
Study population
The subjects were recruited between April 2011 and
March 2012 at The Second Affiliated Hospital of Wenzhou Medical
University in Wenzhou (Zhejiang), China. The Wenzhou Medical
University Ethics Committee approved the study. Informed written
consent was obtained from each subject, i.e., the mother. Samples
were collected from females between the ages of 18 and 42 years old
whose infants were full-term (≥37 weeks), viable without known
genetic disorders and from normal pregnancies. Normal pregnancies
were defined by a lack of hypertension, hepatitis, heart disease,
psychological disorders, gestational diabetes and IGT. Newborns
were immediately weighed following delivery. Infants with birth
weights ≥4,000 g were considered macrosomic infants. An infant with
a normal birth weight was randomly selected as a control within
three days of the birth of the macrosomic infant. In total, 101
infants, including 49 macrosomic babies and 52 control newborns,
were selected.
Placental sampling
Placental biopsies from 101 deliveries were obtained
within 15 minutes of the delivery from mothers who were considered
to be full-term. A chorionic villous biopsy (~1 g) was excised,
obtained from the maternal side of the placenta 2 cm from the
umbilical cord insertion site. Biopsies that were free of maternal
decidua were washed and rinsed in sterile phosphate-buffered
saline. Biopsies were cut into small sections, suspended at a ratio
of 5:1 in RNAlater solution (Ambion, Austin, TX, USA), incubated at
4°C overnight and stored at −80°C until nucleic acid extraction was
performed.
DNA methylation measurements
Genomic DNA was extracted from placental tissues
with the Cell and Tissue DNA kit (BioTek, Beijing, China),
according to the manufacturer’s instructions. DNA quality was based
on purity and concentration, which were determined by measuring the
absorbance at 260 and 280 nm. Genomic DNA (200 ng) from each sample
was treated with bisulfite using the EZ 96-DNA methylation kit
(Zymo Research, Orange, CA, USA), according to the standard
overnight bisulfite treatment instructions. A total of 99 DNA
samples (excluding two degraded DNA samples) were treated with
bisulfite on two 96-well plates. DNA samples from macrosomic and
normal-weight newborns were equally distributed on the plates.
Samples from male and female infants were also equally distributed
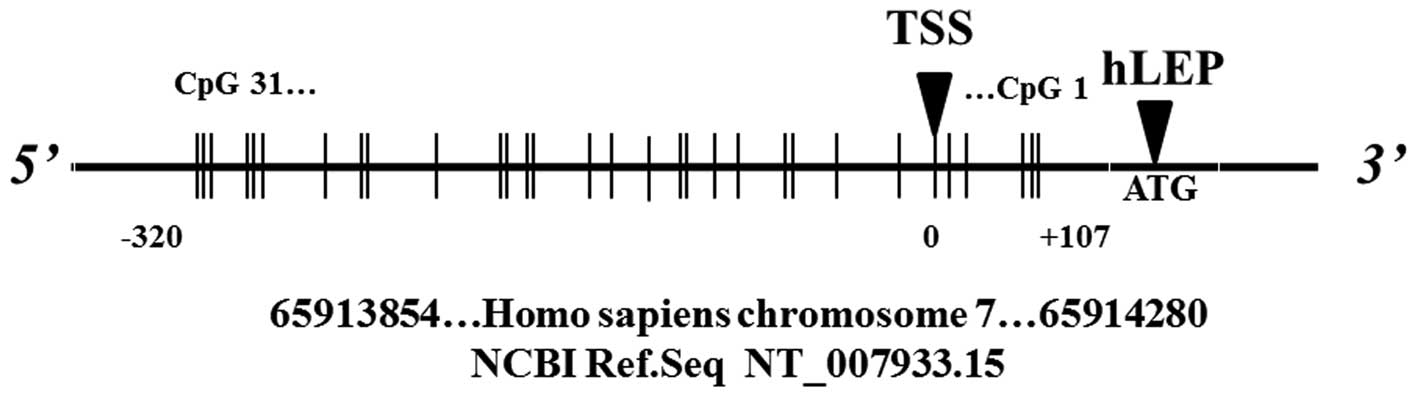
on the two plates. The targeted region of the LEP (gene ID, 3952)
promoter includes several CpG sites of which the methylation status
affects transcription (22) and
associates with glucose levels in females with IGT (21). The methylation level was determined
with the gold standard Sequenom MassARRAY platform (CapitalBio,
Beijing, China). This system combines matrix-assisted laser
desorption/ionization time-of-flight mass spectrometry with RNA
base-specific cleavage. A detectable pattern was analyzed to
determine the methylation status. PCR primers were designed with
Methprimer (http://epidesigner.com) and the
following primers were used based on the reverse complementary
strand of LEP (forward, 5′-ATTTAGAGTTGTGTGGGGTTTTGT-3′; reverse,
5′-CACCTTCCCAAAAAACTAATCCTTA-3′) to amplify base pairs
65913854–65914280 of the Homo sapiens chromosome 7 genomic contig,
GRCh37.p2 reference primary assembly (NCBI reference sequence,
NT_007933.15). A total of 31 CpG sites, which were divided into 16
CpG units, were examined in the LEP promoter, with the exception of
the 15th and 18th CpG sites (Fig.
1), which did not exhibit signals. The spectra methylation
ratios were generated with Epityper software version 1.0 (Sequenom,
San Diego, CA, USA).
mRNA expression measurements
Total RNA was extracted from the placental tissue
using TRIzol reagent (cat. no. 15596-026; Invitrogen, Carlsbad, CA,
USA). RNA quality was assessed by agarose gel electrophoresis and
by measuring the absorbance at 260 and 280 nm. Total RNA (1 μg) was
reverse-transcribed with the ReverTra Ace qPCR RT kit (Toyobo,
Osaka, Japan), following the manufacturer’s instructions. LEP mRNA
levels were quantified with THUNDERBIRD SYBR qPCR mix (Toyobo) and
an Applied Biosystems Step One Plus Real-Time PCR system (Applied
Biosystems, Foster, CA, USA). The following PCR primer sequences
were synthesized by Sangon (Shanghai, China). LEP (NM_000230.2)
forward, 5′-ATTTCACACACGCAGTCAGTCT-3′ and reverse, 5′-TCT
TGGATAAGGTCAGGATGG-3′. LEP expression levels were normalized to
glyceraldehyde-3-phoshate dehydrogenase expression as an internal
control. Expression levels were calculated for macrosomic and
control babies with the mean ± standard deviation (SD)
2−ΔCt method (23).
Statistical analysis
The quantitative data are expressed as the mean ±
SD. Anthropometric and pregnancy characteristics and DNA
methylation levels demonstrated normal distributions and were
analyzed by one-way analysis of variance and unpaired t-tests. LEP
mRNA expression (2−ΔCt) did not exhibit normal
distribution. Thus, differences in LEP expression between
macrosomic and control groups were assessed with the Mann-Whitney
rank sum test, in which the results were presented as the median
and interquartile ranges. Similar results were obtained with
unpaired t-tests following log transformation. Significant
differences in categorical variables were examined by chi-squared
test. A statistically significant difference was indicated by
P<0.05, and all the P-values reported were two-tailed.
Statistical analyses were performed with SPSS version 14.0 software
(SPSS, Inc., Chicago, IL, USA).
Results
Sample characteristics
Variations in the methylation levels of the LEP
promoter region in 99 placental samples and in the LEP mRNA
expression in 101 placental samples obtained from full-term infants
were examined. The demographics data of the total study population
(n=101) are listed in Table I.
Infants were grouped according to birth weights as normal-weight
newborns (n=52) and macrosomic infants (n=49). The distributions of
maternal age, gestational age, infant gender and alcohol and
smoking status during pregnancy were not significantly different
between the groups. As expected, females with higher body weights
prior to pregnancy tended to give birth to macrosomic babies by
cesarean section. The amount of weight gained during pregnancy did
not differ between the groups.
 | Table ICharacteristics at birth and during
pregnancy. |
Table I
Characteristics at birth and during
pregnancy.
| Characteristic | Macrosomia
(n=49) | Control (n=52) | P-value |
|---|
| Maternal age in
years, mean ± SD | 28.9±4.2 | 29.4±4.1 | 0.556 |
| Gestational age in
weeks, mean ± SD | 39.2±1.3 | 39.0±1.1 | 0.352 |
| Maternal
pre-pregnancy weight in kg, mean ± SD | 55.8±6.8 | 53.0±6.8 | 0.049 |
| Height in meters,
mean ± SD | 1.60±0.05 | 1.59±0.04 | 0.433 |
| Body mass index in
kg/m2, mean ± SD | 21.8±2.3 | 20.9±2.5 | 0.075 |
| Education status in
years, mean (%) | | | 0.593 |
| <6 | 12 (24.5) | 16 (30.8) | |
| 6–12 | 8 (16.3) | 7 (13.5) | |
| >12 | 25 (51.0) | 29 (55.7) | |
| Missing | 4 (8.2) | 0 (0.0) | |
| Parity, n (%) | | | 0.432 |
| Primiparity | 34 (69.4) | 31 (59.6) | |
| Multiparity | 15 (30.6) | 21 (40.4) | |
| Birth weight in g,
mean ± SD | 4307.9±206.6 | 3526.6±345.8 | <0.001 |
| Infant gender, n
(%) | | | 0.305 |
| Male | 34 (69.4) | 30 (57.7) | |
| Female | 15 (30.6) | 22 (42.3) | |
| Alcohol during
pregnancy, n (%) | | | 0.593 |
| No | 48 (98.0) | 50 (96.2) | |
| Yes | 1 (2.0) | 2 (3.8) | |
| Tobacco during
pregnancy, n (%) | | | <0.001 |
| No | 49 (100.0) | 52 (100.0) | |
| Yes | 0 (0.0) | 0 (0.0) | |
| Weight gain during
pregnancy in kg, mean ± SD |
| Total | 19.5±4.5 | 17.7±5.6 | 0.093 |
| 1–3 months | 3.8±4.0 | 2.4±2.6 | 0.052 |
| 3–6 months | 9.1±8.6 | 9.3±10.3 | 0.913 |
| 6–9 months | 9.3±10.0 | 9.0±8.9 | 0.868 |
| Delivery method, n
(%) | | | 0.001 |
| C-section | 44 (89.8) | 31 (59.6) | |
| Vaginal | 5 (10.2) | 20 (40.4) | |
LEP DNA methylation
DNA methylation analyses focused on a 426-bp human
LEP promoter region, which included 31 cytosine CpG dinucleotides
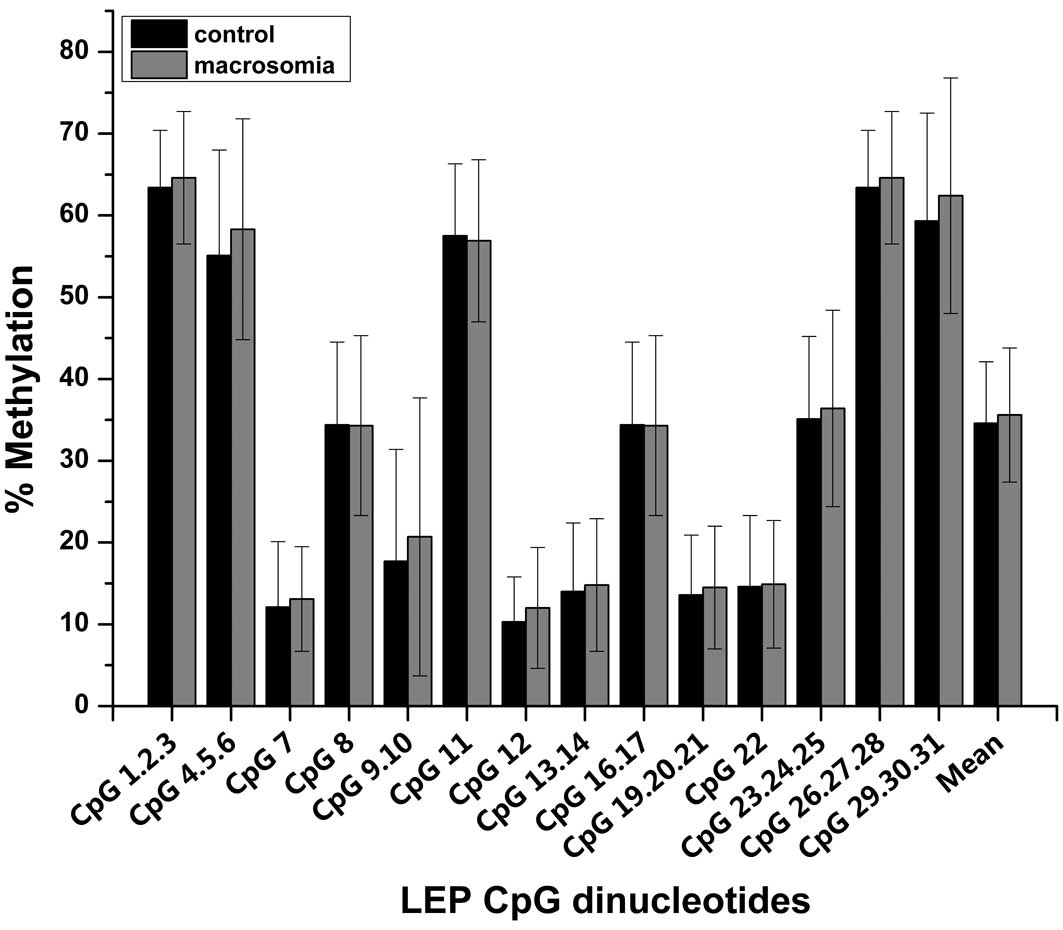
(Fig. 1). Differences in the
methylation levels of these 31 CpG sites are shown for macrosomic
and control placentas (Fig. 2).
The average DNA methylation levels were 35.6% and 34.6% for the
macrosomic and control placentas, respectively, which was not
significantly different (P=0.538). Similarly, the two groups were
not significantly different in the extent of methylation of
individual CpG dinucleotides.
A stratified analysis was performed to reduce the
effect of heterogeneity. First, the analysis was restricted in
parity. Significant differences were identified in the methylation
of certain CpG dinucleotides in the LEP promoter of macrosomic and
control groups of primiparous females. A higher methylation level
was identified in the macrosomic group of primiparous females
(Table II). Second, weekly
stratification of the gestational age from 37 to 41 weeks
demonstrated that no significant differences were identified
between the two groups at any gestational age, with the exception
of 39 weeks. The mean and individual CpG dinucleotide methylation
levels were higher in macrosomic infants than those in the control
group at the gestational age of 39 weeks (P<0.05; Table III).
| Table IIMean level of methylation of four CpG
dinucleotides (only statistically significant data are shown) in
the LEP promoter region in macrosomic (n=34) and control (n=31)
groups within the primiparity group. |
Table II
Mean level of methylation of four CpG
dinucleotides (only statistically significant data are shown) in
the LEP promoter region in macrosomic (n=34) and control (n=31)
groups within the primiparity group.
| CpG
dinucleotides | Macrosomia, % (mean
± SD) | Control, % (mean ±
SD) | P-value |
|---|
| CpG1.2.3 | 65.0±8.0 | 61.3±7.0 | 0.045 |
| CpG9.10 | 21.5±19.0 | 13.3±9.0 | 0.034 |
| CpG26.27.28 | 65.0±8.0 | 61.3±7.0 | 0.045 |
| CpG29.30.31 | 63.6±14.0 | 55.4±11.0 | 0.014 |
| Table IIIMean levels of methylation of seven
CpG dinucleotides (only statistically significant data are shown)
in the LEP promoter region in macrosomic (n=14) and control (n=17)
groups at the gestational age of 39 weeks. |
Table III
Mean levels of methylation of seven
CpG dinucleotides (only statistically significant data are shown)
in the LEP promoter region in macrosomic (n=14) and control (n=17)
groups at the gestational age of 39 weeks.
| CpG
dinucleotides | Macrosomia, % (mean
± SD) | Control, % (mean ±
SD) | P-value |
|---|
| CpG4.5.6 | 60.9±10.0 | 51.6±6.0 | 0.045 |
| CpG7 | 14.0±5.0 | 8.8±6.0 | 0.019 |
| CpG8 | 38.2±8.0 | 32.1±7.0 | 0.035 |
| CpG9.10 | 22.6±15.0 | 11.8±6.0 | 0.008 |
| CpG16.17 | 38.2±8.0 | 32.1±7.0 | 0.035 |
| CpG22 | 18.2±8.0 | 12.3±6.0 | 0.025 |
| CpG23.24.25 | 41.8±11.0 | 32.9±8.0 | 0.015 |
| Mean | 38.0±7.0 | 32.0±6.0 | 0.016 |
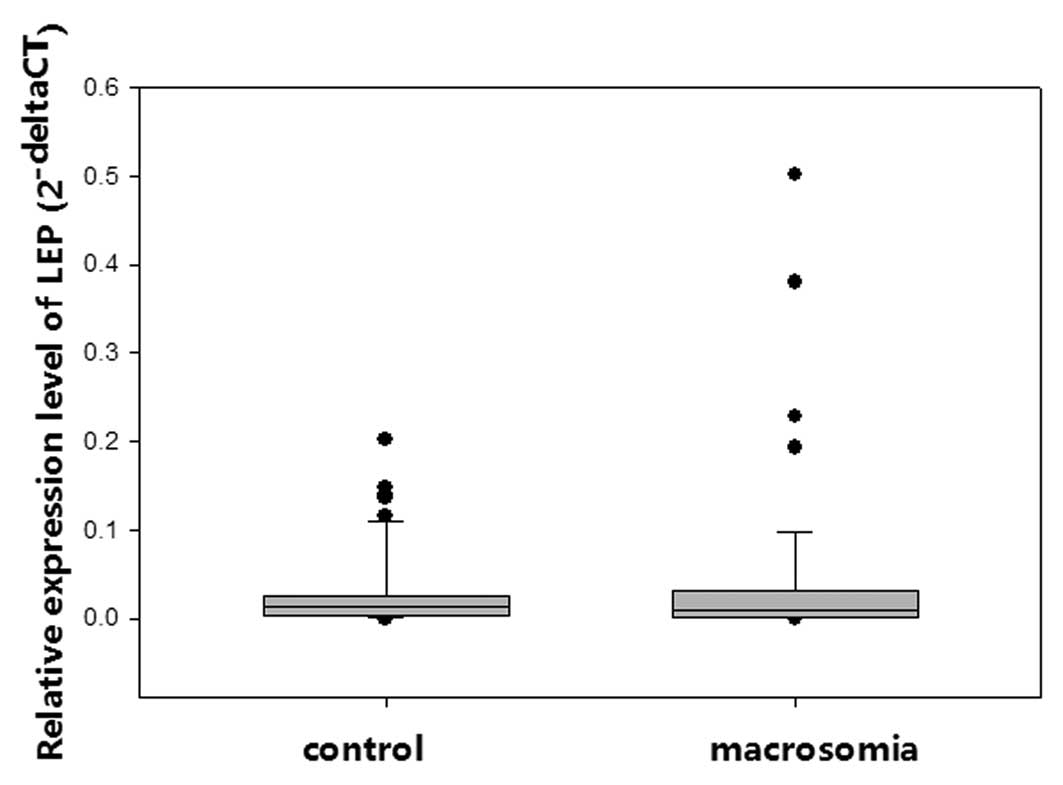
mRNA expression
Quantitative PCR was performed to understand the
contribution of placental LEP to infant birth weight. No
significant differences in LEP mRNA expression levels were
identified between the macrosomic and control groups (Fig. 3). The stratified analysis did not
indicate significant differences, although the methylation status
was significantly different.
Discussion
LEP promotes the proliferation and invasiveness of
trophoblast cells and also affects local angiogenesis. LEP may have
an affect on the outcome of pregnancy and fetal growth by
interfering with placental development (24). In the present study, significant
differences were identified in the DNA methylation of individual
CpG dinucleotides in the placental LEP promoter at the gestational
age of 39 weeks for macrosomic infants who were born to primiparous
females. These results indicate that the methylation status of LEP
in macrosomia may be altered within a specific gestational period
or during certain maternal conditions.
Previous studies have indicated that placental LEP
methylation may be altered by certain maternal conditions during
pregnancy and lactation, including prenatal famine exposure
(25), gestational IGT (21), a low-protein diet (26) or diet-induced obesity (19). Moreover, Hogg et al
(27) demonstrated that placental
LEP is hypomethylated in early-onset pre-eclampsia. However, no
significant differences were identified in the mean methylation
level of the LEP promoter between macrosomic and normal-weight
babies of primiparous mothers. Potential differences in the
methylation of individual CpG dinucleotides may have been diluted
by the overall number of CpG sites, as methylation changes may not
occur at all CpG sites within a promoter. Hogg et al
(27) conjectured that CpG sites
proximal to SP1 or C/EBP binding motifs are more likely to be
methylated. In the present study, the methylation variations
identified occurred proximal to these motifs, supporting this
hypothesis.
In the present study, placental LEP mRNA expression
was not significantly different between the groups. These results
were consistent with previous findings from dually perfused human
placentas (28,29), which indicated that maternal levels
of circulating LEP during pregnancy are determined by placental LEP
production, but do not correlate with fetal weight. Rather, the
fetus was proposed to regulate its own energy metabolism and
appetite (30,31). The present results also indicated
that placental LEP did not directly contribute to fetal growth. In
addition, the results that were obtained in females who had normal
pregnancies differed from findings showing that placental LEP
expression is upregulated during pregnancy-related pathological
conditions, including gestational diabetes (with or without fetal
overgrowth) and pre-eclampsia (with or without fetal growth
restriction) (32,33). These differences may be explained
by the increased demands that are placed on the placenta to deliver
nutrients during pathological conditions.
The present results revealed a lack of
correspondence between LEP promoter methylation and LEP expression.
These data indicated that methylation was not a main factor
contributing to placental LEP expression during normal pregnancies.
However, LEP expression may be regulated by promoter methylation
during other conditions. A previous study identified various
regulatory elements within the LEP promoter, e.g., cAMP and
glucocorticoid response elements and CCATT/enhancer and SP1 binding
sites (34). Methylation at CpG
sites proximal to or within these regulatory elements affected LEP
expression in adipocytes (20,35)
and during pregnancy-related pathological conditions, including IGT
(21), early-onset pre-eclampsia
(27) and prenatal famine exposure
(25). Furthermore, Hogg et
al (27) observed monoallelic
expression of placental LEP during normal pregnancies and a trend
towards biallelic LEP expression during early-onset pre-eclampsia.
This study also indicated that the loss of normal monoallelic LEP
expression was associated with hypomethylation, which increased the
overall level of LEP expression. We hypothesize that placental
methylation is one of the mechanisms regulating LEP expression.
Various degrees of promoter hypomethylation alter the proportion of
cells with biallelic expression, causing variations in the overall
levels of LEP expression. These variations may exist to adapt to
various physiological and pathological conditions.
Epidemiological evidence shows that multiparity is a
risk factor for macrosomia, but that primiparity is associated with
an increased risk of low birth weight (36). The present study identified
hypermethylation of individual CpG dinucleotides in the placental
LEP promoter for macrosomic infants of primiparous females. These
data may provide an epigenetic basis for the aforementioned
epidemiological data discussed. Additionally, a previous anatomical
study demonstrated that the proportion of non-muscular tissues
increased with parity, such that the first pregnancy caused
permanent anatomical changes in the spiral arteries. These changes
may modify vascular remodeling during subsequent pregnancies
(37). Further research is
required to investigate the correlation between variations in LEP
promoter methylation and anatomical changes.
In the present study, the mean methylation level and
methylation of several CpG dinucleotides were significantly
increased in the macrosomia group at the gestational age of 39
weeks. Increased methylation was observed at the SPI and TATA box,
which are significant for LEP transcription (35). However, this difference did not
appear at other gestational ages. Moreover, LEP mRNA levels were
not significantly different between groups according to
stratification by gestational age. A similar result was observed in
the study by Hogg et al (27), in which gestational age was
considered a confounding factor for the direct comparison of LEP
DNA methylation between controls and early-onset pre-eclampsia
cases. The present results indicated that gestational age was not a
predictor of LEP methylation and vice versa.
In conclusion, macrosomia is a multifactorial
condition. The present study indicated that placental LEP
methylation in macrosomic infants may be affected by maternal
conditions or by a specific gestational period. This data provides
valuable information with regard to the contribution of placental
LEP to macrosomia.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81072378) and the Natural
Science Funds of Zhejiang (grant no. Y2101185).
References
|
1
|
Fuchs F, Bouyer J, Rozenberg P and Senat
MV: Adverse maternal outcomes associated with fetal macrosomia:
what are the risk factors beyond birthweight? BMC Pregnancy
Childbirth. 13:902013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bao C, Zhou Y, Jiang L, et al: Reasons for
the increasing incidence of macrosomia in Harbin, China. BJOG.
118:93–98. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ornoy A: Prenatal origin of obesity and
their complications: Gestational diabetes, maternal overweight and
the paradoxical effects of fetal growth restriction and macrosomia.
Reprod Toxicol. 32:205–212. 2011. View Article : Google Scholar
|
|
4
|
Boney CM, Verma A, Tucker R and Vohr BR:
Metabolic syndrome in childhood: association with birth weight,
maternal obesity, and gestational diabetes mellitus. Pediatrics.
115:e290–e296. 2005. View Article : Google Scholar
|
|
5
|
Oral E, Cağdaş A, Gezer A, Kaleli S,
Aydinli K and Oçer F: Perinatal and maternal outcomes of fetal
macrosomia. Eur J Obstet Gynecol Reprod Biol. 99:167–171. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ng SK, Olog A, Spinks AB, Cameron CM,
Searle J and McClure RJ: Risk factors and obstetric complications
of large for gestational age births with adjustments for community
effects: results from a new cohort study. BMC Public Health.
10:4602010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gluckman PD and Hanson MA: Developmental
origins of disease paradigm: a mechanistic and evolutionary
perspective. Pediatr Res. 56:311–317. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hales CN, Barker DJ, Clark PM, et al:
Fetal and infant growth and impaired glucose tolerance at age 64.
BMJ. 303:1019–1022. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gluckman PD, Hanson MA, Cooper C and
Thornburg KL: Effect of in utero and early-life conditions on adult
health and disease. N Engl J Med. 359:61–73. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rich-Edwards JW, Stampfer MJ, Manson JE,
et al: Birth weight and risk of cardiovascular disease in a cohort
of women followed up since 1976. BMJ. 315:396–400. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Giapros V, Papadimitriou P, Challa A and
Andronikou S: The effect of intrauterine growth retardation on
renal function in the first two months of life. Nephrol Dial
Transplant. 22:96–103. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hediger ML, Overpeck MD, McGlynn A,
Kuczmarski RJ, Maurer KR and Davis WW: Growth and fatness at three
to six years of age of children born small- or
large-for-gestational age. Pediatrics. 104:e331999.PubMed/NCBI
|
|
13
|
Ross JA: High birthweight and cancer:
evidence and implications. Cancer Epidemiol Biomarkers Prev.
15:1–2. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Godfrey KM: The role of the placenta in
fetal programming-a review. Placenta. 23(Suppl A): S20–S27. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nelissen EC, van Montfoort AP, Dumoulin JC
and Evers JL: Epigenetics and the placenta. Hum Reprod Update.
17:397–417. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Menon R, Conneely KN and Smith AK: DNA
methylation: an epigenetic risk factor in preterm birth. Reprod
Sci. 19:6–13. 2012. View Article : Google Scholar
|
|
17
|
Christou H, Connors JM, Ziotopoulou M, et
al: Cord blood leptin and insulin-like growth factor levels are
independent predictors of fetal growth. J Clin Endocrinol Metab.
86:935–938. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Grisaru-Granovsky S, Samueloff A and
Elstein D: The role of leptin in fetal growth: a short review from
conception to delivery. Eur J Obstet Gynecol Reprod Biol.
136:146–150. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Milagro FI, Campión J, García-Díaz DF,
Goyenechea E, Paternain L and Martínez JA: High fat diet-induced
obesity modifies the methylation pattern of leptin promoter in
rats. J Physiol Biochem. 65:1–9. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Melzner I, Scott V, Dorsch K, et al:
Leptin gene expression in human preadipocytes is switched on by
maturation-induced demethylation of distinct CpGs in its proximal
promoter. J Biol Chem. 277:45420–45427. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bouchard L, Thibault S, Guay SP, et al:
Leptin gene epigenetic adaptation to impaired glucose metabolism
during pregnancy. Diabetes Care. 33:2436–2441. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stöger R: In vivo methylation patterns of
the leptin promoter in human and mouse. Epigenetics. 1:155–162.
2006.PubMed/NCBI
|
|
23
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
D’Ippolito S, Tersigni C, Scambia G and Di
Simone N: Adipokines, an adipose tissue and placental product with
biological functions during pregnancy. Biofactors. 38:14–23.
2012.PubMed/NCBI
|
|
25
|
Tobi EW, Lumey LH, Talens RP, et al: DNA
methylation differences after exposure to prenatal famine are
common and timing- and sex-specific. Hum Mol Genet. 18:4046–4053.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jousse C, Parry L, Lambert-Langlais S, et
al: Perinatal undernutrition affects the methylation and expression
of the leptin gene in adults: implication for the understanding of
metabolic syndrome. FASEB J. 25:3271–3278. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hogg K, Blair JD, von Dadelszen P and
Robinson WP: Hypomethylation of the LEP gene in placenta and
elevated maternal leptin concentration in early onset
pre-eclampsia. Mol Cell Endocrinol. 367:64–73. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Linnemann K, Malek A, Sager R, Blum WF,
Schneider H and Fusch C: Leptin production and release in the
dually in vitroperfused human placenta. J Clin Endocrinol Metab.
85:4298–4301. 2000.PubMed/NCBI
|
|
29
|
Lepercq J, Challier JC, Guerre-Millo M,
Cauzac M, Vidal H and Hauguel-de Mouzon S: Prenatal leptin
production: evidence that fetal adipose tissue produces leptin. J
Clin Endocrinol Metab. 86:2409–2413. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hauguel-de Mouzon S, Lepercq J and
Catalano P: The known and unknown of leptin in pregnancy. Am J
Obstet Gynecol. 194:1537–1545. 2006.PubMed/NCBI
|
|
31
|
Newbern D and Freemark M: Placental
hormones and the control of maternal metabolism and fetal growth.
Curr Opin Endocrinol Diabetes Obes. 18:409–416. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lepercq J, Cauzac M, Lahlou N, et al:
Overexpression of placental leptin in diabetic pregnancy: a
critical role for insulin. Diabetes. 47:847–850. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hoegh AM, Borup R, Nielsen FC, Sørensen S
and Hviid TV: Gene expression profiling of placentas affected by
pre-eclampsia. J Biomed Biotechnol. 2010:7875452010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang Y, Proenca R, Maffei M, Barone M,
Leopold L and Friedman JM: Positional cloning of the mouse obese
gene and its human homologue. Nature. 372:425–432. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Marchi M, Lisi S, Curcio M, et al: Human
leptin tissue distribution, but not weight loss-dependent change in
expression, is associated with methylation of its promoter.
Epigenetics. 6:1198–1206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Al-Farsi YM, Brooks DR, Werler MM, Cabral
HJ, Al-Shafaee MA and Wallenburg HC: Effect of high parity on
occurrence of some fetal growth indices: a cohort study. Int J
Womens Health. 4:289–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Khong TY, Adema ED and Erwich JJ: On an
anatomical basis for the increase in birth weight in second and
subsequent born children. Placenta. 24:348–353. 2003. View Article : Google Scholar : PubMed/NCBI
|