Introduction
The myocardium is one of the target organs of septic
shock (1–2), which is a major cause of mortality.
This myocardial injury may occur as a result of the release of
proinflammatory cytokines induced by bacterial endotoxin
lipopolysaccharide (LPS) (3).
Furthermore, it appears that LPS may be responsible for multiple
organ failure during septic shock (4). It has also been demonstrated that LPS
reduced myocardial function (5).
Numerous studies have suggested that LPS-induced myocardial
dysfunction is mediated by multiple proinflammatory mediators,
including tumor necrosis factor-α (TNF-α), Toll-like receptor 4
(TLR4) and TLR2 (6–8).
Recently, LPS was reported to stimulate
cardiomyocyte autophagy (4,9),
which may mediate cell death. Autophagy, which has been suggested
to be an essential function for cell homeostasis, as well as cell
defense and adaptation to an adverse environment, is a type of
programmed cell death (10–12).
Autophagy has an important role in the heart, and activation of
autophagy has been observed in a variety of heart diseases,
including cardiac hypertrophy, heart failure and ischemia
reperfusion injury. Therefore, it is important to regulate
cardiomyocyte autophagy in order to reduce myocardial injury
induced by LPS.
It is well established that the incidence of
cardiovascular disease is reduced in females prior to menopause,
which may be due to estrogen (E2) levels (13–14).
Studies have demonstrated that E2 exhibits cardioprotective effects
due to the ability to decrease TNF-α levels (15). However, few studies have
investigated whether E2 may regulate cardiomyocyte autophagy. Based
on these observations, the present study aimed to examine whether
E2 may reduce cardiomyocyte injury by regulating autophagy.
Materials and methods
Animal care
All animal experiments were approved by the Animal
Research Ethics Committee of the Second Military Medical University
(Shanghai, China). The experimental procedures conformed with the
guide for the care and use of laboratory animals published by the
US National Institutes of Health.
Cell culture and experimental
procedures
Neonatal cardiomyocytes were prepared from the
hearts of Sprague-Dawley rats younger than 3 days (16). On the 4th day, the cardiomyocytes
were randomized to three groups: The control group (con), where the
cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)
with 5% CO2 and 95% air for 24 h; the LPS group, where
the cells were treated with 1 μg/ml LPS for 24 h; and the E2+LPS
group, where the cells were treated with 10−8 M E2, and
then were treated with 1 μg/ml LPS 30 min later.
3-(4, 5-dimethylthiazol-2-yl)-2,
5-diphenyl tetrazonium bromide (MTT) assay
For the MTT assay, 10 μl MTT solution was added to
the growing cells and incubated for 4 h. The crystals were then
solubilized by adding 100 μl solubilization solution. The
absorbance of the purple solution was determined at a wavelength of
450 nm with a microtiter plate reader (Bio-Rad, Hercules, CA,
USA).
Lactate dehydrogenase (LDH) assay
LDH release was measured following treatment as a
cellular injury index. The culture media was collected for
determination of LDH activity using an Hitachi 7020 chemistry
analyzer (Hitachi, Ltd., Tokyo, Japan).
Quantitative polymerase chain reaction
(qPCR) of Atg5 and Beclin1
Total RNA of cells was isolated using TRIzol reagent
and reverse transcribed according to the manufacturer’s
instructions (Thermo Scientific, Waltham, MA, USA). Dysregulated
Atg5 and Beclin1 were validated by qPCR in duplicates using the
Mini OPTICON realtime PCR system (Bio-Rad). The annealing
temperature of Atg5 and Beclin1 was set at 56°C. The comparative Ct
(threshold cycle) method with arithmetic formulae
(2−ΔΔCt) was used to determine the relative quantitation
of gene expression of the target and housekeeping genes (β-actin).
The following sense and antisense primers were used: Beclin1
(accession number NM_001034117), forward 5′-GGCAGTGGCTCCTATT-3′ and
reverse 5′-GGCGTGCTGTGCTCTGAAAA-3′; Atg5 (accession number
NM_001014250), forward 5′-AGTGGAGGCAACAGAACC-3′ and reverse
5′-GACACGAACTGGCACATT-3′.
Western blotting of
microtubule-associated protein light chain 3 (LC3)
The protein concentration was determined with a
bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology, Haimen, Jiangsu, China) according the manufacturer’s
instructions. Equal quantities of protein (40 μg) from the
cardiomyocytes were subjected to western blotting analysis to
evaluate LC3 expression (the primary rabbit antibody was purchased
from Sigma, St. Louis, MO, USA) with an enhanced chemiluminescence
detection kit (Amersham Biosciences, Piscataway, NJ, USA). The
results are presented as LC3-II/LC3-I.
Statistical analysis
Quantitative data are presented as the mean ±
standard error. Statistical significance was determined using
one-way analysis of variance. P<0.05 was considered to indicate
a statistically significant difference.
Results
E2 produces a cardioprotective effect
against LPS
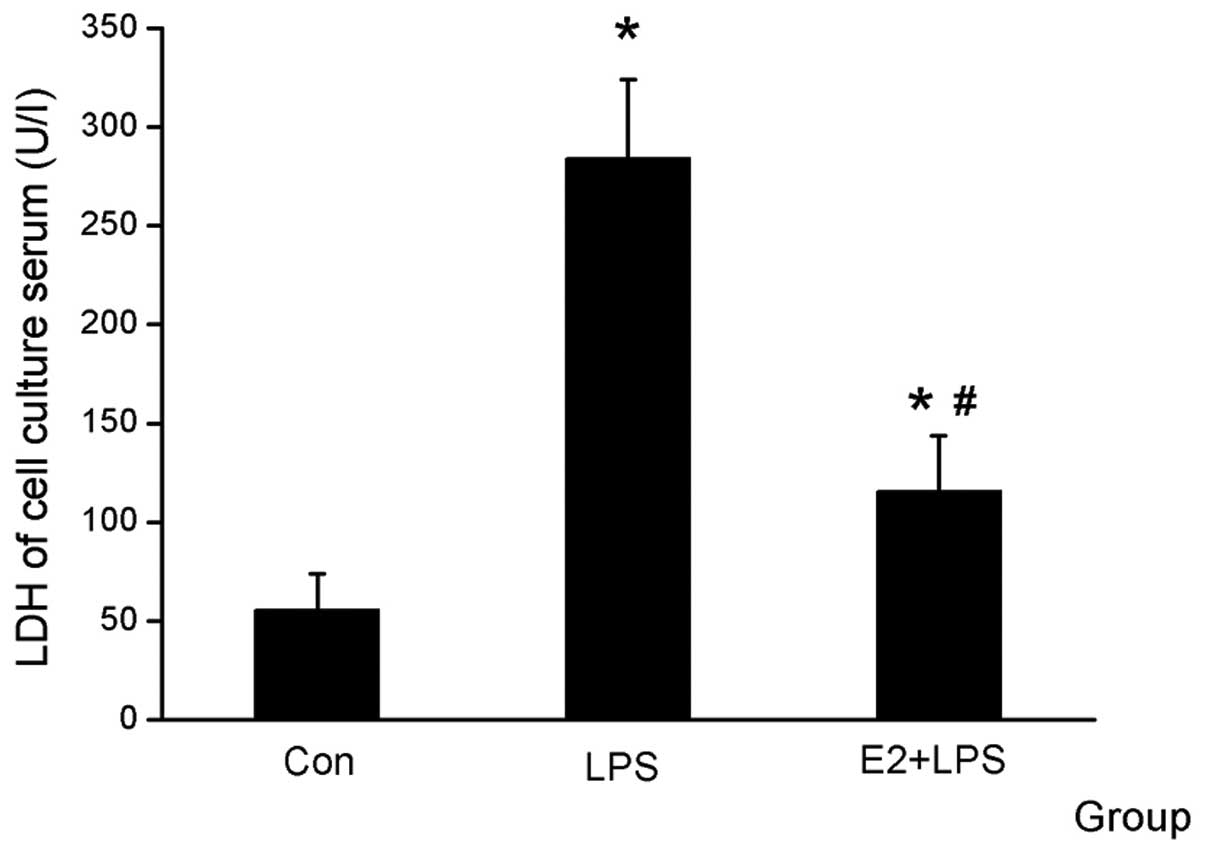
The serum was collected for the LDH and MTT assays
and was used for comparing cardiomyocyte vitality. It was
identified that LDH was higher and the cell vitality was decreased
in the LPS group compared with the Con group, and that E2 was able
to attenuate the effect of LPS (Figs.
1 and 2).
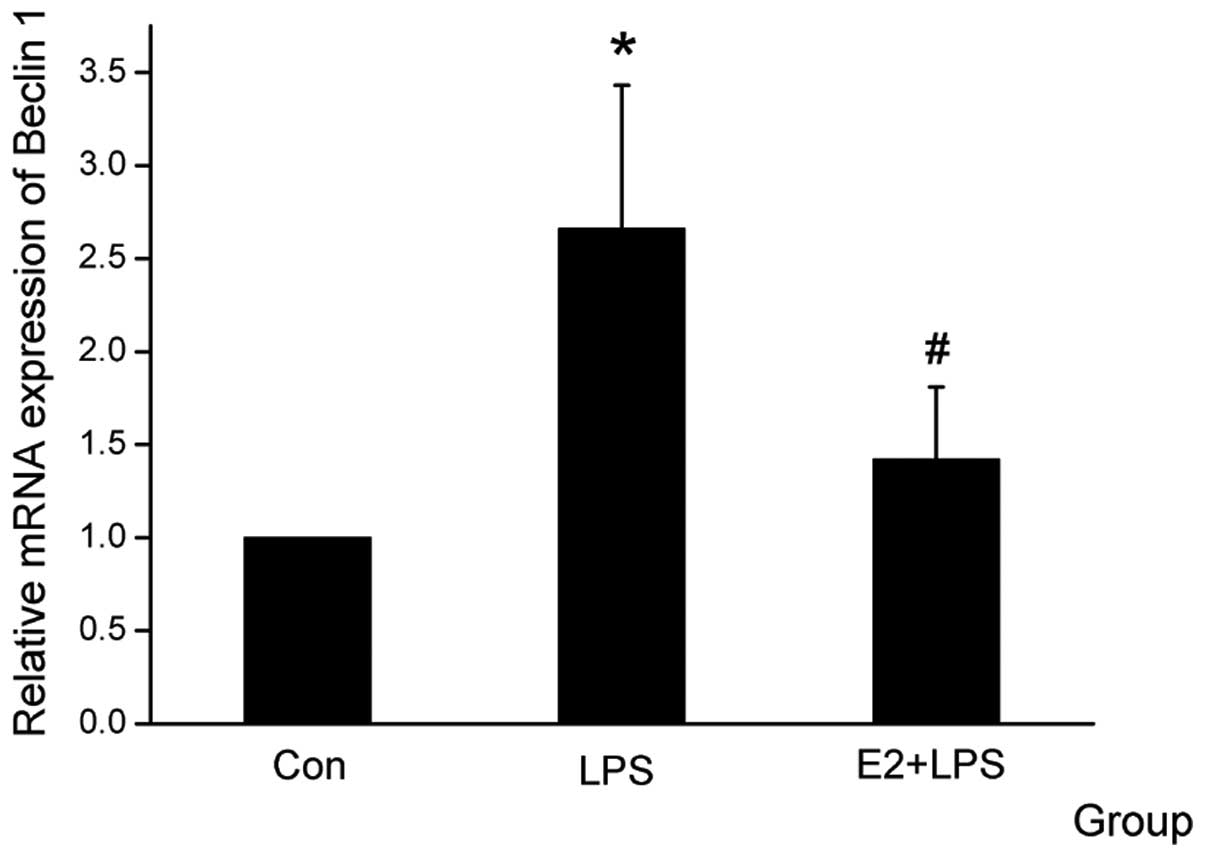
E2 attenuates Atg5 and Beclin1 mRNA
expression level
The Atg family members, particularly Beclin1 and
Atg5, have been reported to have an important role in the
autophagic cell death pathway (17–18).
When the cardiomyocytes were treated with E2, the mRNA expression
of Atg5 and Beclin1 was downregulated, which was upregulated by LPS
(Figs. 3 and 4).
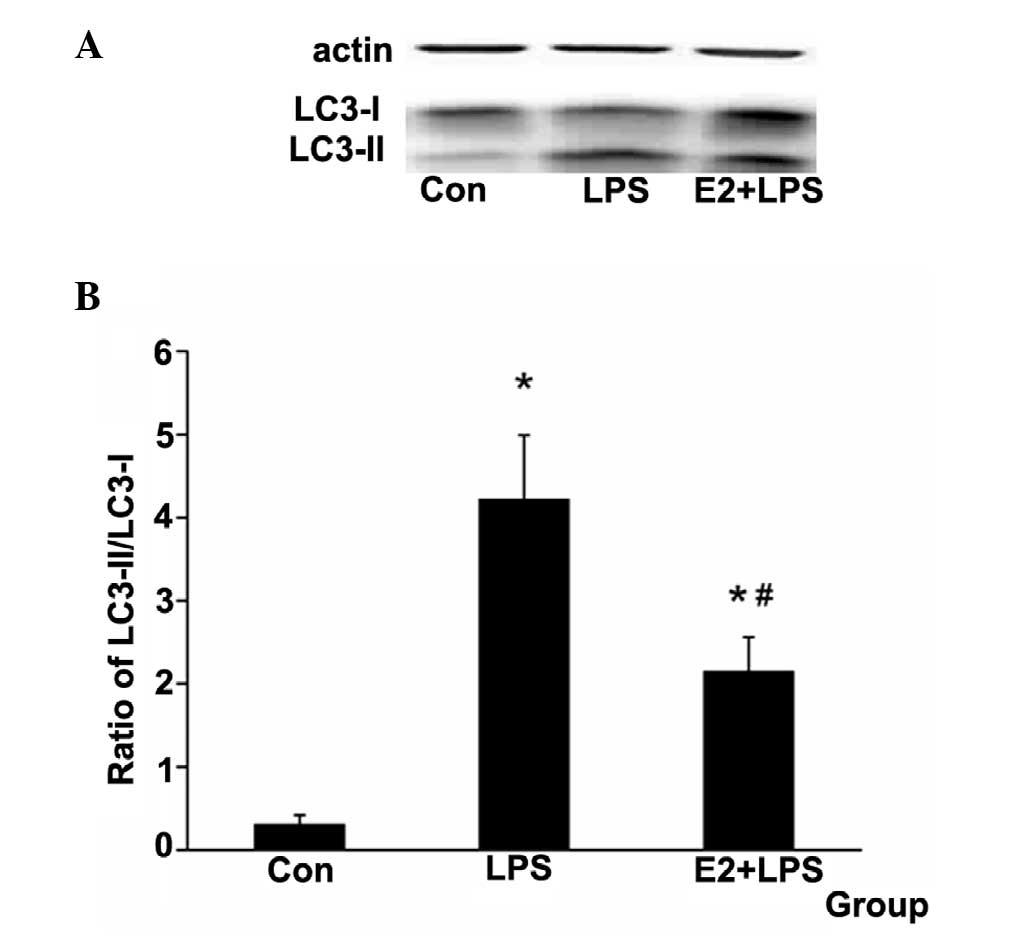
E2 attenuates the LC3-II protein relative
expression level
LC3-II was used as a marker of autophagy (19). The ratio of LC3-II/LC3-I was used
to examine the autophagy level in the present study. It was
identified that the ratio was increased in the LPS group and
decreased in the E2 + LPS group (Fig.
5).
Discussion
LPS, which is a major component of bacterial outer
walls, has been demonstrated to be responsible for the multiorgan
dysfunction that characterizes septic shock (4). It has been found that LPS is able to
stimulate inflammatory mediator production and activate NF-κB
(20–21). The myocardium is one of the main
target organs of septic shock (2).
Therefore, it is clinically beneficial to investigate ways to
attenuate the myocardial injury induced by LPS in patients with
septic shock. In the present study LPS was used to simulate the
heart injury induced by septic shock in vitro. According to
this model, the present study aimed to investigate novel reagents
that may protect the heart against LPS injury. LDH and cell
vitality were generally used to evaluate cell injury (16,22).
It was found that LPS was harmful to the cardiomyocytes and E2
attenuated the injury induced by LPS.
Apoptosis, necrosis and autophagy occur in
cardiomyocytes during cardiac injury (23). The autophagy process is regulated
by Atgs, among which Beclin1 is required for the autophagy vesicle
nucleation step of autophagy. The autophagosome is formatted
through two pathways, the Atg12-Atg5-Atg16 pathway and the
Atg4-Atg7-Atg3 pathway. These conjugations lead to the conversion
of the soluble form of LC3 (LC3-I) to the autophagic
vesicle-associated form (LC3-II), which is used as a marker of
autophagy (19). In the majority
of these studies, the ratio of LC3-II/LC3-I has been used for
examining the autophagy level (24).
Apoptosis and necrosis are well established as
detrimental processes to the heart (25). However, the effect of autophagy in
the heart is controversial (26–27).
At low levels, autophagy removed the damaged proteins and
organelles, that facilitated myocardial survival during periods of
energy deprivation (28).
Therefore, low levels of autophagy are beneficial to cardiomyocytes
(29). However, excessive levels
of autophagy appear to contribute to cardiomyocyte damage (24). Furthermore, accumulation of
autophagic vacuoles precedes apoptotic cell death, and autophagy
induces cell death when apoptosis is inhibited. For instance, it
has been demonstrated that autophagy was marginally increased in
the myocardium during the ischemic period, and it was protective
for the heart, while during the reperfusion period autophagy was
markedly enhanced and was subsequently harmful to the heart
(24,27). Furthermore, it has been identified
that the inhibition of autophagy is cardioprotective against
LPS-induced injury (9). Therefore,
moderate regulation of autophagy may aid in attenuating
cardiomyocyte injury induced by LPS.
Studies have suggested that E2 has important
cardioprotective roles against ischemia-reperfusion (IR) injury
(30–31) and that E2 treatment may upregulate
the expression of anti-apoptotic genes (32). Recently, several studies
demonstrated that Beclin1 was able to downregulate E2ic signaling,
suggesting the importance of the interaction between E2 and
autophagy (33).
The biological effects of E2 are predominantly
mediated via E2 receptors (ERs). The two classic ER isoforms (ERα
and ERβ) are expressed in cardiomyocytes, and there appears to be
no difference in the distribution or abundance between males and
females (30). ERα is mainly
expressed in the ventricles and its activation may result in rapid
cardioprotective signaling (34).
ERβ is evenly distributed throughout the heart and may not be
involved in cardioprotection (35–36).
E2 may protect the heart following ischemia-reperfusion by
decreasing TNF-α levels (15),
which has been associated with cardiomyocyte autophagy induced by
LPS. In the present study, cardiomyocyte autophagy was induced by
LPS, as is consistent with the results of Yuan et al
(4). LDH in the serum was
increased and the cell vitality was decreased following LPS
treatment. This suggested that LPS may be harmful to cardiomyocytes
by inducing autophagy. When the cardiomyocytes were treated with E2
1 h prior to LPS, the autophagy level and LDH in the serum were
attenuated and the cell vitality was increased. Therefore, E2 may
protect cardiomyocytes by attenuating autophagy against LPS,
mediated by the ERα subtype receptor.
In conclusion, the results demonstrated that E2 has
an important protective role against LPS-induced injury by
regulating autophagy. However, further studies are required to
investigate the mechanisms underlying the interaction between E2
and the regulation of autophagy.
Acknowledgements
The present study was supported by the Nature
Science Foundation of Science and Technology Commission of Shanghai
municipality (grant nos. 12ZR1438000 and 12ZR1454600) and the
National Nature Science Foundation of China (grant no.
81200181).
References
|
1
|
Rudiger A and Singer M: The heart in
sepsis: from basic mechanisms to clinical management. Curr Vasc
Pharmacol. 11:187–195. 2013.PubMed/NCBI
|
|
2
|
Zhang M, Wang X, Wang X, et al: Oxymatrine
protects against myocardial injury via inhibition of JAK2/STAT3
signaling in rat septic shock. Mol Med Rep. 7:1293–1299.
2013.PubMed/NCBI
|
|
3
|
Hickson-Bick DL, Jones C and Buja LM:
Stimulation of mitochondrial biogenesis and autophagy by
lipopolysaccharide in the neonatal rat cardiomyocyte protects
against programmed cell death. J Mol Cell Cardiol. 44:411–418.
2008. View Article : Google Scholar
|
|
4
|
Yuan H, Perry CN, Huang C, et al:
LPS-induced autophagy is mediated by oxidative signaling in
cardiomyocytes and is associated with cytoprotection. Am J Physiol
Heart Circ Physiol. 296:H470–H479. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Smeding L, Plötz FB, Lamberts RR, van der
Laarse WJ, Kneyber MC and Groeneveld AB: Mechanical ventilation
with high tidal volumes attenuates myocardial dysfunction by
decreasing cardiac edema in a rat model of LPS-induced peritonitis.
Respir Res. 13:232012.
|
|
6
|
Tien YC, Lin JY, Lai CH, et al:
Carthamus tinctorius L. prevents LPS-induced TNFalpha
signaling activation and cell apoptosis through JNK1/2-NFkappaB
pathway inhibition in H9c2 cardiomyoblast cells. J Ethnopharmacol.
130:505–513. 2010. View Article : Google Scholar
|
|
7
|
Lorne E, Dupont H and Abraham E: Toll-like
receptors 2 and 4: initiators of non-septic inflammation in
critical care medicine? Intensive Care Med. 36:1826–1835. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Leon CG, Tory R, Jia J, Sivak O and Wasan
KM: Discovery and development of toll-like receptor 4 (TLR4)
antagonists: a new paradigm for treating sepsis and other diseases.
Pharm Res. 25:1751–1761. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Turdi S, Han X, Huff AF, et al:
Cardiac-specific overexpression of catalase attenuates
lipopolysaccharide-induced myocardial contractile dysfunction: role
of autophagy. Free Radic Biol Med. 53:1327–1338. 2012. View Article : Google Scholar
|
|
10
|
Meléndez A, Tallóczy Z, Seaman M,
Eskelinen EL, Hall DH and Levine B: Autophagy genes are essential
for dauer development and life-span extension in C. elegans.
Science. 301:1387–1391. 2003.PubMed/NCBI
|
|
11
|
Stromhaug PE and Klionsky DJ: Approaching
the molecular mechanism of autophagy. Traffic. 2:524–531. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Otto GP, Wu MY, Kazgan N, Anderson OR and
Kessin RH: Macroautophagy is required for multicellular development
of the social amoeba Dictyostelium discoideum. J Biol Chem.
278:17636–17645. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vitale C, Mendelsohn ME and Rosano GM:
Gender differences in the cardiovascular effect of sex hormones.
Nat Rev Cardiol. 6:532–542. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wenger NK: Clinical characteristics of
coronary heart disease in women: emphasis on gender differences.
Cardiovasc Res. 53:558–567. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu Y, Arenas IA, Armstrong SJ, Plahta WC,
Xu H and Davidge ST: Estrogen improves cardiac recovery after
ischemia/reperfusion by decreasing tumor necrosis factor-alpha.
Cardiovasc Res. 69:836–844. 2006. View Article : Google Scholar
|
|
16
|
Jian X, Xiao-yan Z, Bin H, et al: MiR-204
regulate cardiomyocyte autophagy induced by hypoxia-reoxygenation
through LC3-II. Int J Cardiol. 148:110–112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yue Z, Jin S, Yang C, Levine AJ and Heintz
N: Beclin 1, an autophagy gene essential for early embryonic
development, is a haploinsufficient tumor suppressor. Proc Natl
Acad Sci USA. 100:15077–15082. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Song X, Zhang X, Wang X, et al: Tumor
suppressor gene PDCD4 negatively regulates autophagy by inhibiting
the expression of autophagy-related gene ATG5. Autophagy.
9:743–755. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nishida K, Kyoi S, Yamaguchi O, Sadoshima
J and Otsu K: The role of autophagy in the heart. Cell Death
Differ. 16:31–38. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu MJ, Wang L, Ding HY, Weng CY and Yen
JH: Glossogyne tenuifolia acts to inhibit inflammatory
mediator production in a macrophage cell line by downregulating
LPS-induced NF-kappa B. J Biomed Sci. 11:186–199. 2004.
|
|
21
|
Hou RC, Chen YS, Chen CH, Chen YH and Jeng
KC: Protective effect of 1,2,4-benzenetriol on LPS-induced NO
production by BV2 microglial cells. J Biomed Sci. 13:89–99. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
He B, Xiao J, Ren AJ, et al: Role of miR-1
and miR-133a in myocardial ischemic postconditioning. J Biomed Sci.
18:222011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Whelan RS, Kaplinskiy V and Kitsis RN:
Cell death in the pathogenesis of heart disease: mechanisms and
significance. Annu Rev Physiol. 72:19–44. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Matsui Y, Takagi H, Qu X, et al: Distinct
roles of autophagy in the heart during ischemia and reperfusion:
roles of AMP-activated protein kinase and Beclin 1 in mediating
autophagy. Circ Res. 100:914–922. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wong A, Grubb DR, Cooley N, Luo J and
Woodcock EA: Regulation of autophagy in cardiomyocytes by
Ins(1,4,5)P(3) and IP(3)-receptors. J Mol Cell Cardiol. 54:19–24.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gurusamy N and Das DK: Is autophagy a
double-edged sword for the heart? Acta Physiol Hung. 96:267–276.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Martinet W, Agostinis P, Vanhoecke B,
Dewaele M and De Meyer GR: Autophagy in disease: a double-edged
sword with therapeutic potential. Clin Sci (Lond). 116:697–712.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gottlieb RA and Mentzer RM: Autophagy
during cardiac stress: joys and frustrations of autophagy. Annu Rev
Physiol. 72:45–59. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gottlieb RA, Finley KD and Mentzer RM Jr:
Cardioprotection requires taking out the trash. Basic Res Cardiol.
104:169–180. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Deschamps AM, Murphy E and Sun J: Estrogen
receptor activation and cardioprotection in ischemia reperfusion
injury. Trends Cardiovasc Med. 20:73–78. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Booth EA and Lucchesi BR:
Estrogen-mediated protection in myocardial ischemia-reperfusion
injury. Cardiovasc Toxicol. 8:101–113. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nikolic I, Liu D, Bell JA, Collins J,
Steenbergen C and Murphy E: Treatment with an estrogen
receptor-beta-selective agonist is cardioprotective. J Mol Cell
Cardiol. 42:769–780. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Straface E, Vona R, Gambardella L, et al:
Cell sex determines anoikis resistance in vascular smooth muscle
cells. FEBS Lett. 583:3448–3454. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jeanes HL, Tabor C, Black D, Ederveen A
and Gray GA: Oestrogen-mediated cardioprotection following
ischaemia and reperfusion is mimicked by an oestrogen receptor
(ER)alpha agonist and unaffected by an ER beta antagonist. J
Endocrinol. 197:493–501. 2008. View Article : Google Scholar
|
|
35
|
Lizotte E, Grandy SA, Tremblay A, Allen BG
and Fiset C: Expression, distribution and regulation of sex steroid
hormone receptors in mouse heart. Cell Physiol Biochem. 23:75–86.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Booth EA, Obeid NR and Lucchesi BR:
Activation of estrogen receptor-alpha protects the in vivo rabbit
heart from ischemia-reperfusion injury. Am J Physiol Heart Circ
Physiol. 289:H2039–H2047. 2005. View Article : Google Scholar : PubMed/NCBI
|