Introduction
Methamphetamine (MA) is a highly addictive
psychostimulant that belongs to the phenethylamine and amphetamine
class of psychoactive drugs. Its hydrochloride is a colorless
transparent crystal, commonly termed ‘ice’. MA is a potent and
addictive drug of abuse that has long been associated with a myriad
of adverse effects. It is known to cause harm to the human brain,
heart, liver and kidney. The majority of studies have focused on
the neurotoxicity and neuropsychological deficits caused by MA
(1–3). MA was reported to cause widespread
apoptosis in the mouse brain, including in the striatum, cortex and
hippocampus indicating that apoptosis is an important molecular
mechanism involved in MA-induced neurotoxicity (4). Previous studies have demonstrated
that MA is able to cause neuropathological alterations in the
rodent brain via apoptotic mechanisms similar to those reported in
various models of neuronal death (5–7). The
accumulated data support that MA-induced cell death is accompanied
by activation of several pathways whose roles have been well
documented in other models of neuronal apoptosis.
There has been increasing concern that prescription
stimulant use may be linked to adverse cardiovascular
complications. Chronic use or acute overdose of MA may lead to, not
only psychological but also physical harm, primarily consisting of
cardiovascular damage (8).
Previous studies have demonstrated evidence of MA-associated acute
and chronic cardiovascular pathology (9). To date, MA-induced acute myocardial
infarction has been overwhelmingly reported (10–15).
In addition to this, much of the evidence for MA-induced
cardiomyopathy is also present in humans. Cardiomyopathy is
typically a chronic disease of gradual onset that is often
associated with chronic MA use (16–18).
Cardiomyocyte apoptosis is important in the progression of heart
failure. In patients with end-stage cardiomyopathy, loss of
cardiomyocytes due to apoptosis is observed leading to the
progression of cardiac dysfunction and ultimately heart failure
(19–22).
However, whether MA is involved in regulating
cardiomyocyte apoptosis in MA-induced myocardial injury remains to
be elucidated. The mechanisms underlying MA-induced cardiotoxicity
have not yet been clearly elucidated with respect to the apoptotic
pathway. In addition, no effective treatment exists for the adverse
effects of MA. Therefore, in the present study, it was hypothesized
that identifying a novel target to inhibit apoptosis could
partially reduce the symptoms caused by MA and protect
cardiomyocytes from apoptosis.
To elucidate the possible mechanism underlying
MA-induced toxicity, our laboratory performed gene expression
microarray analysis and revealed that insulin-like growth factor
binding protein-5 (IGFBP5) was significantly upregulated in cells
(data not shown). Insulin-like growth factor binding proteins
comprise a family of proteins that bind and regulate the functions
of insulin-like growth factors (IGFs). IGFBP5 is involved in
apoptotic processes in various cell types. In prostate and mammary
glands and in the rat brain following hypoxic ischemic injury,
increased expression of IGFBP5 has been associated with apoptotic
processes (23–25).
The aim of the present study was to investigate
cardiomyocyte apoptosis induced by MA. It was hypothesized that
IGFBP5 may also be involved in the regulation of cardiomyocyte
apoptosis induced by MA. However, it is not known whether silencing
IGFBP5 is able to protect against the loss of cardiomyocytes due to
apoptosis. Silencing through small interfering (si)RNA is a natural
mechanism for the suppression of specific gene activity that is
highly conserved in evolution. The use of siRNAs in mammals has
been extremely useful for understanding the biological role of
several genes using a relatively simple and efficient technique
(26). In the present study,
siRNA-mediated gene silencing was performed to investigate the
knockdown of IGFBP5 expression.
Materials and methods
Cell culture
NRVMs were prepared from 0–1 day-old neonatal
Sprague-Dawley rats (Experimental Animal Center of Southern Medical
University, Gangzhou, Guangdong, China). Rats were sacrificed by
immersion in 75% alcohol. Ventricles were removed and washed in
Hank’s solution, then minced and incubated with 0.25% trypsinase at
4°C for 12–16 h. Dulbecco’s modified Eagle’s medium (Sigma-Aldrich,
Shanghai, China) containing 10% fetal bovine serum (Corning Inc.,
New York, NY, USA) was added to terminate digestion for 5 min at
37°C. The supernatant was discarded. Hank’s solution (25 ml;
Gibo-BRL, Paisley, UK) was supplemented with 25 mg collagenase type
II (Sigma-Aldrich) and 125 mg bovine serum albumin (Roche Applied
Science, Indeanapolis, IN, USA) as digestive solution. Digestive
solution was added and placed in a water bath for 1 min at 37°C.
The supernatant was discarded. Subsequently, fresh digestive
solution was added and placed in a water bath on the top of a hot
plate stirrer stirring the tissue fragments with a magnetic bar for
15 min at 37°C and the supernatant was collected. The latter
digestion step was repeated four times. Cells in the supernatant
were isolated by centrifugation for 10 min at 2,000 × g at room
temperature. In order to reduce fibroblast contamination, cells
resuspended in NRVMs culture medium were pre-plated for 1 h. The
supernatant was aspirated gently and cells were plated onto
six-well plates. Cells were incubated at 37°C in a humidified
atmosphere containing 5% CO2 during the experiments. All
animal procedures were conducted in accordance with the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals (Bethesda, MA, USA) and were approved by the Animal Care
and Use Committees of the Southern Medical University.
Transfection of IGFBP5 siRNA
NRVMs were transfected with siRNA using
Lipofectamine 2000 reagent (Invitrogen, Shanghai, China), according
to the manufacturer’s instructions. NRVMs were grown to 80–90%
confluency. The cells were then transfected with IGFBP5 siRNA and
Lipofectamine for 6 h in OptiMEM (Gibco-BRL, Paisley, UK). The siNC
was added as a nonspecific control. The complex medium was replaced
after 6 h incubation at 37°C with the same volume of fresh culture
medium. The cells were maintained in an incubator for 48 h. At 48
h, NRVMs of the MA-exposure groups were treated with 1.5 mM MA for
48 h.
The following two sequences targeting IGFBP5 by the
siRNA method were used: siIGFBP5-1, 5′-CGC GTC CCC GGA AGG AAT TCT
GGA AGA TAT TTC AAG AGA ATA TCT TCC AGA ATT CCT TCC TTT TTG GAA
AT-3′; siIGFBP5-2, 5′-CGC GTC CCC GGA AGA TAT GCC TGT GGA TCC TTC
AAG AGA GGA TCC ACA GGC ATA TCT TCC TTT TTG GAA AT-3′.
RNA extraction and quantitative
(q)PCR
Total RNA was extracted using RNAiso Plus (Takara
Bio, Inc., Shiga, Japan) according to the manufacturer’s
instructions. The concentration and OD260/OD280 value of total RNA
were measured using a Beckman Coulter DU 520UV/Vis
spectrophotometer (Beckman Coulter, Miami, FL, USA). Reverse
transcription was conducted starting from 1 μg of total RNA using a
PrimeScript RT reagent kit with gDNA Eraser (Takara Bio, Inc.) and
excluded any potential genomic DNA contamination. The mRNA levels
were measured by RT-PCR with an Applied Biosystems 7500 fast
real-time PCR system (Applied Biosystems, Foster City, CA, USA)
using SYBR Premix Ex Taq II (Takara Bio, Inc.). The PCR cycling
conditions were as follows: 2 min at 50°C, 30 sec at 95°C, 40
cycles of 15 sec at 95°C and 34 sec at 60°C, followed by a
dissociation stage. The IGFBP5 fragment was amplified using the
following primers: IGFBP5, forward 5′-ATG AAG CTG CCG GGC-3′ and
reverse 5′-TCA ACG TTA CTG CTG TCG AAG-3′. RNA content was
normalized to 18S ribosomal RNA and relative changes in gene
expression were quantified using the threshold cycle
(2−ΔΔCt) method with RQ software (Applied
Biosystems).
Western blotting
Total protein was extracted for western blotting.
Samples were homogenized in ice-cold RIPA buffer (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA) containing protease
inhibitors. The protein concentration of the samples was determined
using a BCA protein assay kit (Thermo Fisher Scientific, Waltham,
MA, USA). Protein samples were separated by sodium dodecyl sulfate
polyacrylamide gel electrophoresis and transferred onto
polyvinylidene difluoride membranes (Millipore, Billerica, MA,
USA). Non-specific binding was blocked with 5% non-fat skim milk in
Tris-buffered saline with Tween 20 (TBST) at room temperature for 1
h. Membranes were incubated with rabbit polyclonal anti-IGFBP5
antibody (1:1,000; Abcam, Cambridge, UK) and rabbit polyclonal
anti-caspase-3 antibody (1:200, Santa Cruz Biotechnology, Inc.) at
4°C overnight. Following being washed with TBST, membranes were
reacted with horseradish peroxidase-conjugated goat anti-rabbit IgG
(1:2,000; Amersham Biosciences, Tokyo, Japan) at room temperature
for 1 h. The bound secondary antibodies were visualized with Thermo
Scientific Pierce ECL Western Blotting Substrate (Thermo Scientific
Pierce, Rockford, IL, USA) according to the manufacturer’s
instructions. Signal intensities of bands were analyzed and the
relative protein levels were calculated by comparison with the
quantity of β-actin as a loading control.
Evaluation of apoptosis
Apoptotic cells were quantified using a terminal
deoxyribonucleotide transferase-mediated dUTP nick end-labeling
(TUNEL) assay (Roche Applied Science, Indianapolis, IN, USA)
according to the manufacturer’s instructions. NRVMs were fixed in
4% paraformaldehyde (Dingguo Changsheng, Beijing, China) in fresh
phosphate-buffered saline (pH 7.4; Dingguo Changsheng) at 4°C for 1
h, incubated with fluorescein-conjugated terminal deoxynucleotidyl
transferase enzyme for 1 h at 37°C in the dark, and then mounted
with 4′,6′-diamidino-2-phenylindole (Dingguo Changsheng) for
nuclear counterstaining. Cross-sections were imaged (20× and 40×
objective) using a fluorescent microscope (Nikon, Tokyo, Japan).
The apoptotic rate was calculated as follows: (number of apoptosis
cells / total number of cells) × 100%.
Statistical analysis
Statistical analyses of the data were performed
using SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA). Statistics
were performed using one way analysis of variance and Student’s
t-test. The results are presented as the mean ± standard deviation
and P<0.05 was considered to indicate a statistically
significant difference.
Results
MA induces apoptosis in NRVMs
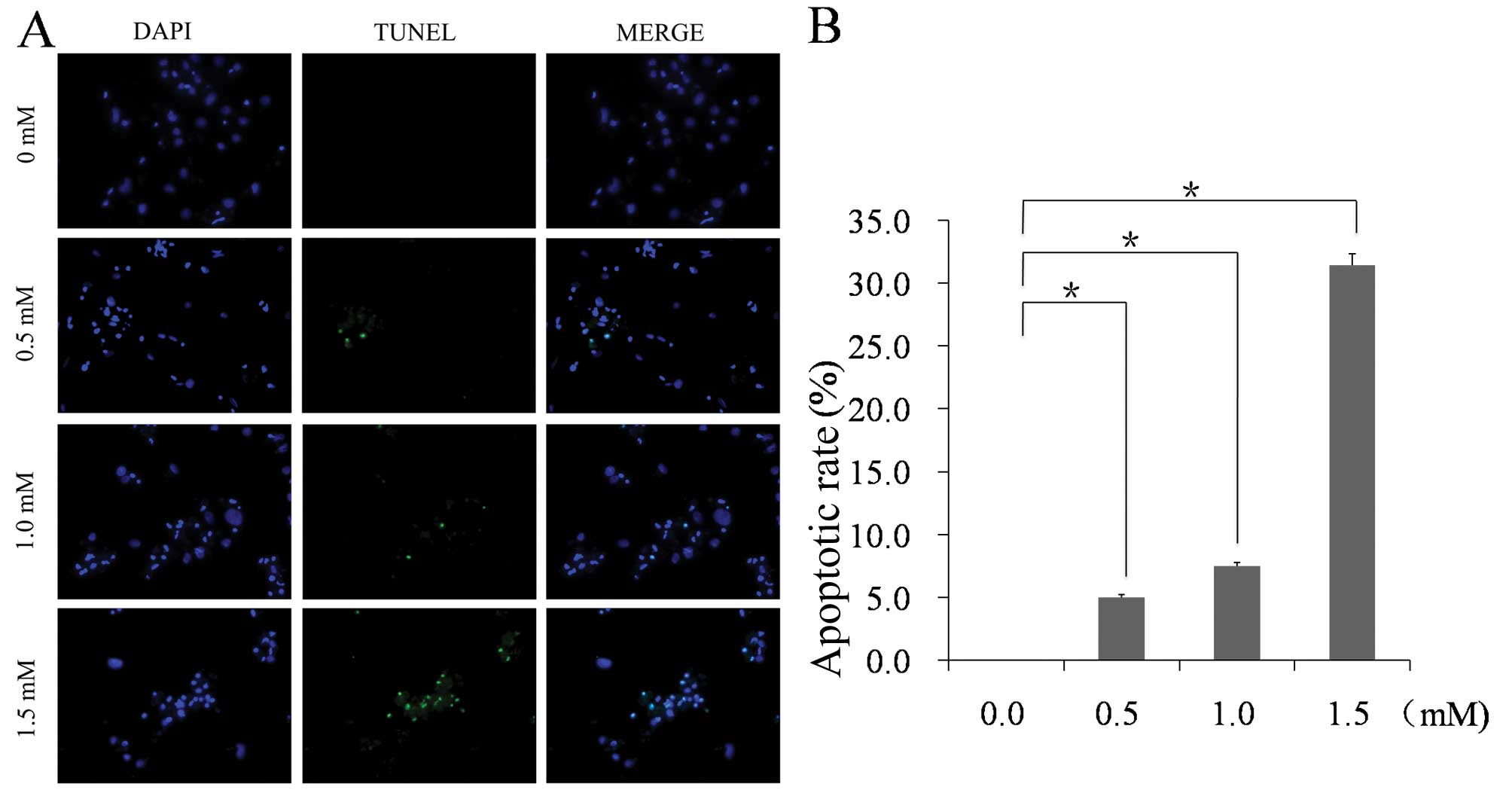
A TUNEL assay was performed to detect DNA damage in
NRVMs. The results indicated that the apoptotic rate increased in
NRVMs exposed to 1.5 mM MA relative to the control cells
(31.43±0.50 vs. 0.00±0.00%; n=5; P<0.01). No
TUNEL-positive cells were observed in the control group, whereas a
small amount of TUNEL-positive cells were observed in the 0.5 mM MA
group (5.00±0.23%), a moderate amount in the 1.0 mM MA group
(7.50±0.30%) and a significantly increased amount of TUNEL-positive
cells were observed in the 1.5 mM MA group (Fig. 1). The images show representative
TUNEL staining from three independent experiments.
MA increases IGFBP5 mRNA and protein
expression in NRVMs
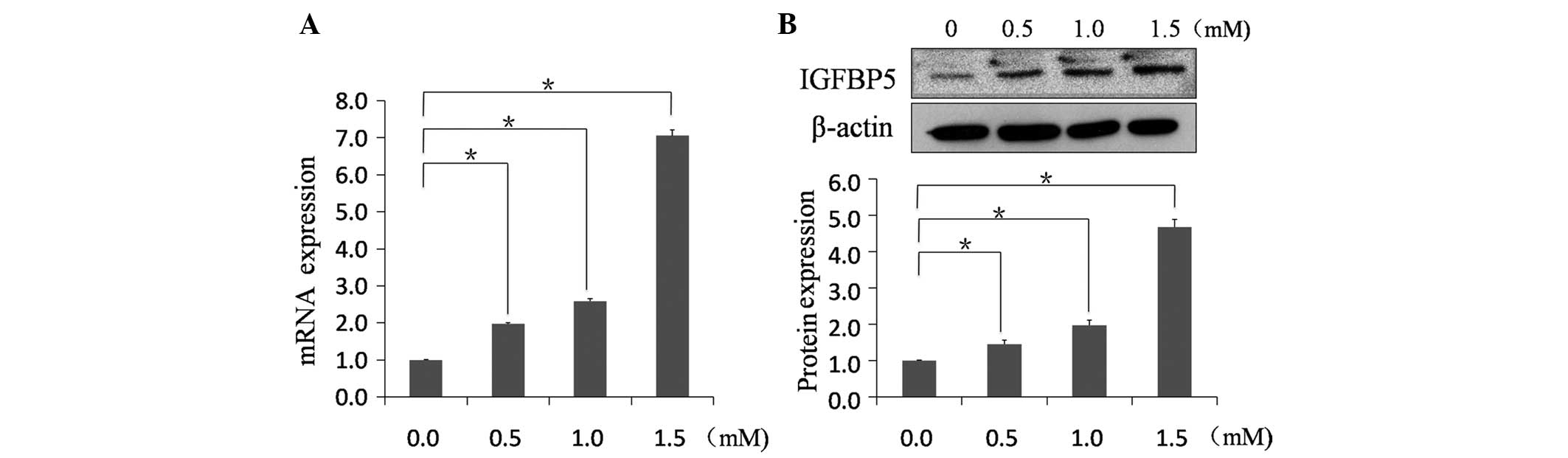
IGFBP5 mRNA was identified in NRVMs using qPCR
(Fig. 2A). Melting curve analysis
confirmed the specificity of transcripts of IGFBP5 in NRVMs. To
evaluate how MA affects IGFBP5 expression, NRVMs were exposed to
varying concentrations of MA. MA increased the mRNA expression of
IGFBP5 in a dose-dependent manner. The mRNA expression of IGFBP5
significantly increased in the 1.5 mM MA group by 7.06±0.16 fold
(n=6; P<0.01). IGFBP5 protein expression was also
evaluated in MA-treated NRVMs (Fig.
2B). Immunoblotting using anti-IGFBP5 antibody indicated that
the molecular weight of the IGFBP5 protein was 31 kDa. MA increased
IGFBP5 protein expression in MA-treated NRVMs to 4.67±0.21 fold
(n=6; P<0.01).
siIGFBP5 knockdown of the IGFBP5 protein
in NRVMs
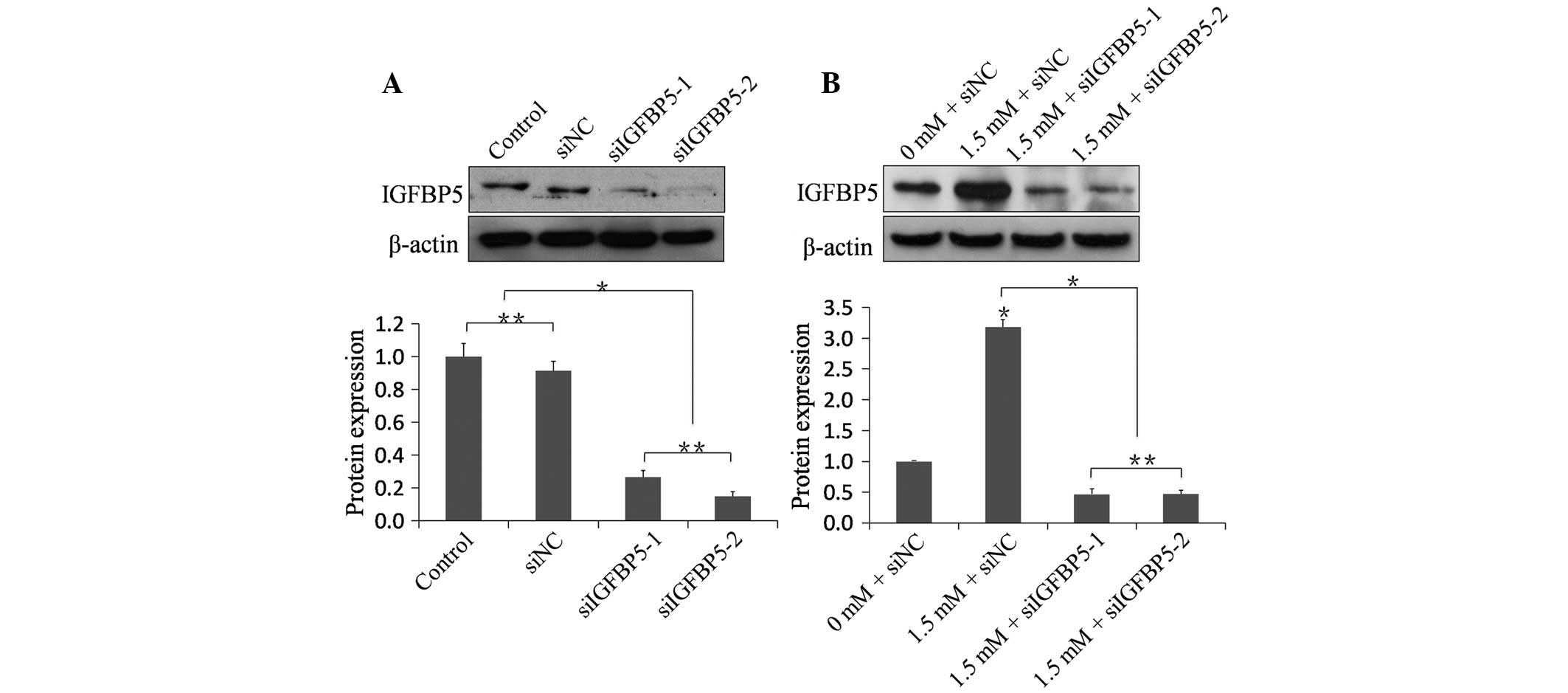
The knockdown efficacy of siIGFBP5 on the protein
level was investigated. To verify the efficacy of synthetic
siRNA-mediated silencing of IGFBP5, two sequences (siIGFBP5-1 and
siIGFBP5-2) that target IGFBP5 were selected. The siNC was used as
a nonspecific control. Western blot analysis demonstrated that siNC
had no silencing effect. The siIGFBP5-1 and siIGFBP5-2 groups
exhibited a significant decrease in IGFBP5 compared with the siNC
group and the control group, demonstrating that siIGFBP5-1 and
siIGFBP5-2 could efficiently knockdown IGFBP5 at the protein level
(Fig. 3A). Whether the knockdown
efficacy of siIGFBP5 could be affected following MA treatment was
also investigated. Following transfection, NRVMs of MA-treated
groups were treated with 1.5 mM MA for 48 h. The 1.5 mM + siNC
group exhibited a significant increase in IGFBP5 compared with the
0 mM + siNC group (n=6; P<0.01). The 1.5 mM + siIGFBP5-1
group and the 1.5 mM + siIGFBP5-2 group exhibited a significant
decrease in IGFBP5 compared with the 1.5 mM + siNC group,
respectively (n=6; P<0.01), demonstrating that siIGFBP5-1
and siIGFBP5-2 could efficiently knockdown IGFBP5 at the protein
level even following treatment with MA (Fig. 3B).
Transfection with siIGFBP5 reduces the
apoptotic rate in NRVMs induced by MA
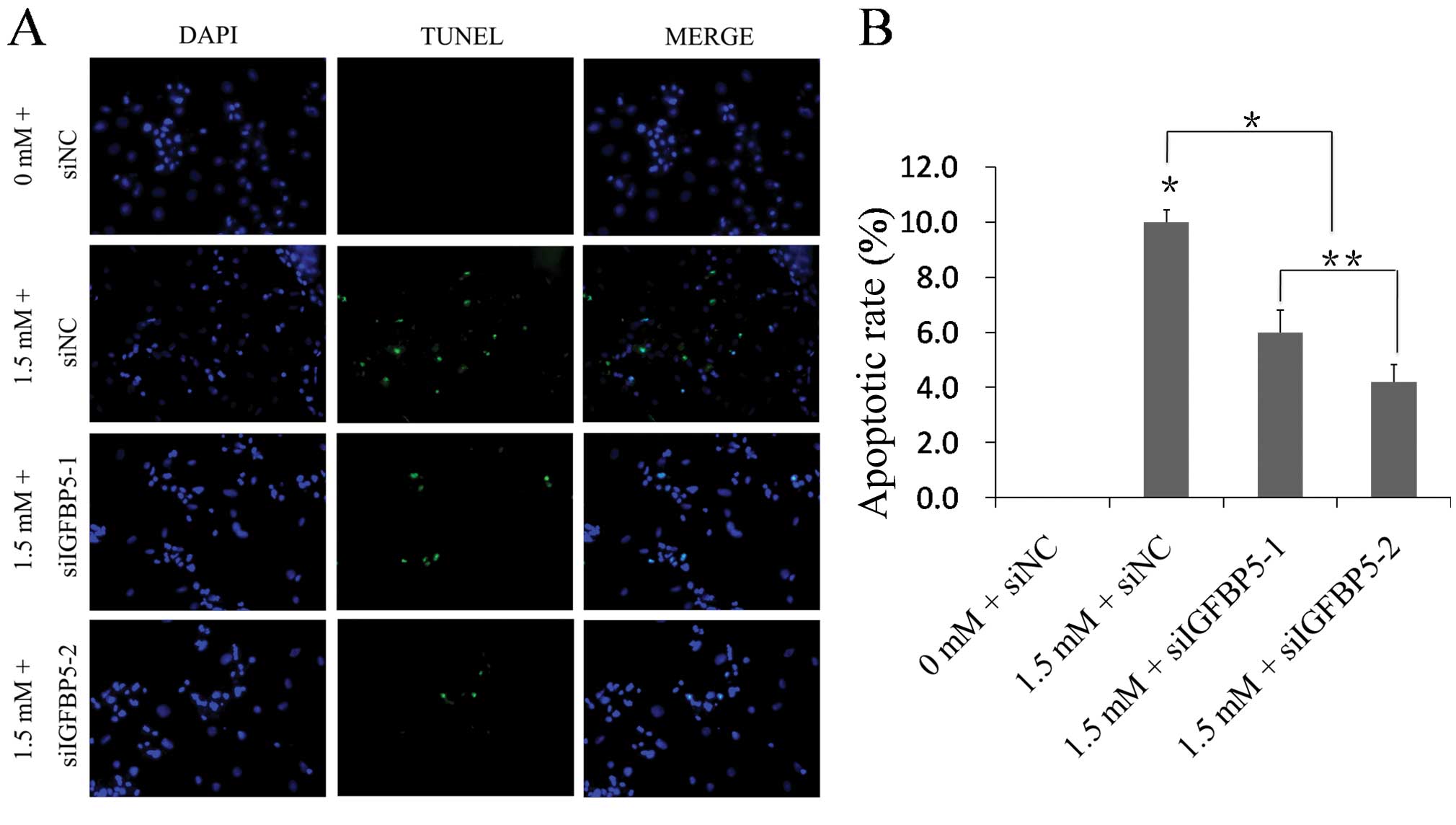
Following showing that MA induces apoptosis in
NRVMs, it was examined whether silencing the IGFBP5 gene is able to
reduce MA-associated impairment in vitro. A TUNEL assay was
performed to detect DNA damage in NRVMs. The results indicated that
the apoptotic rate increased in the 1.5 mM + siNC group relative to
the 0 mM + siNC group (10.00±0.45 vs. 0.00±0.00%; n=5;
P<0.01; Fig. 4). The apoptotic
rate decreased in the 1.5 mM + siIGFBP5-1 group and the 1.5 mM +
siIGFBP5-2 group relative to the 1.5 mM + siNC group, respectively
(6.00±0.80 vs. 10.00±0.45%; 4.20±0.64 vs. 10.00±0.45%; n=5;
P<0.01; Fig. 4). The images
show representative TUNEL staining from three independent
experiments.
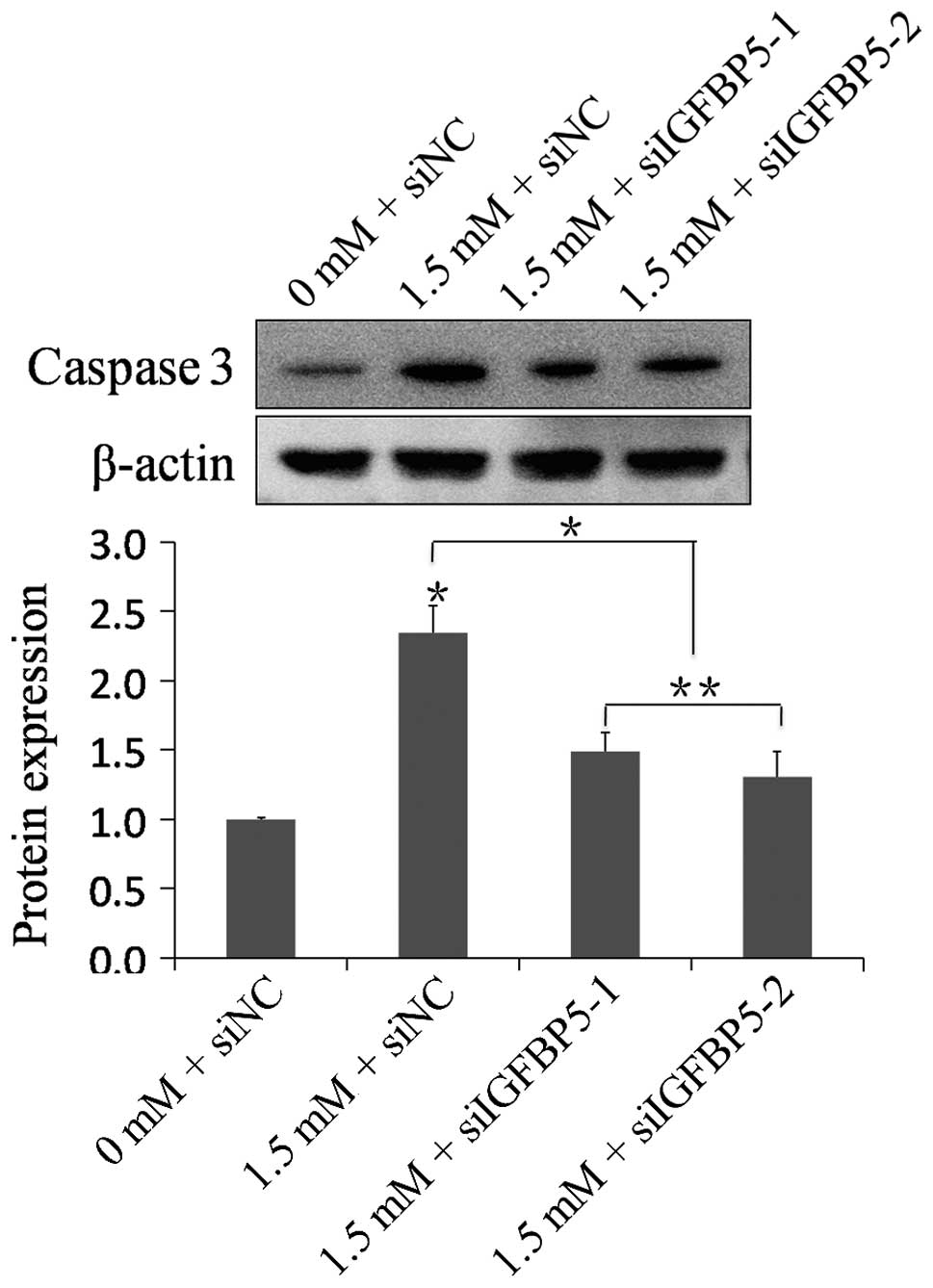
Expression of caspase-3
To investigate whether apoptosis occurred through
caspase-3, the expression of caspase-3 was analyzed by western
blotting. Compared with the 0 mM + siNC group, the expression of
caspase-3 was significantly increased in the 1.5 mM + siNC group
(n=6, P<0.01). The 1.5 mM + siIGFBP5-1 group and the 1.5
mM + siIGFBP5-2 group exhibited a significant decrease in caspase-3
expression compared with the 1.5 mM + siNC group, respectively
(n=6, P<0.01; Fig. 5).
Silencing IGFBP5 with siRNA significantly suppressed the expression
of caspase-3 in NRVMs following MA treatment.
Discussion
The present study examined whether silencing IGFBP5
is able to protect against the loss of cardiomyocytes due to
MA-induced apoptosis. The accumulated evidence supports the idea
that MA can lead to the death of cells via apoptosis. MA is known
to trigger neuronal, splenic and thymic damage via apoptosis
(27,28). The cardiotoxic action of MA has
been of particular interest since standardized doses consistently
produces myocardial lesions (29,30).
To date, studies have focused on MA abusers that almost always have
severe damage in their cardiovascular system, yet less is known
regarding cardiac damage via cardiomyocyte apoptosis.
The effect of MA on cardiomyocyte apoptosis, an
important component of cardiac remodeling leading to heart failure,
was analyzed (19–22). Our data using TUNEL staining
demonstrated that MA induces apoptosis in NRVMs. With varying
concentrations (0, 0.5, 1.0 or 1.5 mM) of MA treatment, the
apoptotic rate increased as the MA concentration increased. A
significant increase in the apoptotic rate was detected after the
NRVMs were treated with 1.5 mM MA (Fig. 1).
Identifying proteins associated with MA-induced
apoptosis and silencing them is an essential step in developing
therapeutics. In a previous study, our laboratory identified
proteins associated with the pathogenesis of MA-induced toxicity by
proteomic profiling in the rat brain, including oxidative stress,
degeneration, apoptosis, mitochondrial pathway and energy
metabolism (31). To probe more
possible proteins, our laboratory also performed gene expression
microarray analysis and revealed that IGFBP5 was significantly
upregulated following MA exposure (data not shown).
Regulation of IGFs by IGFBP5 was known to affect
cell proliferation, differentiation and survival. In particular,
IGFBP5 has cell-type and tissue-type specific effects and its role
in cell growth is complicated. IGFBP5 affects cell survival and
apoptosis in a variety of cells, and the response to IGFBP5 depends
on the cell type. IGFBP5 induces apoptosis in mammary epithelial
cells (24), breast cancer cells
(32) and osteosarcoma cells
(33) but prevents apoptosis in
neuroblastoma cells (34), C2
myoblasts (35) and human stellate
cells (36). The increased
expression of IGFBP5 may be involved in MA-induced toxicity,
therefore we further postulate that IGFBP5 is one of the novel
target involved in MA-induced apoptosis.
The results from the present study provided evidence
that IGFBP5 expression at the transcriptional and translational
levels were increased by MA treatment (Fig. 2), which is consistent with our
hypothesis that IGFBP5 is involved in MA-induced cardiotoxicity.
The mRNA and protein levels of IGFBP5 increased in a dose-dependent
manner in NRVMs, and were significantly upregulated following
treatment with 1.5 mM MA. The concentration of 1.5 mM MA was
selected for our subsequent studies. To the best of our knowledge,
this is the first study to report the increased expression of
IGFBP5 in MA-treated NRVMs.
Previous studies have demonstrated that the
expression of IGFBP5 is implicated in cardiovascular disease and
even heart failure, which further suggests a novel role for IGFBP5
in the heart (37–40). Silencing of IGFBP5 expression
allowed examination of its distinct cellular, physiological and
pathophysiological functions (38,40).
Synthetic siRNA targeting of the IGFBP5 gene was applied to silence
IGFBP5 expression. Our data demonstrated that synthetic siIGFBP5
could efficiently knockdown IGFBP5 expression, and the nonspecific
silencing control siNC demonstrated no effect on the expression of
IGFBP5 (Fig. 3). Observations from
the present study demonstrated that MA significantly increased the
expression of IGFBP5 compared with the group not treated with MA.
The efficient knockdown of IGFBP5 expression also presented in
NRVMs with MA treatment was preceded by siIGFBP5 transfection
(Fig. 3B).
IGFBP5 may induce or inhibit apoptosis in a variety
of cells, and in the present study, the link between IGFBP5 and
apoptosis induced by MA in NRVMs was observed. IGFBP5 was found to
trigger apoptosis of NRVMs following exposure to MA. Furthermore,
the results from the TUNEL assay demonstrated that silencing IGFBP5
expression protected NRVMs from MA-induced apoptosis to a certain
degree (Fig. 4). IGFBP5 knockdown
attenuated MA-induced apoptosis of cardiomyocytes. Our findings are
consistent with the hypothesis that IGFBP5 may be involved in the
loss of cardiomyocytes via apoptotic processes, and this evidence
further suggested that IGFBP5 may have an important pathological
role in MA-induced cardiotoxicity. However, the mechanisms
contributing to this protective effect require further elucidation
in future studies.
Activation of the caspase family is known to be a
crucial mechanism for inducing apoptotic death signals.
Pro-apoptotic signals trigger a cascade of caspases, which lead to
the cleavage of a set of proteins, resulting in disassembly of the
cell (41,42). Caspase-3 is the primary activator
of apoptotic DNA fragmentation (43). The present study indicated that MA
increased the expression of caspase-3. Silencing IGFBP5 with siRNA
significantly suppressed the expression of caspase-3 in NRVMs
following MA treatment. This indicated that the suppression or
induction of apoptosis was mediated by a caspase-3-dependent
pathway.
In conclusion, it is clear that IGFBP5 is involved
in MA-induced apoptosis. The present study investigated
cardiomyocyte apoptosis and the expression of IGFBP5 in MA-exposed
NRVMs. To the best of our knowledge, the present study provided the
first evidence that IGFBP5 is a potential regulator of MA-induced
apoptosis. Additionally, silencing the expression of IGFBP5 is able
to significantly protect cardiomyocytes from MA-induced apoptosis.
Accordingly, the present study suggested that IGFBP5 is a potential
target for the treatment of MA-induced apoptosis. It was further
postulated that IGFBP5 provides a new mode for the regulation of
apoptosis associated with MA-associated cardiac diseases.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 81102301 and 81172907).
References
|
1
|
Scott JC, Woods SP, Matt GE, et al:
Neurocognitive effects of methamphetamine: a critical review and
meta-analysis. Neuropsychol Rev. 17:275–297. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shrem MT and Halkitis PN: Methamphetamine
abuse in the United States: contextual, psychological and
sociological considerations. J Health Psychol. 13:669–679. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carvalho M, Carmo H, Costa VM, et al:
Toxicity of amphetamines: an update. Arch Toxicol. 86:1167–1231.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Deng X, Wang Y, Chou J and Cadet JL:
Methamphetamine causes widespread apoptosis in the mouse brain:
evidence from using an improved TUNEL histochemical method. Brain
Res Mol Brain Res. 93:64–69. 2001. View Article : Google Scholar
|
|
5
|
Jayanthi S, Deng X, Noailles PA, Ladenheim
B and Cadet JL: Methamphetamine induces neuronal apoptosis via
cross-talks between endoplasmic reticulum and
mitochondria-dependent death cascades. FASEB J. 18:238–251. 2004.
View Article : Google Scholar
|
|
6
|
Cadet JL, Jayanthi S and Deng X: Speed
kills: cellular and molecular bases of methamphetamine-induced
nerve terminal degeneration and neuronal apoptosis. FASEB J.
17:1775–1788. 2003. View Article : Google Scholar
|
|
7
|
Li Y, Hu Z, Chen B, et al: Taurine
attenuates methamphetamine-induced autophagy and apoptosis in PC12
cells through mTOR signaling pathway. Toxicol Lett. 215:1–7. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Darke S, Kaye S, McKetin R and Duflou J:
Major physical and psychological harms of methamphetamine use. Drug
Alcohol Rev. 27:253–262. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaye S, McKetin R, Duflou J and Darke S:
Methamphetamine and cardiovascular pathology: a review of the
evidence. Addiction. 102:1204–1211. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Costa GM, Pizzi C, Bresciani B, et al:
Acute myocardial infarction caused by amphetamines: a case report
and review of the literature. Ital Heart J. 2:478–480.
2001.PubMed/NCBI
|
|
11
|
Waksman J, Taylor RN Jr, Bodor GS, et al:
Acute myocardial infarction associated with amphetamine use. Mayo
Clin Proc. 76:323–326. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sztajnkrycer MD, Hariharan S and Bond GR:
Cardiac irritability and myocardial infarction in a 13-year-old
girl following recreational amphetamine overdose. Pediatr Emerg
Care. 18:E11–E15. 2002.PubMed/NCBI
|
|
13
|
Hung MJ, Kuo LT and Cherng WJ:
Amphetamine-related acute myocardial infarction due to coronary
artery spasm. Int J Clin Pract. 57:62–64. 2003.PubMed/NCBI
|
|
14
|
Brennan K, Shurmur S and Elhendy A:
Coronary artery rupture associated with amphetamine abuse. Cardiol
Rev. 12:282–283. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Watts DJ and McCollester L:
Methamphetamine-induced myocardial infarction with elevated
troponin I. Am J Emerg Med. 24:132–134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hong R, Matsuyama E and Nur K:
Cardiomyopathy associated with the smoking of crystal
methamphetamine. JAMA. 265:1152–1154. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wijetunga M, Seto T, Lindsay J and Schatz
I: Crystal methamphetamine-associated cardiomyopathy: tip of the
iceberg? J Toxicol Clin Toxicol. 41:981–986. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Crean AM and Pohl JE: ‘Ally McBeal heart?’
- drug induced cardiomyopathy in a young woman. Br J Clin
Pharmacol. 58:558–559. 2004.
|
|
19
|
Olivetti G, Abbi R, Quaini F, et al:
Apoptosis in the failing human heart. N Engl J Med. 336:1131–1141.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Takemura G and Fujiwara H: Role of
apoptosis in remodeling after myocardial infarction. Pharmacol
Ther. 104:1–16. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Narula J, Haider N, Virmani R, et al:
Apoptosis in myocytes in end-stage heart failure. N Engl J Med.
335:1182–1189. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Narula J, Kolodgie FD and Virmani R:
Apoptosis and cardiomyopathy. Curr Opin Cardiol. 15:183–188. 2000.
View Article : Google Scholar
|
|
23
|
Nickerson T, Pollak M and Huynh H:
Castration-induced apoptosis in the rat ventral prostate is
associated with increased expression of genes encoding insulin-like
growth factor binding proteins 2,3,4 and 5. Endocrinology.
139:807–810. 1998. View Article : Google Scholar
|
|
24
|
Tonner E, Barber MC, Allan GJ, et al:
Insulin-like growth factor binding protein-5 (IGFBP-5) induces
premature cell death in the mammary glands of transgenic mice.
Development. 129:4547–4557. 2002.PubMed/NCBI
|
|
25
|
Beilharz EJ, Klempt ND, Klempt M, et al:
Differential expression of insulin-like growth factor binding
proteins (IGFBP) 4 and 5 mRNA in the rat brain after transient
hypoxic-ischemic injury. Brain Res Mol Brain Res. 18:209–215. 1993.
View Article : Google Scholar
|
|
26
|
McManus MT and Sharp PA: Gene silencing in
mammals by small interfering RNAs. Nat Rev Genet. 3:737–747. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Iwasa M, Maeno Y, Inoue H, Koyama H and
Matoba R: Induction of apoptotic cell death in rat thymus and
spleen after a bolus injection of methamphetamine. Int J Legal Med.
109:23–28. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Davidson C, Gow AJ, Lee TH and Ellinwood
EH: Methamphetamine neurotoxicity: necrotic and apoptotic
mechanisms and relevance to human abuse and treatment. Brain Res
Brain Res Rev. 36:1–22. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Smith HJ, Roche AH, Jausch MF and Herdson
PB: Cardiomyopathy associated with amphetamine administration. Am
Heart J. 91:792–797. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kalant H and Kalant OJ: Death in
amphetamine users: causes and rates. Can Med Assoc J. 112:299–304.
1975.PubMed/NCBI
|
|
31
|
Li X, Wang H, Qiu P and Luo H: Proteomic
profiling of proteins associated with methamphetamine-induced
neurotoxicity in different regions of rat brain. Neurochem Int.
52:256–264. 2008. View Article : Google Scholar
|
|
32
|
Butt AJ, Dickson KA, Jambazov S and Baxter
RC: Enhancement of tumor necrosis factor-alpha-induced growth
inhibition by insulin-like growth factor-binding protein-5
(IGFBP-5), but not IGFBP-3 in human breast cancer cells.
Endocrinology. 146:3113–3122. 2005. View Article : Google Scholar
|
|
33
|
Su Y, Wagner ER, Luo Q, et al:
Insulin-like growth factor binding protein 5 suppresses tumor
growth and metastasis of human osteosarcoma. Oncogene.
30:3907–3917. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tanno B, Vitali R, De Arcangelis D, et al:
Bim-dependent apoptosis follows IGFBP-5 down-regulation in
neuroblastoma cells. Biochem Biophys Res Commun. 351:547–552. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cobb LJ, Salih DA, Gonzalez I, et al:
Partitioning of IGFBP-5 actions in myogenesis: IGF-independent
anti-apoptotic function. J Cell Sci. 117:1737–1746. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sokolović A, Sokolović M, Boers W,
Elferink RP and Bosma PJ: Insulin-like growth factor binding
protein 5 enhances survival of LX2 human hepatic stellate cells.
Fibrogenesis Tissue Repair. 3:32010.PubMed/NCBI
|
|
37
|
Fischer F, Schulte H, Mohan S, et al:
Associations of insulin-like growth factors, insulin-like growth
factor binding proteins and acid-labile subunit with coronary heart
disease. Clin Endocrinol (Oxf). 61:595–602. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kim KS, Seu YB, Baek SH, et al: Induction
of cellular senescence by insulin-like growth factor binding
protein-5 through a p53-dependent mechanism. Mol Biol Cell.
18:4543–4552. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Godard MP, Whitman SA, Song YH and
Delafontaine P: Skeletal muscle molecular alterations precede
whole-muscle dysfunction in NYHA Class II heart failure patients.
Clin Interv Aging. 7:489–497. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lee DH, Kim JE and Kang YJ: Insulin like
growth factor binding protein-5 regulates excessive vascular smooth
muscle cell proliferation in spontaneously hypertensive rats via
ERK 1/2 phosphorylation. Korean J Physiol Pharmacol. 17:157–162.
2013. View Article : Google Scholar
|
|
41
|
Enari M, Sakahira H, Yokoyama H, et al: A
caspase-activated DNase that degrades DNA during apoptosis, and its
inhibitor ICAD. Nature. 391:43–50. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Thornberry NA and Lazebnik Y: Caspases:
enemies within. Science. 281:1312–1316. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wolf BB, Schuler M, Echeverri F and Green
DR: Caspase-3 is the primary activator of apoptotic DNA
fragmentation via DNA fragmentation factor-45/inhibitor of
caspase-activated DNase inactivation. J Biol Chem. 274:30651–30656.
1999. View Article : Google Scholar : PubMed/NCBI
|