Introduction
Chronic obstructive pulmonary disease (COPD) is
characterized by the progressive obstruction of airflow, which is
accompanied by an increased chronic inflammatory response in the
airway and lung parenchyma (1).
The inflammatory response is induced by deleterious particles or
gases (1). Airway inflammation and
remodeling induced by cigarette smoke, particularly the enlargement
of alveolar airspaces and remodeling of small airways, are the
pathological basis of airflow obstruction (2–4).
However, the inflammatory response in the airway precedes typical
pathological changes (5).
Therefore, investigation of the cellular and molecular mechanisms
underlying the early involvement of chronic smoking on the
inflammatory response and its pathological damage to lung tissue is
important for understanding the pathogenesis of COPD and for
providing novel treatment strategies for early intervention.
Smoking cessation relieves airflow obstruction and
the inflammatory response (6,7).
However, previous human and animal experiments have demonstrated
that, once COPD is initiated, the pulmonary inflammatory response
continues (8–10) and the enlarged alveolar airspace
cannot be reversed following smoking cessation (11). The outcome of collagen deposition
in the walls of the small airways remains to be elucidated.
Furthermore, the pathological and inflammatory outcomes in the lung
following cessation in the early stages of chronic smoking, prior
to the complete development of emphysema, remain unclear.
The present study observed the levels of
inflammation and lesions in the lungs of mice exposed to cigarette
smoke, and the changes following smoking cessation. The study also
aimed to investigate the effects of early smoking cessation, and to
examine the pathogenic mechanisms occurring at a stage when COPD
may have an increased possibility of being reversed.
Materials and methods
Ethics
The present study was approved by the Ethics
Committee of Shengjing First Affiliated Hospital of China Medical
University (Shenyang, China).
Animals and study procedures
A total of 25 (5 in each group) female specific
pathogen-free (SPF) C57BL/6 J mice (8–12-weeks-old; 20.2±2.1 g)
were purchased from the Experimental Animal Centre, Shengjing
Hospital of China Medical University. The mice were bred in SPF
conditions and were maintained at a constant temperature and
humidity, with ad libitum access to food and water.
The mice were randomized into five groups. In the
subacute smoking group, the mice were exposed to cigarette smoke
for 4 weeks. In the 1-week smoking cessation group, the mice were
exposed to cigarette smoke for 4 weeks followed by 1 week exposure
to room air. In the 4-week smoking cessation group, the mice were
exposed to cigarette smoke for 4 weeks followed by 4 week exposure
to room air. In the normal control group, the mice were exposed to
room air for 4 weeks. In the chronic smoking control group, the
mice were exposed to cigarette smoke for 8 weeks (5). The mice were sacrificed within 24 h
following the final exposure to air or smoke.
Exposure to cigarette smoke
As described previously (12,13),
exposure to cigarette smoke was performed in a chamber (160×50×50
cm; 20–24°C; 12-h light/dark cycle) attached to a cigarette-smoke
generator, which was constructed for the present study by burning a
cigarette in a sterile bottle, then blowing the smoke into the
chamber with an electromagnetic air pump. The smoke from 10
sequentially ignited cigarettes (Red Dragon™; Wuhan Red Dragon Co.
Ltd., Wuhan, China), each containing13 mg tar and 1.3 mg nicotine,
and fresh air were introduced simultaneously into the chamber. Each
session of cigarette smoke exposure lasted ~90 min, twice daily for
five days each week (Monday to Friday).
Histological and morphometric
analyses
The mice (n=8), used for morphometric analyses were
anesthetized with 10% chloral hydrate (0.03 ml/10 g) and sacrificed
by exsanguination of the abdominal aorta. The lungs were isolated
and fixed by infusion with 4% paraformaldehyde (Sinopharm Chemical
Reagent Co., Ltd., Shanghai, China) through a tracheal cannula at a
constant pressure of 25 cm H2O, part of the left lung
was frozen and embedded in OCT Compound (#4583; Sakura Finetek USA,
Inc., Torrance, CA, USA), and other parts were then immersed in
paraformaldehyde. Between 24 and 48 h later, a mid-sagittal slice
of the left lung was embedded in paraffin (Kangtai Clinical Reagent
Co., Ltd., Beijing, China), and 5 μm sections were dewaxed
and hydrated prior to histological analyses. The sections were then
stained with hematoxylin and eosin and Sirius Red (Sigma-Aldrich
China, Inc., Shanghai, China) for detection of collagen fibers.
Airspace enlargement
Airspace enlargement was evaluated, as previously
described by Biselli et al (14). Briefly, the sections were placed
under an Eclipse E800 light microscope (Nikon Corporation, Tokyo,
Japan) connected to a R1 Wireless Close-Up Speedlight System video
camera (Nikon Corporation) at x400 magnification. Images were
captured and displayed on a monitor. A frame of five concentric
semi-circles, with radii of 50, 100, 150, 200 and 250 μm,
was attached to the monitor screen with the center of the
semi-circles placed over a terminal bronchiole, thereby allowing
the semi-circle lines to lie over the lung parenchyma. The length
of each of the five semi-circles was divided by the number of
intersections between the line and the lung tissue. A total of 10
fields per slide were selected according to methods described by
Biselli et al (14) then
analyzed, producing one measurement of the degree of airspace
enlargement at each of the five distances from the terminal
bronchiole.
Collagen measurement
The collagen in the airway walls was stained using
Sirius Red and scored quantitatively. A ttoal of three lung
sections in each mouse were examined. Referring to the method of
Bracke et al (15), the
length of the basement membrane (Pbm) and the area in the airway
wall covered by the stain were determined using Image-Pro Plus 6.0
software (Media Cybernetics, Inc., Rockville, MD, USA). The area of
collagen was normalized to Pbm. All airways with a Pbm <2,000 mm
and cut at reasonable cross-sections, defined by a ratio of
minimal-to-maximal internal diameter of 0.5, were included.
Immunohistochemistry
For immunohistochemical analyses, the lung sections
were deparaffinized and hydrated and endogenous peroxidase was
blocked by 3% hydrogen peroxide (Jiutian Pharmaceutical Co., Ltd.,
Anshan, China). Antigen retrieval was performed as follows: Slices
were dipped in 0.01 M citrate buffer (pH 6.0) and microwave heated
to boiling for 10 min, then this was repeated twice further. The
sections were then blocked with rabbit serum blocking reagent
(Boster Biological Technology, Ltd., Wuhan, China) at room
temperature for 20 min, and were incubated overnight at 4°C with
rat anti-mouse Mac-3 monoclonal antibody (clone M3/84; 1:150
dilution; #553322; BD Biosciences, San Jose, CA, USA), or rabbit
anti-mouse transforming growth factor (TGF)-β1 polyclonal antibody
(1:100 dilution; sc-146; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA). The sections were then incubated with goat anti-rat
(1:300 dilution; sc-2041) or goat anti-rabbit (1:300 dilution;
sc-2040) biotin-conjugated secondary antibodies (Santa Cruz
Biotechnology, Inc.) at 37°C for 2 h. Diaminobenzidine (Boster
Biological Technology, Ltd.)was used as a chromogen, and the nuclei
were stained with hematoxylin. Negative controls were obtained by
omitting the primary antibody.
Analyses of alveolar macrophages (AMs) were
performed by counting the number of cells and dividing this number
by the tissue area in the same visual field. Analyses of the
expression levels of TGF-β1 were performed in the airways and lung
parenchyma. Images were captured of 10 fields of parenchyma and
five airways in each mouse. The stained area was calculated using
Image-Pro Plus software and divided by the tissue area.
Western blotting
Lung tissues were homogenized on ice by cytoplasmic
protein extraction using a Vibra-Cell VCX130 ultrasonic processor
(Sonics & Materials, Inc., Newtown, CT, USA). Protein samples
were extracted from lung-tissue homogenates using a Total
Cytoplasm-Nuclei Protein Extract kit (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA), according to the manufacturer’s instructions.
The samples were normalized for total lung protein content using a
BCA Protein Assay kit (Beyotime Institute of Biotechnology,
Jiangsu, China) prior to further analyses. A total of 60 μg
protein samples were separated by 10% SDS-PAGE and transferred to
polyvinylidene difluoride membranes (Beyotime Institute of
Biotechnology). The membranes were then incubated for 2 h in a
blocking solution (1X TBS with 5% defatted milk powder and 0.1%
Tween-20), followed by incubation overnight at 4°C with rabbit
anti-mouse matrix metalloproteinase (MMP)-12 polyclonal antibody
(1:400 dilution; sc-30072) and rabbit anti-mouse β-actin polyclonal
antibody (1:400 dilution; sc-130656) (Santa Cruz Biotechnology,
Inc.). Following washing with Tris-buffered saline with 0.1%
Tween-20 (TBST), the membranes were incubated with goat anti-rabbit
horseradish peroxidase conjugated secondary antibodies (1:5,000
dilution; Santa Cruz Biotechnology, Inc.) for 2 h at room
temperature. Each step was concluded by three rinses with TBST.
BeyoECL Plus electrochemiluminescence substrate (Beyotime Institute
of Biotechnology) was used for photographic development and
fixation of the blots. Following light exposure, the membranes were
chemically stripped to remove the antibodies and then reprocessed,
but with antibodies targeting the internal control, β-actin. Images
were captured and gray values calculated using Quantity One
software, version 4.6.2 (Bio-Rad Laboratories, Inc.). The
quantities of target protein were normalized to the value for
β-actin in that sample.
Immunofluorescence
To confirm the activation and expression of MMP-12
in the macrophages, double immunofluorescent staining was performed
in the frozen lung sections. A combination of the rat anti-mouse
Mac-3 and rabbit anti-mouse MMP-12 primary antibodies was used, and
phosphate-buffered saline (PBS) was used as the negative control.
The working dilutions of the Mac-3 and MMP-12 antibodies were 1:50
and 1:100, respectively. Working concentrations of goat anti-rat
fluorescein isothiocyanate (FITC)-conjugated IgG (bsF-0293G) and
goat anti-rabbit tetramethylrhodamine (TRITC)-conjugated IgG
(bsf-0295G) (BIOSS, Beijing, China) were 1:100. The sections were
incubated with primary antibodies at 4°C overnight, followed by
incubation with the goat anti-rabbit horseradish
peroxidase-conjugated secondary antibodies (1:5,000 dilution;
sc-2004; Santa Cruz Biotechnology, Inc.) for 2 h at room
temperature in the dark. Following primary and secondary antibody
incubation, the sections were washed with PBS twice then incubated
with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; BIOSS)
for 20 min at room temperature in the dark. Microscopic red
fluorescence indicated the expression of MMP-12 antigen, labeled by
TRITC; green fluorescence indicated macrophages, labeled by FITC;
and blue fluorescence indicated DAPI-stained nuclei. Images of the
three fluorescence channels were superimposed using Nikon EZ-C1
FreeViewer image analysis software, version 3.0 (Nikon
Corporation), with yellow fluorescence indicating colocalization of
the MMP-12 antigen and macrophages.
Statistical analyses
Statistical analyses were performed using SPSS 11.5
(SPSS, Inc., Chicago, IL, USA). The data are presented as the mean
± standard error of the mean. The results were analyzed by one-way
analysis of variance. A least significant difference post
hoc test was used to determine significant differences between
the groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Enlargement of alveolar airspaces
The alveolar airspaces ~100 μm from the
terminal bronchioles were significantly enlarged in the mice
exposed to cigarette smoke for 4 weeks compared with the control
mice (P=0.004). This enlargement was not reversed following smoking
cessation, however, exposure to cigarette smoke for 8 weeks did not
result in further enlargement of the alveolar airspaces (Table I). Sizes of the alveolar airspaces
~50, 150, 200 and 250 μm from the terminal bronchioles were
not significantly different between the groups of mice.
 | Table IComparison of the number of alveolar
airspaces at a distance of ~100 μm from the terminal
bronchioles in each group. |
Table I
Comparison of the number of alveolar
airspaces at a distance of ~100 μm from the terminal
bronchioles in each group.
| Group | Number | Mean | STD |
|---|
| Control | 5 | 25.60 | 1.97 |
| S4w | 5 | 31.62b | 2.65 |
| SC1w | 5 | 29.24b | 1.42 |
| SC4w | 5 | 29.88a | 2.66 |
| S8w | 5 | 32.94b | 2.74 |
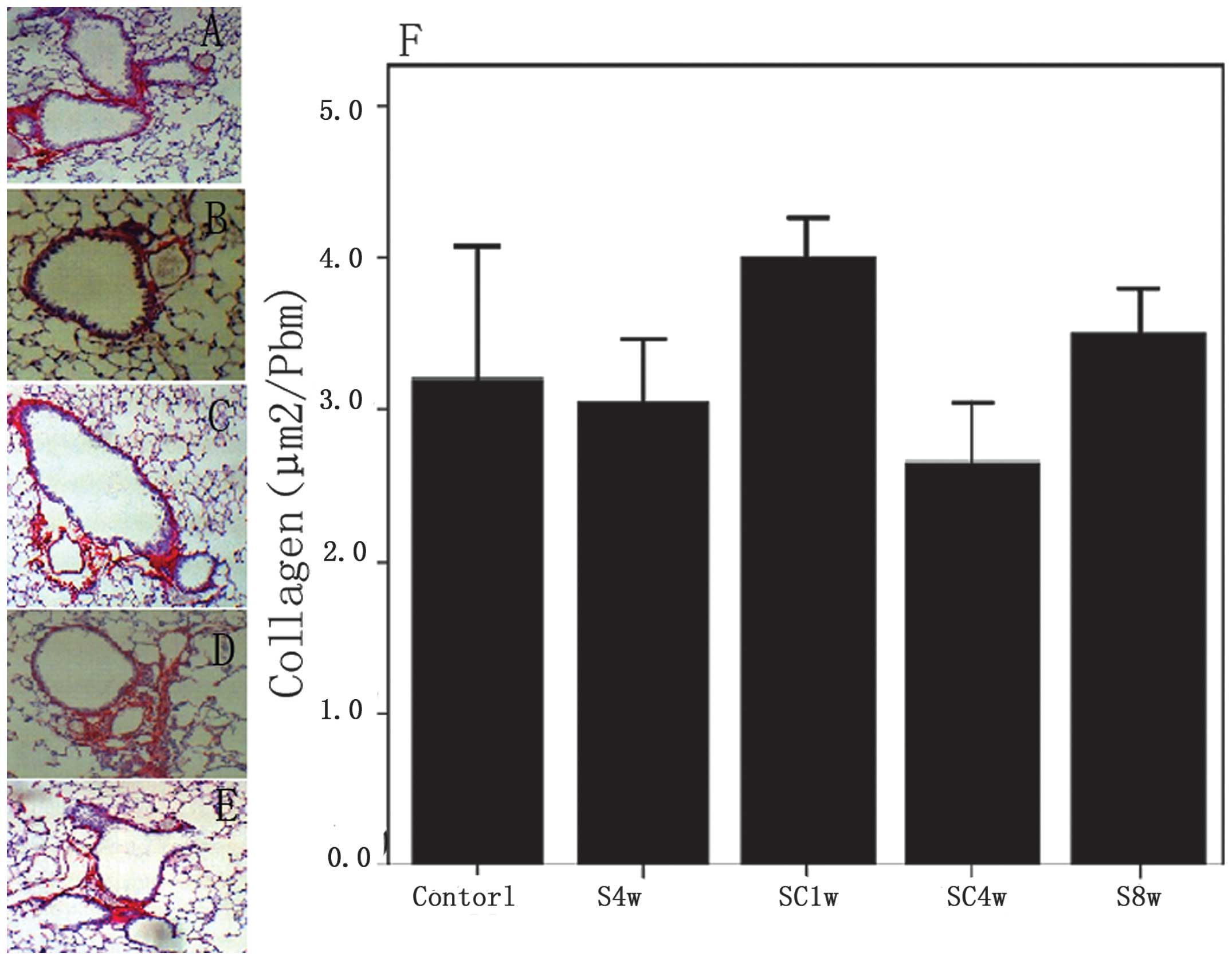
Collagen deposition around the small
airways
The deposition of collagen around the small airways
was not significantly increased in the mice exposed to cigarette
smoke for 4 or 8 weeks compared with the normal control group. In
addition, no significant difference was observed in the collagen
deposition prior to or following smoking cessation (Fig. 1).
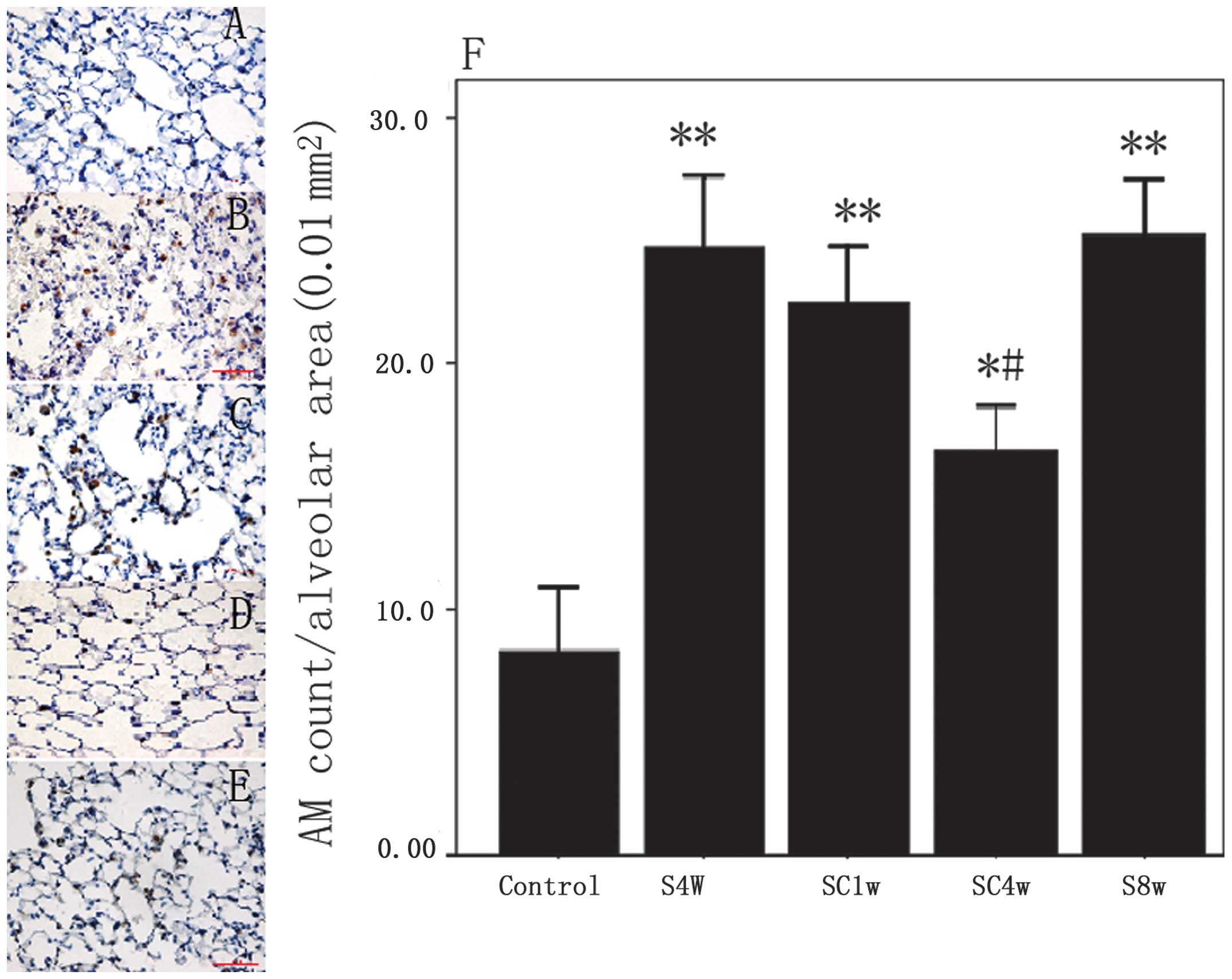
Macrophage infiltration
Macrophage infiltration into the lungs was
significantly increased in the mice exposed to cigarette smoke for
4 weeks compared with the normal control group (P=0.001). The
increased pulmonary infiltration of macrophages was significantly
decreased after 4 weeks of smoking cessation (SC4w, vs. S4w;
P=0.022); however, the levels did not return completely to the
those observed in the normal control group (SC4w vs. control;
P=0.015). Furthermore, mice exposed to cigarette smoke for 8 weeks
did not exhibit increased macrophage infiltration compared with the
mice exposed to cigarette smoke for 4 weeks (Fig. 2).
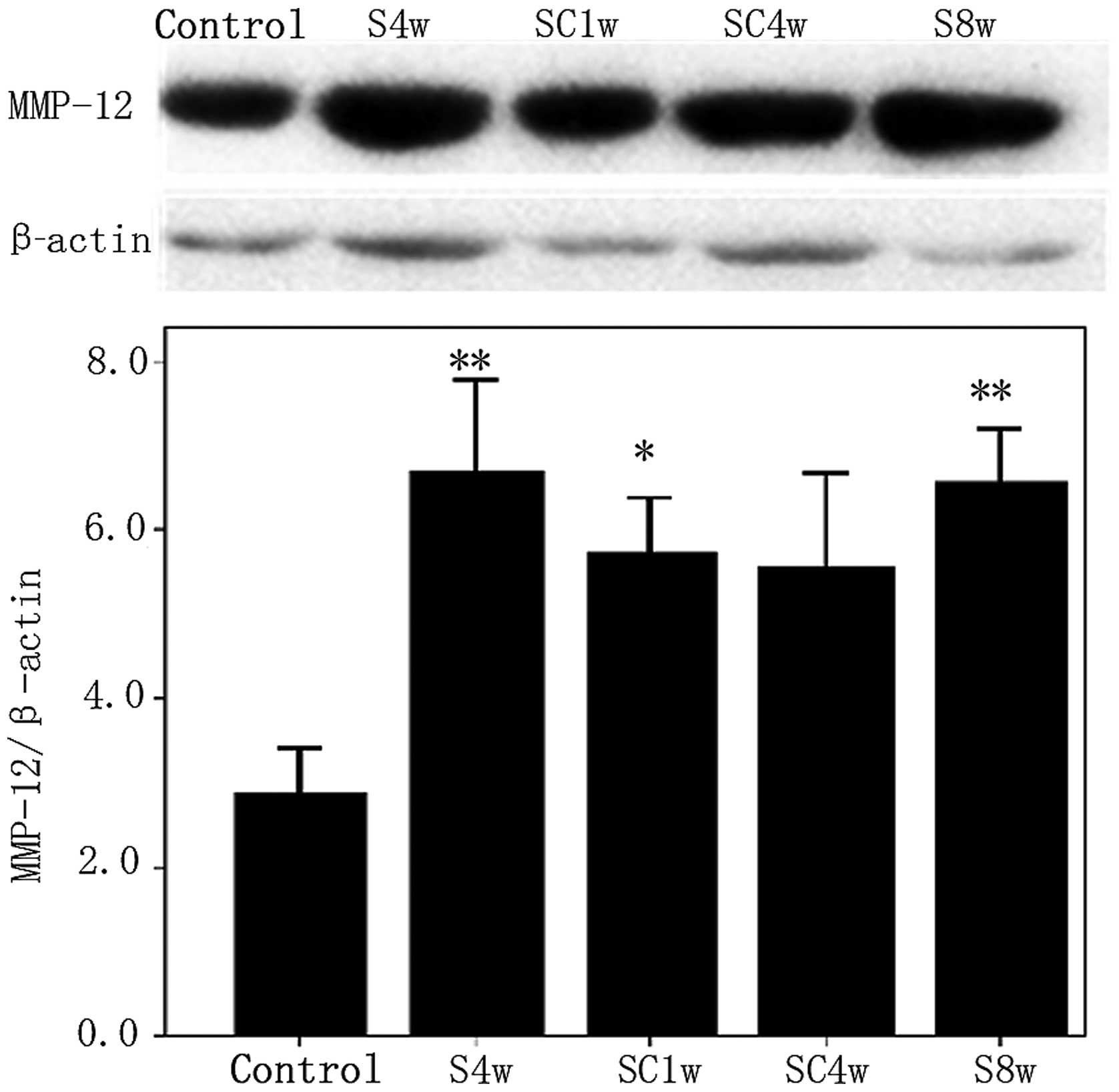
Expression and distribution of MMP-12
protein
The protein expression of MMP-12 was upregulated in
the lungs of the mice exposed to cigarette smoke for 4 weeks
compared with the control mice (P= 0.014); however, no significant
differences were observed between the mice exposed to cigarette
smoke for 4 weeks and those exposed for 8 weeks (P=0.92). The
protein expression levels of MMP-12 were significantly higher in
the mice 1 week following smoking cessation compared with the
control group (P=0.009; Fig. 3).
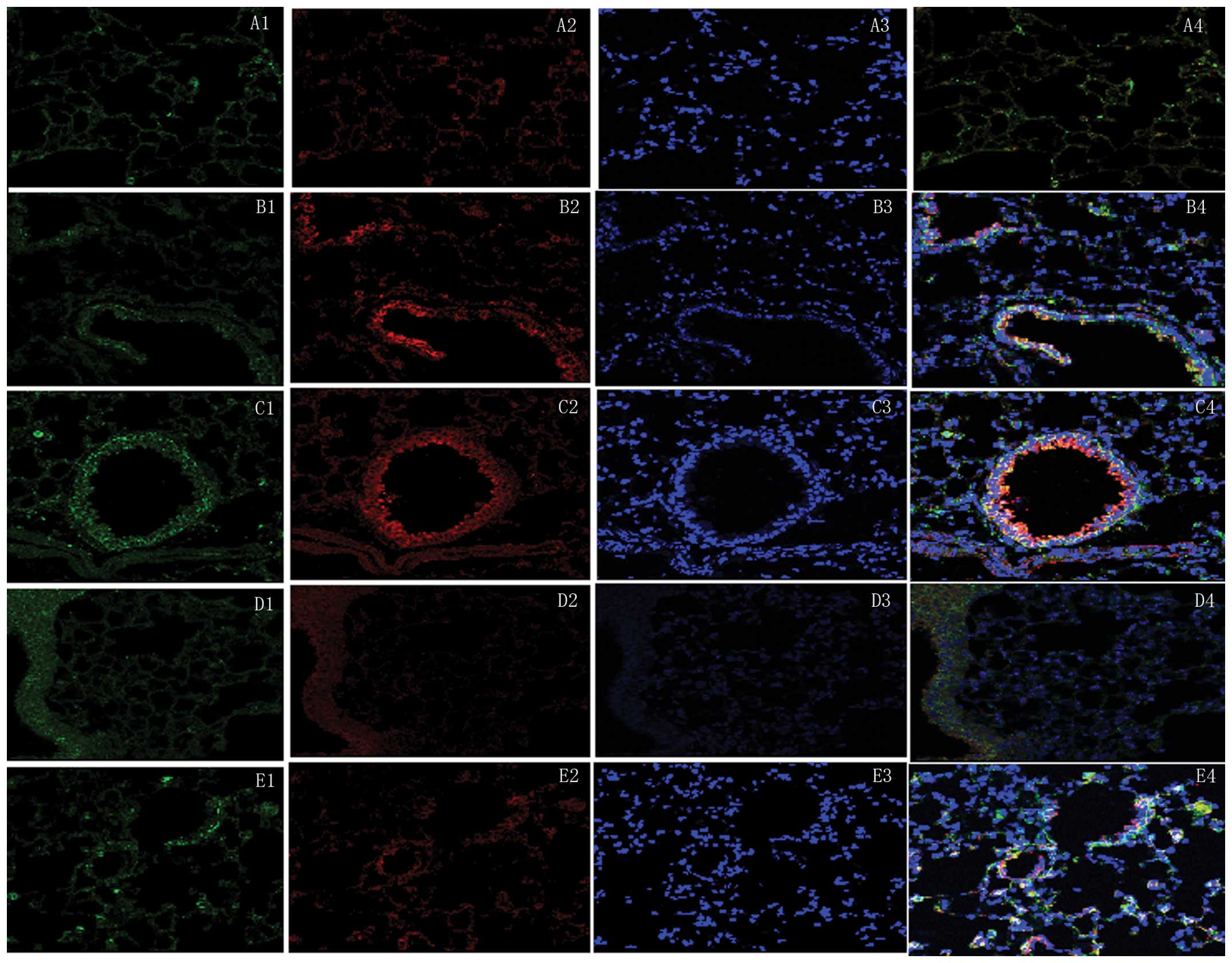
Double immunofluorescence labeling demonstrated that exposure to
cigarette smoke induced macrophages to secrete MMP-12 and increased
their recruitment into the lung parenchyma and airways. However,
macrophages, which secreted MMP-12 following smoking cessation were
rarely seen in the lung parenchyma and were located predominantly
around the bronchial walls (Fig.
4). MMP-12 antigens (red fluorescence) were located in the
cytoplasm and Mac-3 (green fluorescence) was distributed
predominantly in the cytomembrane and cytoplasm. DAPI staining
(blue fluorescence) was used to stain the cell nuclei. In the
three-channel composite images, yellow fluorescence represented the
colocalization of MMP-12 and Mac-3.
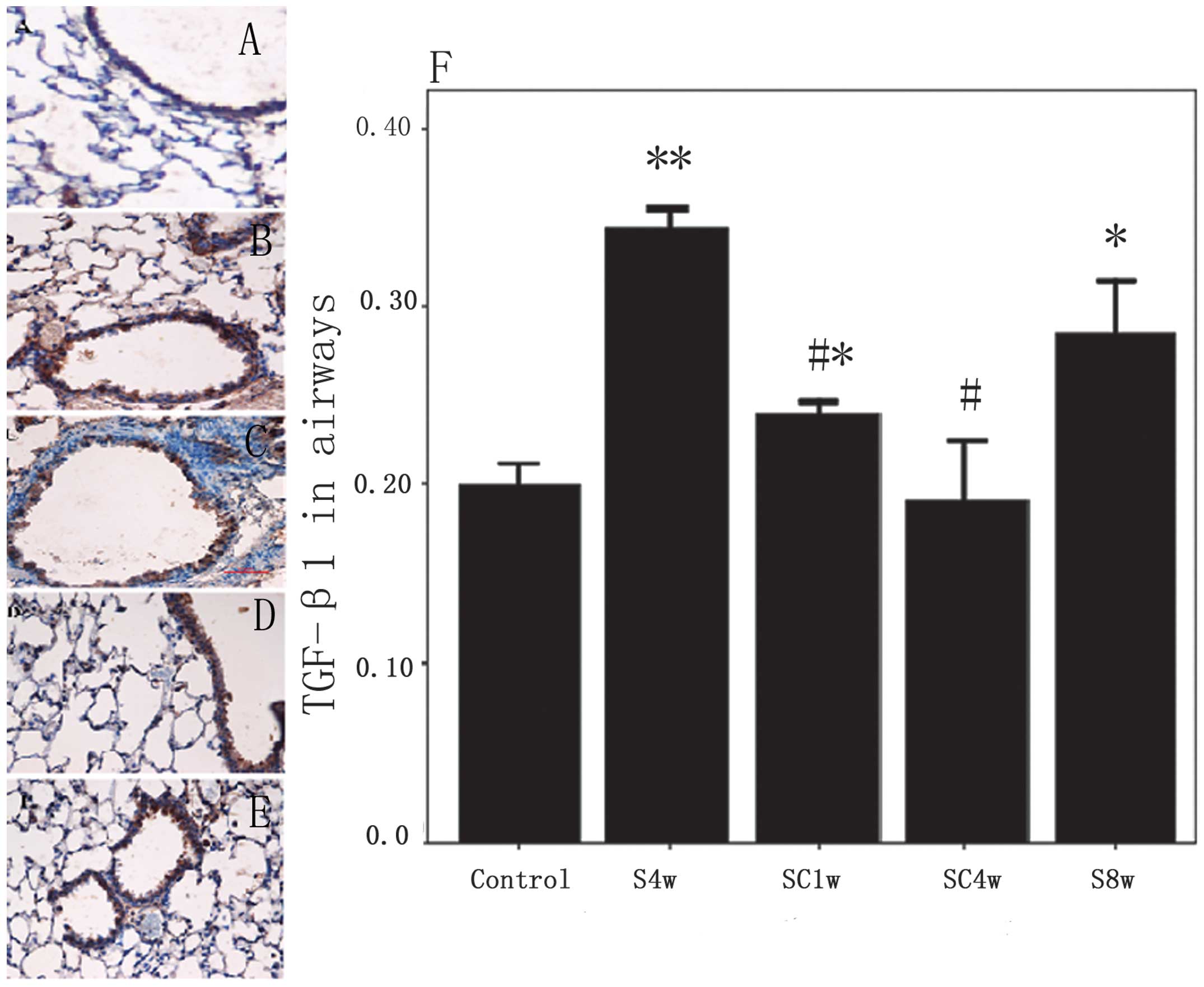
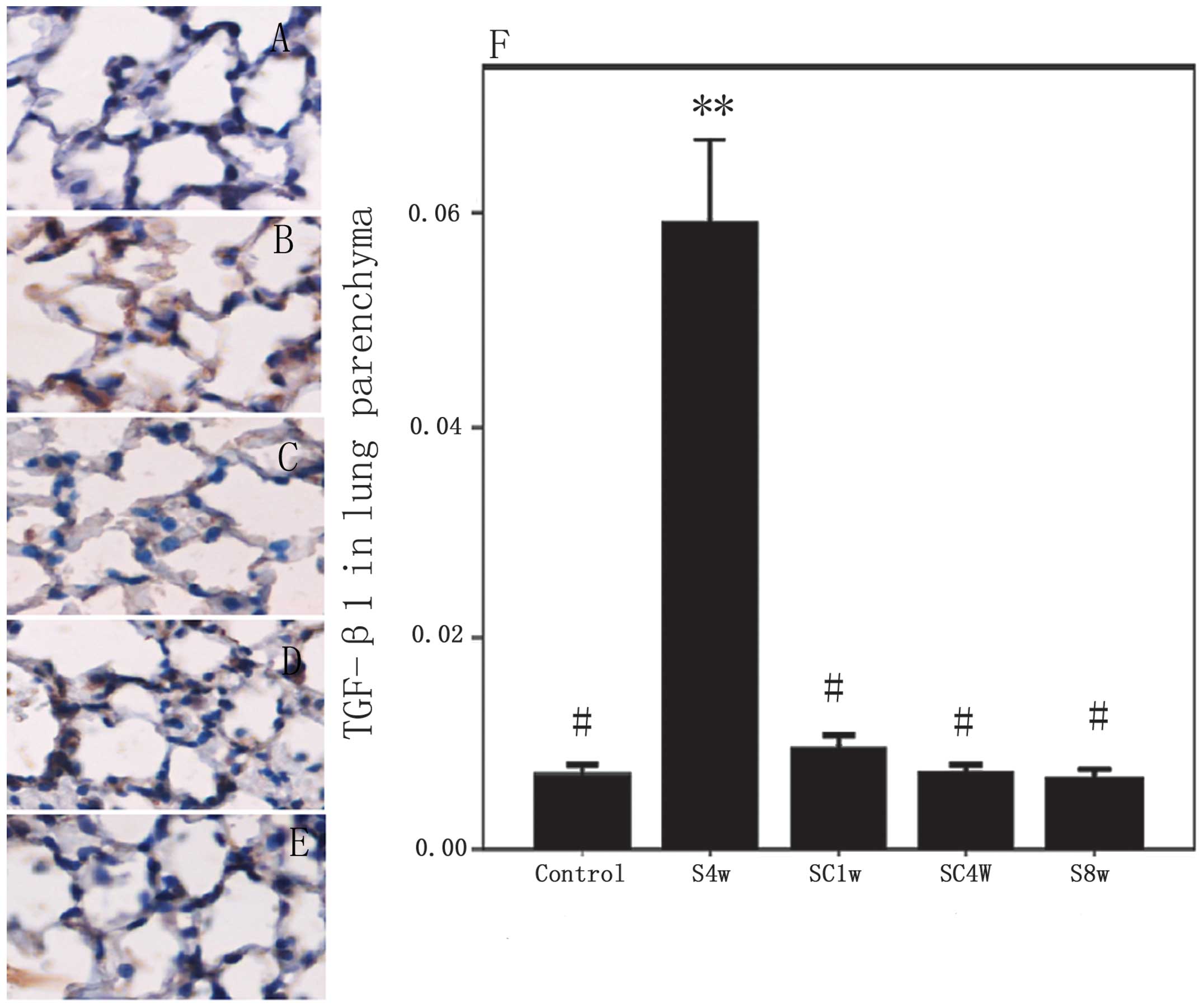
Expression and distribution of TGF-β1
protein
The protein expression of TGF-β1 was upregulated in
the airways and lung parenchyma of the mice exposed to cigarette
smoke for 4 weeks compared with the control mice (P<0.001). No
further increase was observed in the protein expression of TGF-β1
in the airways (P=0.077), but the levels were significantly
decreased in the lung parenchyma (P=0.000) of the mice exposed to
cigarette smoke for 8 weeks compared with those exposed for 4
weeks. Following smoking cessation for 4 weeks and 1 week,
respectively, the protein expression levels of TGF-β1 were returned
to those observed in the normal control group in the airways and
lung parenchyma (Figs. 5 and
6).
Discussion
COPD is a chronic inflammatory airway disease, which
is characterized by airflow limitations that are not fully
reversible. The typical pathological changes associated with COPD
are enlargement of the alveolar airspaces and remodeling of the
small airways, which are closely associated with airflow
obstruction (2–4). Smoking cessation is an effective way
of relieving the inflammation and airflow obstruction associated
with COPD, and has been advocated worldwide (1). However, investigation of the benefits
of smoking cessation on lung tissue is predominantly based on
patients in whom COPD has developed (18,17).
Therefore, the impact of smoking cessation on the inflammatory
response and lung lesions prior to the establishment of typical
pathological changes remains to be elucidated.
Establishment of emphysema in the majority of murine
models requires exposure to cigarette smoke for ≥24 weeks (14,15).
The present study established a group of mice subjected to subacute
exposure to cigarette smoke for 4 weeks (18), an early cessation group, in which
smoking was terminated after 4 weeks, and control groups, in which
the mice were exposed to filtered air for 4 weeks and to cigarette
smoke for 8 weeks. These groups were established to observe the
impact of early cessation of smoking on the pathological changes
and inflammatory responses in the lung.
AMs are considered the main inflammatory cells of
the lower respiratory tract and the first line of defense against
invasion of the lungs by foreign microorganisms (19,20).
AMs ingest, transport and remove exogenous particulate substances
and pathogenic microorganisms through phagocytosis, pinocytosis and
digestion. AMs are also involved in antigen presentation and the
regulation of inflammatory reactions (19,21–23).
There is evidence that AMs activate and release
proteolytic enzymes, an action that is closely associated with the
development of emphysema (19,24,21).
There is also a correlation between the severity of COPD and the
number of AMs (20,25,26).
MMP-12 is predominantly derived from AMs. MMP-12 was first
identified in the AMs of smokers (27), and degrades the protein components
of the extracellular matrix (ECM), including elastin, fibronectin,
laminin and gelatin (28,29). Furthermore, MMP-12 is involved in
the metabolism, cell migration, tissue repair and remodeling of the
ECM and high expression levels of MMP-12 can lead to degradation of
pathological proteins in the ECM (30). Previous studies have demonstrated
that MMP-12 is important in the establishment of a cigarette
smoke-induced murine model of emphysema, and that MMP-12 gene
knockout can result in the avoidance of emphysema (19,28).
Quantitative analyses of the size of alveolar
airspaces in experimental animal models usually involve the mean
linear intercept (31–33). However, due to the heterogeneity of
pulmonary lesions and the early stage of lung damage, the mean
linear intercept may not be sufficiently sensitive to reveal
localized enlargements of alveolar airspaces (14). The present study calculated the
sizes of the alveolar airspaces at different distances from the
terminal bronchiole, according to the method of Biselli et
al (14). The present study
observed, similar to Biselli et al, that the alveolar septum
100 μm from the terminal bronchiole was enlarged in mice
following subacute exposure to cigarette smoke. Furthermore,
infiltration of AMs was increased and the protein expression of
MMP-12 was upregulated. Increased infiltration of AMs during
exposure to cigarette smoke is a normal defense mechanism of the
body, but an excessive increase and release of proteolytic enzymes
induces local damage to the lung parenchyma (19,20,24,25).
However, the alveolar airspaces, pulmonary infiltration of AMs and
protein expression of MMP-12 were not significantly different
between the 8 and 4 week smoke-exposure groups. Therefore, the
inflammatory response appeared to decrease over time. Similar to
emphysema, locally enlarged alveolar airspaces did not recover
following smoking cessation and, although the infiltration of AMs
and expression of MMP-12 improved, they remained higher compared
with the normal control group. It has been reported that degraded
fragments of elastin are a powerful type of chemokine, which
recruits AMs to lesions (34).
Immunofluorescent localization demonstrated that, following smoking
cessation, the AMs secreting MMP-12 were predominantly distributed
in the walls of the small airways. However, the correlation between
the expression of MMP-12 and the generation of locally degraded
fragments of elastin requires further investigation.
Remodeling of the small airways is associated with
damage and abnormal repair of the ECM (35,36),
however, the pathogenesis remains to be elucidated. TGF-β1 promotes
tissue repair by improving the expression of protein components of
the ECM, including collagen and fibronectin, and inhibiting
degradation of the ECM (37).
MMP-12 degrades the ECM or inhibits TGF-β1 directly, indirectly
downregulates the synthesis of the ECM (30) and prevents excessive repair of
fibrosis. Interactions between TGF-β1 and MMP-12 are closely
associated with the remodeling of small airways (30). Cigarette smoke-induced inflammation
has previously been shown to promote the production and release of
TGF-β1, and the expression of TGF-β1 is increased in the airway
epithelial cells from smokers and patients with COPD (35). Increased expression of TGF-β1 have
been correlated with the dysfunction of bronchial basal membranes
and the number of peribronchiolar fibroblasts (38). The present study demonstrated that
the expression levels of TGF-β1 in the small airways and lung
parenchyma were upregulated after 4 weeks of smoking exposure and
returned to normal 1 week after smoking cessation; however,
abnormally increased collagen deposition was not observed, which
may be due to the early stage of disease or upregulation in the
expression of MMP-12. Compared with the mice exposed to cigarette
smoke for 4 weeks, the expression levels of TGF-β1 in the small
airways of the mice exposed to cigarette smoke for 8 weeks
exhibited no further increase. Furthermore, no abnormally increased
deposition of collagen around the small airways was observed and
the expression of TGF-β1 in the lung parenchyma was decreased.
These results are concordant with those of Churg et al
(36), which suggested that
distinctly different mechanisms regulate ECM metabolism in the
airway wall and lung parenchyma. High expression levels of TGF-β1
and abnormal repair may lead to remodeling of the small airways and
airway obstruction, whereas reduced expression levels of TGF-β1 and
insufficient repair of tissue may contribute to the development of
emphysema. Therefore, TGF-β1 is an important regulator of
inflammation and remodeling, and may offer a novel potential
therapeutic strategy for COPD.
In conclusion, the inflammatory reaction and damage
to the lung parenchyma, induced by subacute exposure to cigarette
smoke, occurred prior to remodeling of the walls of the small
airways. Lung tissue has a self-limiting ability in response to
chronic exposure to smoke, and the pulmonary inflammatory reaction
and pathological damage were not increased by prolonging the
exposure to cigarette smoke for a certain period of time. Local
damage to the lung parenchyma was not reversed by early cessation
of smoking, whereas the pulmonary inflammatory response was
partially downregulated. In addition, macrophage infiltration and
the expression of MMP-12 did not return to normal following smoking
cessation. Following smoking cessation, the expression of MMP-12
was distributed predominantly in the airways, which may prevent the
excessive deposition of collagen or may be due to excessive locally
degraded fragments of elastin. Regulation of the expression levels
of MMP-12 and TGF-β1, particularly regulation of the distribution
in the airways and lung parenchyma, may be a potential strategy for
the early treatment of COPD.
Acknowledgments
The current study was supported by the Shenyang
Science and Technology Program (grant no. F10-149-9-02).
References
|
1
|
Vestbo J, Hurd SS, Agusti AG, et al:
Global strategy for the diagnosis, management, and prevention of
chronic obstructive pulmonary disease: GOLD executive summary. Am J
Respir Crit Care Med. 187:347–365. 2013. View Article : Google Scholar
|
|
2
|
Wright JL, Postma DS, Kerstjens HA, Timens
W, Whittaker P and Churg A: Airway remodeling in the smoke exposed
guinea pig model. Inhal Toxicol. 19:915–923. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cosio M, Ghezzo H, Hogg JC, et al: The
relations between structural changes in small airways and
pulmonary-function tests. N Engl J Med. 298:1277–1281. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hasegawa M, Nasuhara Y, Onodera Y, et al:
Airflow limitation and airway dimensions in chronic obstructive
pulmonary disease. Am J Respir Crit Care Med. 173:1309–1315. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
D’hulst AI, Vermaelen KY, Brusselle GG, et
al: Time course of cigarette smoke-induced pulmonary inflammation
in mice. Eur Respir J. 26:204–213. 2005. View Article : Google Scholar
|
|
6
|
Simmons MS, Connett JE, Nides MA, et al:
Smoking reduction and the rate of decline in FEV(1): results from
the Lung Health Study. Eur Respir J. 25:1011–1017. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Willemse BW, Postma DS, Timens W and ten
Hacken NH: The impact of smoking cessation on respiratory symptoms,
lung function, airway hyperresponsiveness and inflammation. Eur
Respir J. 23:464–476. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Willemse BW, ten Hacken NH, Rutgers B,
Lesman-Leegte IG, Postma DS and Timens W: Effect of 1-year smoking
cessation on airway inflammation in COPD and asymptomatic smokers.
Eur Respir J. 26:835–845. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gamble E, Grootendorst DC, Hattotuwa K, et
al: Airway mucosal inflammation in COPD is similar in smokers and
ex-smokers: a pooled analysis. Eur Respir J. 30:467–471. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lapperre TS, Postma DS, Gosman MM, et al:
Relation between duration of smoking cessation and bronchial
inflammation in COPD. Thorax. 61:115–121. 2006. View Article : Google Scholar
|
|
11
|
Braber S, Henricks PA, Nijkamp FP,
Kraneveld AD and Folkerts G: Inflammatory changes in the airways of
mice caused by cigarette smoke exposure are only partially reversed
after smoking cessation. Respir Res. 11:992010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Z, Zhang JN, Hu XF, et al: Effects of
pentoxifylline on Wnt/β-catenin signaling in mice chronically
exposed to cigarette smoke. Chin Med J (Engl). 123:2688–2694.
2010.
|
|
13
|
Zhang JN, Wang Z, Shi W, et al: Mechanism
responsible for pu lmonary fibrosis induced by concomitant chronic
smoke exposure and pentoxifylline administration. Chin J
Pathophysiol (Chin). 25:333–337. 2009.
|
|
14
|
Biselli PJ, Lopes FD, Moriya HT, et al:
Short-term exposure of mice to cigarette smoke and/or residual oil
fly ash produces proximal airspace enlargments and airway
epithelium remodeling. Braz J Med Biol Res. 44:460–468. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bracke KR, Dentener MA, Papakonstantinou
E, et al: Enhanced deposition of low-molecular-weight hyaluronan in
lungs of cigarette smoke-exposed mice. Am J Respir Cell Mol Biol.
42:753–761. 2010. View Article : Google Scholar
|
|
16
|
Tønnesen P, Carrozzi L, Fagerström K, et
al: Smoking cessation in patients with respiratory diseases: a high
priority, integral component of therapy. Eur Respir J. 29:390–417.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kotz D, Wesseling G, Huibers MJ and van
Schayck OC: Efficacy of confronting smokers with airflow limitation
for smoking cessation. Eur Respir J. 33:754–762. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Demoor T, Bracke KR, Dupont LL, et al: The
role of ChemR23 in the induction and resolution of cigarette
smoke-induced inflammation. J Immunol. 186:5457–5467. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hautamaki RD, Kobayashi DK, Senior RM and
Shapiro SD: Requirement for macrophage elastase for cigarette
smoke-induced emphysema in mice. Science. 277:2002–2004. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shapiro SD: The macrophage in chronic
obstructive pulmonary disease. Am J Respir Crit Care Med.
160:S29–S32. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bitterman PB, Wewers MD, Rennard SI,
Adelberg S and Crystal RG: Modulation of alveolar macrophage-driven
fibroblast proliferation by alternative macrophage mediators. J
Clin Invest. 77:700–708. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Houghton AM, Quintero PA, Perkins DL, et
al: Elastin fragments drive disease progression in a murine model
of emphysema. J Clin Invest. 116:753–759. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Venet A, Hance AJ, Saltini C, Robinson BW
and Crystal RG: Enhanced alveolar macrophage-mediated
antigen-induced T-lymphocyte proliferation in sarcoidosis. J Clin
Invest. 75:293–301. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shapiro SD and Ingenito EP: The
pathogenesis of chronic obstructive pulmonary disease: advances in
the past 100 years. Am J Respir Cell Mol Biol. 32:367–372. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hogg JC, Chu F, Utokaparch S, et al: The
nature of small-airway obstruction in chronic obstructive pulmonary
disease. N Engl J ed. 350:2645–2653. 2004. View Article : Google Scholar
|
|
26
|
Finkelstein R, Fraser RS, Ghezzo H and
Cosio MG: Alveolar inflammation and its relation to emphysema in
smokers. Am J Respir Crit Care Med. 152:1666–1672. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shapiro SD, Kobayashi DK and Ley TJ:
Cloning and characterization of a unique elastolytic
metalloproteinase produced by human alveolar macrophages. J Biol
Chem. 268:23824–23829. 1993.PubMed/NCBI
|
|
28
|
Chandler S, Cossins J, Lury J and Wells G:
Macrophage metalloelastase degrades matrix and myelin proteins and
processes a tumour necrosis factor-alpha fusion protein. Biochem
Biophys Res Commun. 228:421–429. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gronski TJ Jr, Martin RL, Kobayashi DK, et
al: Hydrolysis of a broad spectrum of extracellular matrix proteins
by human macrophage elastase. J Biol Chem. 272:12189–12194. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
England KA, Price AP, Tram KV, Shapiro SD,
Blazar BR and Panoskaltsis-Mortari A: Evidence for early fibrosis
and increased airway resistance in bone marrow transplant recipient
mice deficient in MMP12. Am J Physiol Lung Cell Mol Physiol.
301:L519–L526. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thurlbeck WM, Galaugher W and Mathers J:
Adaptive response to pneumonectomy in puppies. Thorax. 36:424–427.
1981. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yao H, Chung S, Hwang JW, et al: SIRT1
protects against emphysema via FOXO3-mediated reduction of
premature senescence in mice. J Clin Invest. 122:2032–2045. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Podowski M, Calvi C, Metzger S, et al:
Angiotensin receptor blockade attenuates cigarette smoke-induced
lung injury and rescues lung architecture in mice. J Clin Invest.
122:229–240. 2012. View
Article : Google Scholar :
|
|
34
|
Houghton AM, Quintero PA, Perkins DL, et
al: Elastin fragments drive disease progression in a murine model
of emphysema. J Clin Invest. 116:753–759. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Takizawa H, Tanaka M, Takami K, et al:
Increased expression of transforming growth factor-beta1 in small
airway epithelium from tobacco smokers and patients with chronic
obstructive pulmonary disease (COPD). Am J Respir Crit Care Med.
163:1476–1483. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Churg A, Tai H, Coulthard T, Wang R and
Wright JL: Cigarette smoke drives small airway remodeling by
induction of growth factors in the airway wall. Am J Respir Crit
Care Med. 174:1327–1334. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Philipp K, Riedel F, Germann G, Hörmann K
and Sauerbier M: TGF-beta antisense oligonucleotides reduce mRNA
expression of matrix metalloproteinases in cultured
wound-healing-related cells. Int J Mol Med. 15:299–303.
2005.PubMed/NCBI
|
|
38
|
Königshoff M, Kneidinger N and Eickelberg
O: TGF-beta signaling in COPD: deciphering genetic and cellular
susceptibilities for future therapeutic regimen. Swiss Med Wkly.
139:554–563. 2009.PubMed/NCBI
|