Introduction
Mesangial proliferative glomerulonephritis is the
most common type of primary glomerular disease in China. It is
characterized by the proliferation of mesangial cells (MCs) and
deposition of extracellular matrix (ECM), which results in
glomerular sclerosis, and end-stage renal disease (1). MCs are involved in various types of
glomerular injury via the proliferation and secretion of cytokines,
including transforming growth factor-β (TGF-β). TGF-β stimulates
the expression of ECM proteins, including collagen type IV (col-IV)
and interstitial collagen, including collagen type I (col-I)
(2). The dysregulation of cell
apoptosis also contributes to the proliferation of MCs and ECM
deposition (3). B-cell lymphoma 2
(Bcl-2) family members, including the Bcl-2 anti-apoptotic and
Bcl-2-associated X protein (Bax) a pro-apoptotic proteins, are
important regulators of cell apoptosis (4). However, whether these apoptotic
proteins are involved in MCA-stimulated MC proliferation remains to
be elucidated.
Hyperlipemia is responsible for a number of renal
diseases (5), and the
3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase
inhibitors exert modulatory effects on a number of cell signaling
cascades by preventing the synthesis of various isoprenoids derived
from the mevalonate (MVA) pathway (6). Mitogen-activated protein kinases
(MAPKs), including extracellular signal-regulated kinase (ERK),
c-Jun NH2-teminal kinase (JNK)/stress-activated protein kinase,
(SAPK), P38 MAPK and ERK5/big MAPK 1 (BMK1) are key regulators of
MC proliferation and ECM deposition, and are thus closely
associated with the development of mesangial proliferative
glomerulonephritis (7,8). However, the effect of MVA on MCs, its
underlying mechanisms, its effect on MAPKs and downstream
transcription factors and the association between MAPKs and MCs
remain to be elucidated. The aim of the present study was to
investigate the effects of MVA on human mesangial cell (HMC)
proliferation, apoptosis, cell cycle and ECM deposition, as well as
the role of TGF-β1 and the MAPKs in the process, in order to
examine the mechanism of MVA in the development of mesangial
proliferative glomerulonephritis.
Materials and methods
HMC culture
The T-SV40 HMC cell line was provided by Dr Li
Xuewang (Peking Union Medical College Hospital, Beijing, China).
The cells were routinely maintained in RPMI-1640 (Sigma-Aldrich,
St. Louis, MO, USA), containing 10% fetal calf serum (FCS; Sijiqing
Biological Engineering Materials Co., Ltd., Hangzhou, China) and
supplemented with 100 U/ml penicillin and 100 μg/ml
streptomycin (Sijiqing Biological Engineering Materials Co., Ltd.)
at 37°C. The culture medium was replaced every 2 days. When the
cells reached confluence, they were subcultured at a ratio of 1:4,
using the same incubation medium.
Experimental design
The HMCs (60% confluent) were trypsinized (Sijiqing
Biological Engineering Materials Co., Ltd.) and seeded
(4×104 cells/cm2) into petri dishes at 37°C
and were cultured with MVA (Sigma-Aldrich) at various
concentrations (0, 10−9, 10−8,
10−7, 10−6, 10−5, 10−4
and 10−3 M) for 24 h, and at 10−7 M for 12,
24 or 48 h, to evaluate the effects of dose and time on HMC
proliferation.
The HMCs (1×105 cells/ml) were then
cultured with MVA (1×10−7 mol/l), either alone or in the
presence of 50 μmol/l PD98059, an ERK inhibitor; 50
μmol/l SP600125, a JNK inhibitor or 50 μmol/l
SB203580, a P38 MAPK inhibitor (all Axxora-Boppard, Shanghai,
China). The cells were cultured for 24 h at 37°C with RPMI-1640
supplemented with 10% FCS and were then serum-starved for 24 h. The
cells were centrifuged at 500 x g for 5 min at 25°C, the
supernatants were then collected and the total protein was
extracted from the cells. A
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT)
reduction assay was performed to measure the proliferation of the
HMCs. A flow cytometric assay was performed to assess the
proliferative index (PI) and an ELISA was performed to determine
the secretion of TGF-β1, Col-IV and Col-I. The expression levels of
Bcl-2 and Bax were analyzed using western blot analysis. In certain
experiments, p-ERK1/2, p-JNK and p-p38 were measured using western
blot analysis.
MTT assay
The number of living cells in the HMC cultures was
assayed using an MTT assay (Sigma-Aldrich). The MTT-formazan
product was dissolved in phosphate-buffered saline (PBS;
Sigma-Aldrich). Following the addition of RPMI-1640 medium
containing 10% MTT, the cells (1×105 cells/ml) were
incubated at 37°C for 4 h, the medium was aspirated and the cells
were lysed by the addition of 100 μl dimethyl sulfoxide
(DMSO; Sigma-Aldrich). Subsequently, 10 μlof each sample was
diluted in 90 μl fresh DMSO. The samples were then mixed on
a mechanical plate mixer, and the optical density of each sample at
the test and reference wavelengths of 550 and 650 nm, respectively
was measured using a microplate-reader (Model 550; Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Analysis of PI and apoptosis using flow
cytometry
The HMCs were cultured in 10-cm dishes
(1×105 cells/ml) RPMI-1640 medium containing 10% FCS and
then incubated for 24 h in 10% FCS. The cells were collected and
fixed with 1% methanol-free formaldehyde (Sigma-Aldrich) for 20
min. The samples were washed twice with PBS and then resuspended in
70% ethanol (Sijiqing Biological Engineering Materials Co., Ltd.)
for 1 h at 4°C. The fixed and permeated cells were collected by
centrifugation at 500 x g for 5 min at 25°C, washed with PBS and
incubated with RNase (50 μg/ml; Sigma-Aldrich) for 10 min at
room temperature. Finally, 200 μl propidium iodide (65
μg/ml; Sigma-Aldrich) was added to each dish for 10 min on
ice, in order to stain the nuclei. The numbers of cells in the
G1/S, and G2/M phases of the cell cycle were the analyzed using
flow cytometry (BD Accuri™ C6; BD Biosciences, San Jose, CA, USA).
The above experiments were repeated six times. Each sample was
analyzed using CellQuest version 3.0 software (BD Biosciences) to
assess the percentage of cells in each phase of the cell cycle and
the apoptotic rate. The PI was calculated according to the
following equation: PI = (S + G2/M) / (G0/G1 + S + G2/M).
Measurement of TGF-β1, Col-I and Col-IV
using ELISA
The culture supernatant (250 μl) was removed
from each well and incubated with 5 μl 1 N HCl for 60 min to
activate the latent TGF-β1, Col-I or Col-IV. Following
neutralization of the supernatants with 1 N NaOH, the samples were
analyzed using a commercial human TGF-β1, Col-I or Col-IV ELISA kit
(Promega, Madison, WI, USA), according to the manufacturer’s
instructions. A standard curve was constructed using serial
dilutions of ultrapure human TGF-β1, Col-I or Col-IV (Promega). The
cells were harvested by trypsinization and counted using a
microscope (BX51; Olympus Corporation, Tokyo, Japan). Each sample
was measured in duplicate.
Western blot analysis of the expression
levels of Bcl-2, Bax, p-ERK1/2, p-JNK and p-p38
The cells (1×105 cells/ml) were
homogenized in radioimmunoprecipitation assay buffer (Thermo Fisher
Biochemical, Shanghai, China). The homogenates were centrifuged at
12,000 g for 10 min at 4°C, and the supernatants were collected.
The total fractions were denatured in sample buffer [20 μl
protein samples + 5 μl 5X loading buffer (Sigma-Aldrich)] at
100°C for 5 min. The protein (60 mg) was electrophoresed on 10%
SDS-polyacrylamide gels, then transferred onto nitrocellulose
membranes (Merck Millipore, Shanghai, China), blocked with 5%
non-fat milk in Tris-buffered saline with 0.05% Tween 20 (TBST)
buffer overnight at 4°C. The membranes were then incubated with
mouse monoclonal anti-Bax (1:1,000; cat. no. ab5714; Abcam,
Cambridge, UK), mouse monoclonal anti-Bcl-2 (1:1,000; cat. no.
ab201566; Abcam), rabbit monoclonal anti-p-p38 MAPK (1:1,000; cat.
no. 4092; Cell Signaling Technology, Inc., Danvers, MA, USA), mouse
monoclonal anti-p-JNK (1:1,000; cat. no. 9255; Cell Signaling
Technology, Inc.) or rabbit monoclonal anti-p-ERK (1:1,000; cat.
no. 9101; Cell Signaling Technology, Inc.). The blots were washed
with TBST buffer and subsequently incubated with
peroxidase-conjugated anti-mouse or anti-rabbit immunoglobulin G
(1:2,000; cat. nos. sc-2030 or sc-2031; Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA). Following washing with TBST, the blots
were developed with enhanced chemiluminescence reagents (Santa Cruz
Biotechnology, Inc.). Rabbit polyclonal anti-β-actin antibody
(1:500; cat. no. sc-13065; Santa Cruz Biotechnology, Inc.) was used
as a control for each sample.
Statistical analysis
The results are presented as the mean ± standard
deviation. The data were analyzed using a one-way analysis of
variance and a post hoc multiple-comparison test for comparisons of
the mean values among multiple treatment groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
MVA promotes HMC proliferation
To elucidate the role of MVA in HMC proliferation,
the cultured HMCs were treated with MVA at various concentrations
for 24 h, or for various durations. The results demonstrated that
MVA produced a significant dose-dependent increase in the PI at
concentrations <10−7 M, compared with the control
(P<0.05). At concentrations of MVA >10−7 M, the PI
was significantly decreased. MVA treatment also promoted HMC
proliferation in a time-dependent manner (P<0.05; Tables I and II). Following culture of the HMCs with
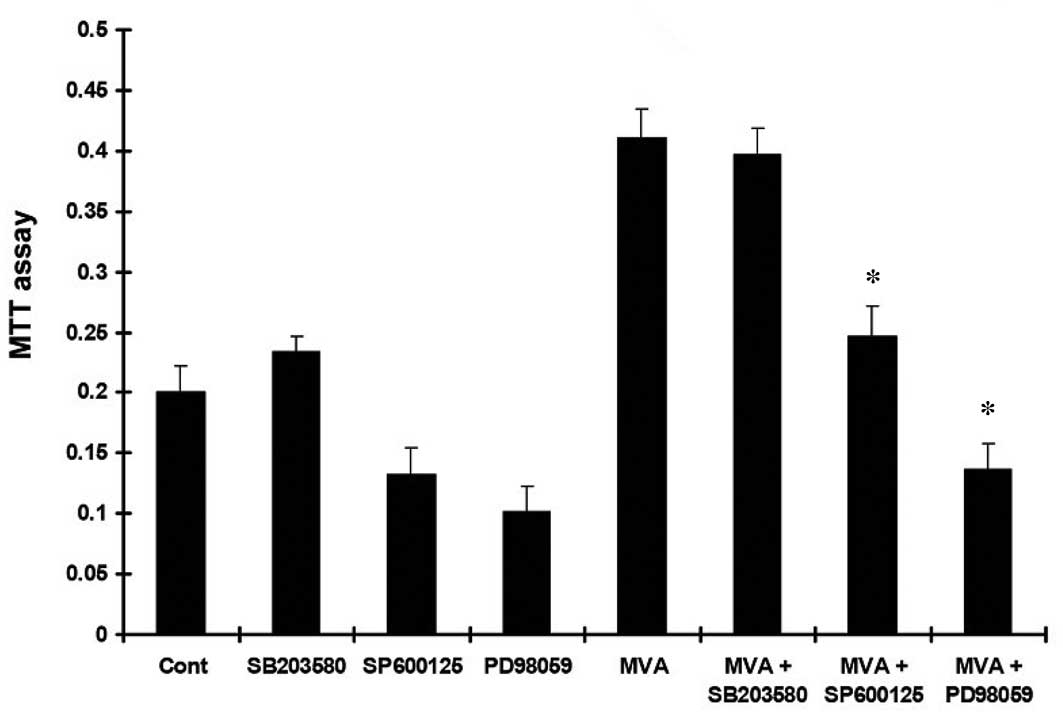
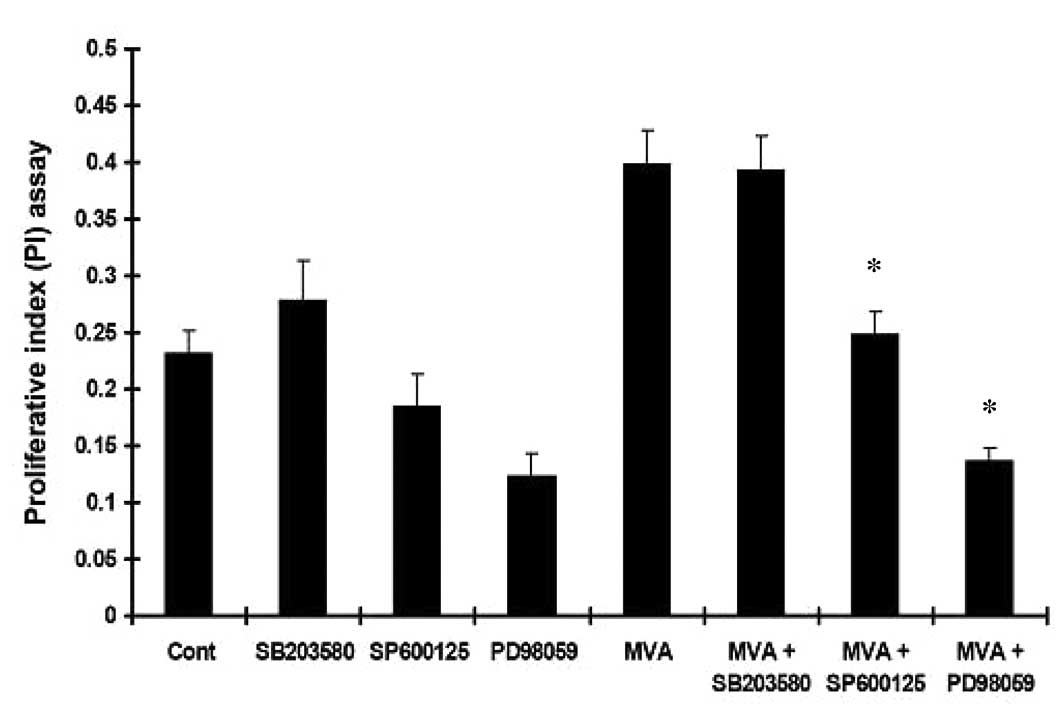
MVA in the presence or absence of PD98059, SP600125 or SB203580,
the increased MTT absorbance (Fig.
1) and PI (Fig. 2) in the HMCs
were inhibited by PD98059 and SP600125, but not by SB203580.
 | Table IEffects of different concentrations of
MVA on human mesangial cell proliferation. |
Table I
Effects of different concentrations of
MVA on human mesangial cell proliferation.
| MVA concentration
(mol/l) | Absorbance at 490 nm
|
|---|
| 12 h | 24 h | 48 h |
|---|
| 0 | 0.119±0.024 | 0.206±0.015a | 0.400±0.019b |
|
1×10−9 | 0.153±0.047c | 0.284±0.033a,c | 0.409±0.017b,c |
|
1×10−8 | 0.171±0.053c,d | 0.302±0.041a,c,d | 0.499±0.010b,c,d |
|
1×10−7 | 0.201±0.040c,e | 0.326±0.019a,c,e | 0.509±0.011b,c,e |
|
1×10−6 | 0.184±0.060c,f | 0.317±0.016c,a,f | 0.406±0.007b,f |
|
1×10−5 | 0.149±0.028c,g | 0.263±0.029c,a,g | 0.390±0.010b,g |
|
1×10−4 | 0.121±0.025h | 0.201±0.031a,h | 0.306±0.005b,h |
|
1×10−3 | 0.088±0.015i | 0.175±0.027a,i | 0.301±0.011b,i |
| Table IIEffects of different concentrations
of MVA on proliferative index. |
Table II
Effects of different concentrations
of MVA on proliferative index.
| MVA concentration
(mol/l) | Proliferation index
|
|---|
| 12 h | 24 h | 48 h |
|---|
| 0 | 14.3±3.4 | 19.1±2.6a | 25.5±2.8b |
|
1×10−9 | 21.9±2.2c | 25.1±3.3a,c | 31.9±3.6b,c |
|
1×10−8 | 23.2±2.0c,d | 27.1±.2.6a,c,d | 33.9±3.8b,c,d |
|
1×10−7 | 27.1±3.14c,e | 31.2±3.2a,c,e | 38.3±4.4b,c,e |
|
1×10−6 | 19.7±2.1c,f | 23.4±1.6c,a,f | 29.5±2.5b,f |
|
1×10−5 | 17.1±2.2c,g | 20.5±2.7a,g | 25.8±1.6b,g |
|
1×10−4 | 14.6±1.5h | 17.4±2.0a,h | 21.6±1.5b,h |
|
1×10−3 | 12.7±1.2i | 14.5±2.1a,i | 17.3±1.3b,i |
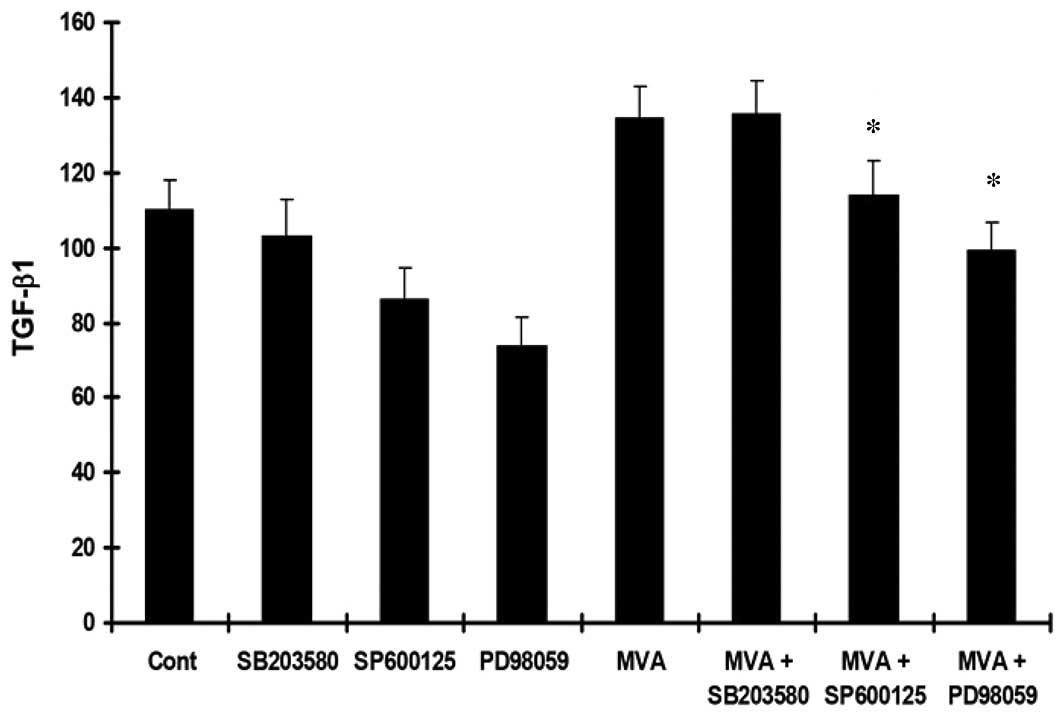
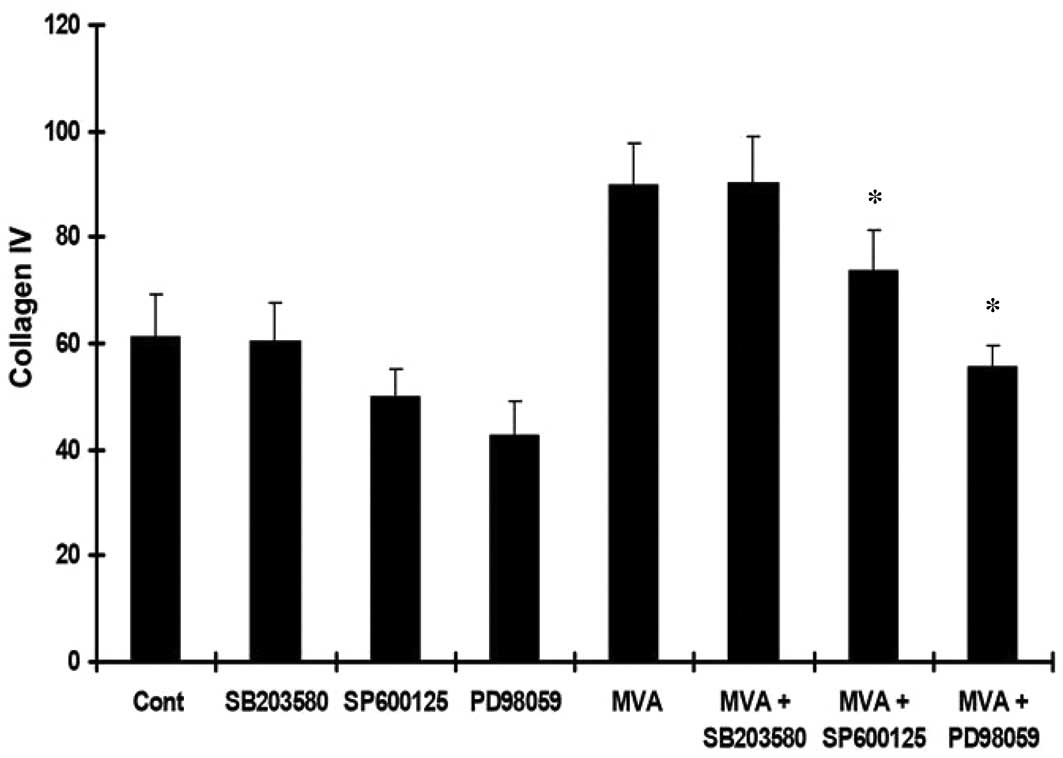
MVA stimulates the expression of ECM
proteins
The HMCs were cultured with MVA alone or in the
presence of PD98059, SP600125 or SB203580. An ELISA was performed
to determine the levels of TGF-β1, Col-IV and Col-I. Treatment with
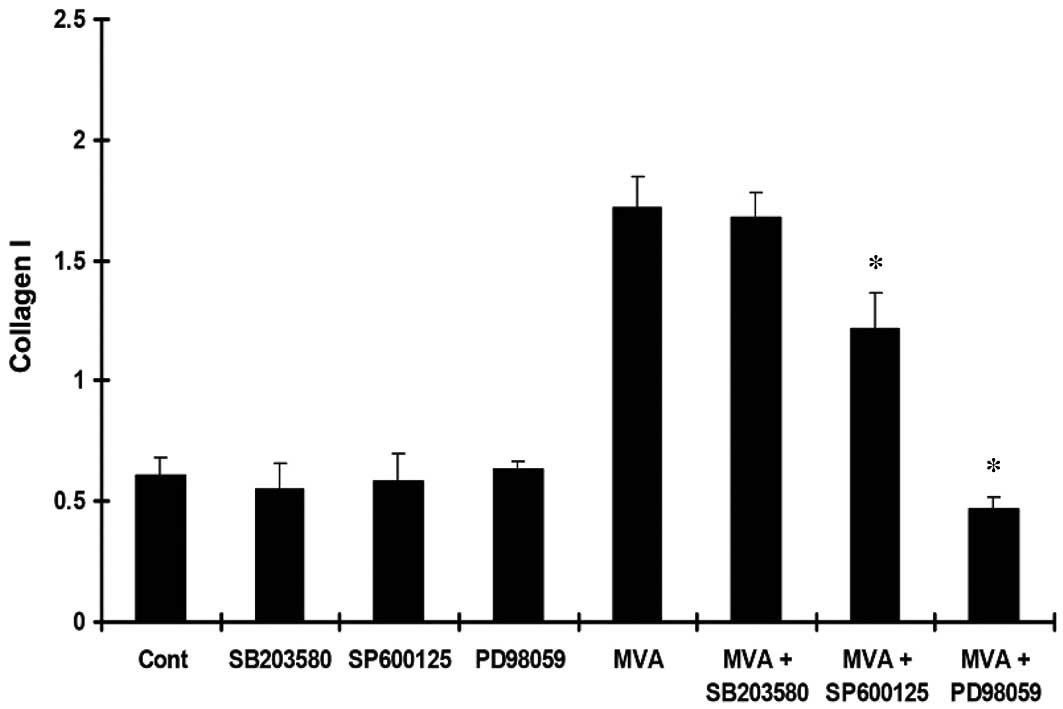
MVA significantly increased the expression of TGF-β1 (Fig. 3), Col-IV (Fig. 4) and Col-I (Fig. 5). However, the effects were
reversed by PD98059 and SP600125, while no significant effect was
observed following treatment with SB203580.
MVA suppresses apoptotic signaling in the
HMCs
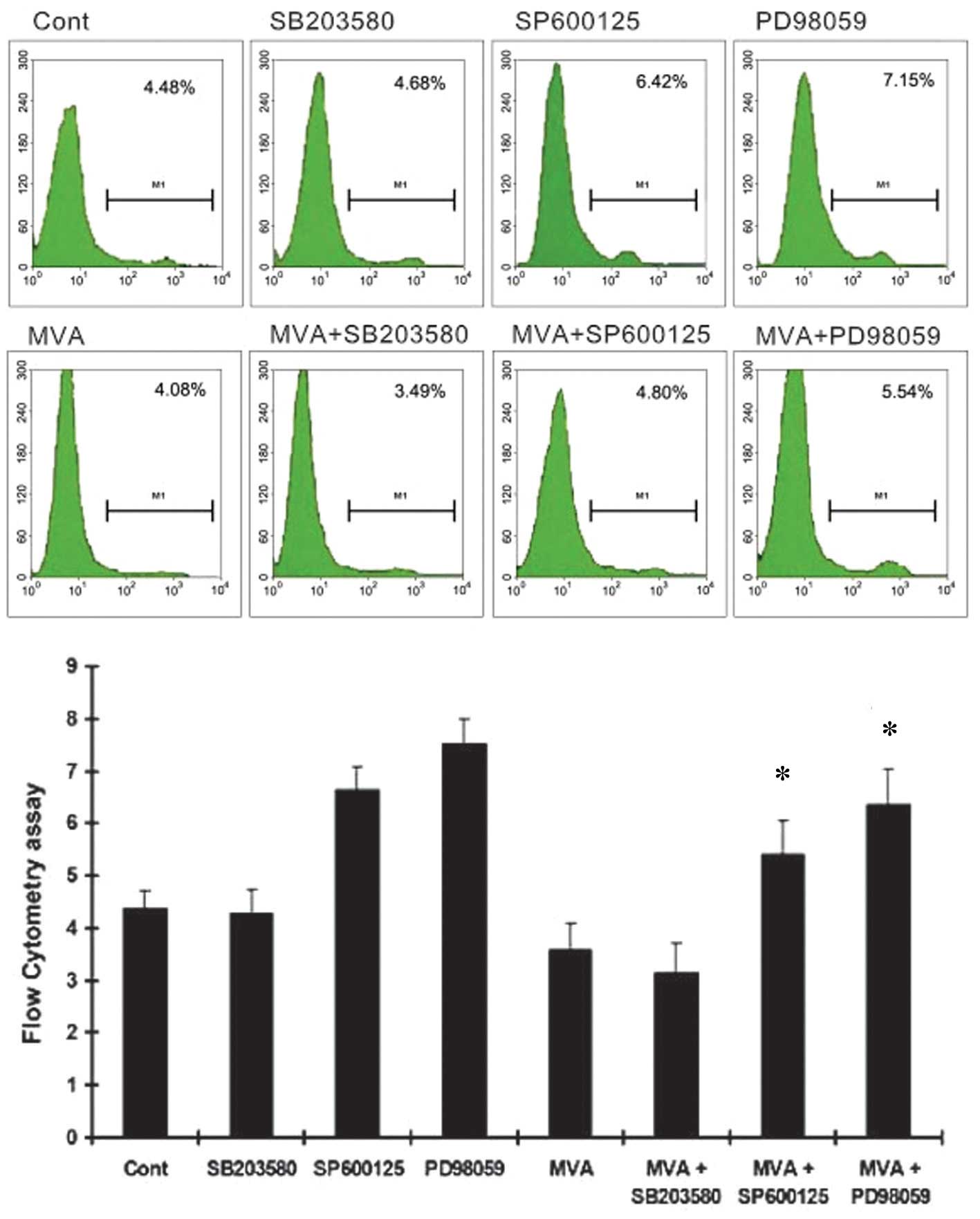
To elucidate the effect of MVA on HMC apoptosis, the
HMCs were cultured with MVA alone or in the presence of PD98059,
SP600125 or SB203580. Flow cytometric analysis was performed to
assess HMC apoptosis. The expression levels of Bcl-2 and Bax were
analyzed using western blot analysis. The data revealed that MVA
treatment significantly inhibited HMC apoptosis. PD98059 and
SP600125 markedly increased the number of apoptotic cells, while
SB203580 had no effect (Fig. 6).
In addition, the expression of Bcl-2 was significantly increased
following treatment with MVA. PD98059 and SP600125 inhibited the
upregulation of Bcl-2, while SB203580 had no effect on the
expression of Bcl-2 (Fig. 7A). The
expression of Bax was significantly downregulated by MVA treatment,
and PD98059 and SP600125 inhibited this downregulation, No
significant effect was observed following treatment with SB203580
(Fig. 7B). The ratio of Bcl-2/Bax
was significantly increased following MVA treatment, however, this
increased Bcl-2/Bax ratio was inhibited by treatment with PD98059
and SP600125, while no significant effect was observed following
treatment with SB203580 (Fig.
7C).
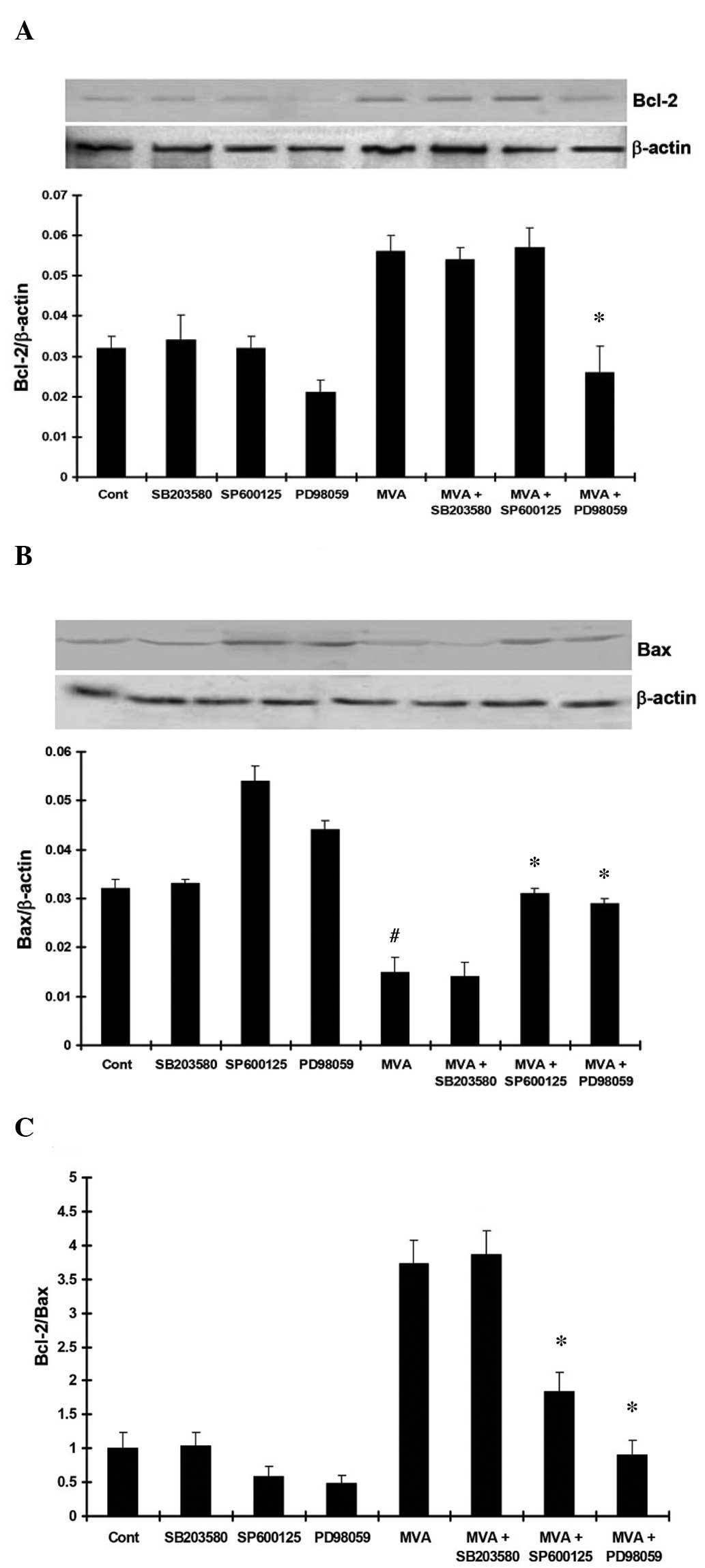 | Figure 7(A) MVA treatment significantly
upregulated the expression of Bcl-2 (P<0.05, vs. control).
PD98059 and SP600125 inhibited the upregulation of Bcl-2, while no
effect was observed following treatment with SB203580. (B) MVA
significantly downregulated the expression of Bax (P<0.05, vs.
control). PD98059 and SP600125 inhibited the downregulation of Bax,
while no effect was observed following treatment with SB203580. (C)
MVA significantly upregulated the Bcl-2/Bax ratio (P<0.05, vs.
control). PD98059 and SP600125 inhibited the upregulation of the
Bcl-2/Bax ratio, while no effect was observed following treatment
with SB203580. The results are presented as the mean ± standard
deviation. *P<0.05, vs. MVA only;
#P<0.05, vs. the control. MVA, mevalonate; Cont,
control; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2-associated X
protein. |
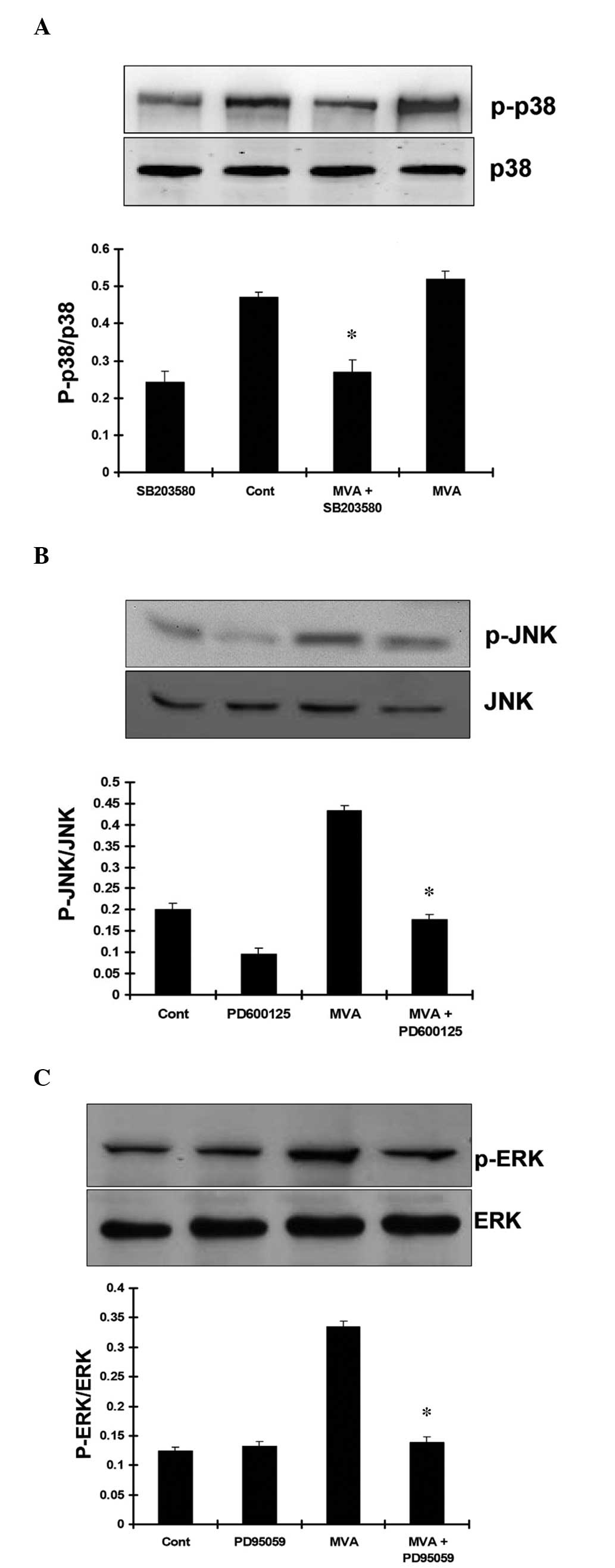
JNK and ERK mediate MVA-induced HMC
proliferation
The HMCs were cultured with MVA alone or in the
presence of PD98059, SP600125 and SB203580. Western blot analysis
was performed to analyze the protein expression levels of p-ERK1/2,
p-JNK and p-p38. The results demonstrated that treatment with MVA
had no significant effect on the expression of p-p38. Treatment
with SB203580 markedly inhibited the expression of p-p38 MAPK
(Fig. 8A). However, MVA
significantly stimulated the expression levels of JNK (Fig. 8B) and ERK (Fig. 8C). As expected, SP600125 and
PD98059 markedly inhibited the phosphorylation of JNK and ERK,
respectively.
Discussion
MC proliferation and ECM deposition are important in
renal fibrosis (9). Emerging
evidence has demonstrated that hyperlipidemia, particularly of
low-density lipoprotein, is involved in the proliferation of MCs,
and HMG-CoA inhibitors can inhibit this process (10). It has been suggested that the
HMG-CoA inhibitors inhibit this process by inhibiting MVA
metabolism. Thus, the present study hypothesized that MVA promotes
MC proliferation and mesangial matrix deposition. The results of
the present study demonstrated that MVA promoted HMC proliferation
in a dose-dependent manner (1×10−9–1×10−7
mol/l), with maximal stimulatory effects at a concentration of
1×10−7. However, higher concentrations
(>1×10−6 mol/l) inhibited HMC proliferation, which
may be due to higher concentrations exerting cytotoxic effects and
inhibiting HMC proliferation. The PI is an index reflecting DNA
synthesis. The results revealed that MVA increased the PI in the
dose-dependent manner, which coincided with its stimulatory effects
on MC proliferation.
Subsequently, the mechanisms of MVA on HMC
proliferation and ECM deposition were investigated. TGF-β is a
major factor in stimulating the expression of fibrotic protein, and
it has been observed that TGF-β promotes ECM accumulation (11,12).
In vitro, TGF-β promotes MC proliferation and matrix
synthesis. The present study demonstrated that MVA treatment
promoted the expression of TGF-β1 and thus promoted the expression
of ECM-associated proteins, including Col-IV and Col-I. These
responses may contribute to glomerular sclerosis and fibrosis.
Dysregulation of apoptotic pathways may also
contribute to renal damage and fibrosis (13). Bcl-2 family members, including
Bcl-2, an anti-apoptotic protein, and Bax, a pro-apoptotic protein,
tightly regulate cell apoptosis. The ratio of Bcl-2 to Bax
determines the survival of the cells. The present study revealed
that MVA treatment upregulated the expression of Bcl-2, while
downregulating the expression of Bax, which increased the Bcl-2/Bax
ratio and inhibited HMC apoptosis. This may have contributed to the
increase in the number of HMCs and ECM accumulation.
The molecular mechanisms underlying the effects MVA
on the HMCs were investigated. MAPKs, including ERK, JNK/SAPK, P38
MAPK and ERK5/BMK1, are key regulators of MC proliferation and ECM
deposition and are thus closely associated with the development of
mesangial proliferative glomerulonephritis. In the present study,
the MAPKs were investigated using their specific inhibitors. The
results demonstrated that MVA treatment significantly stimulated
JNK MAPK. SP600125, a specific inhibitor of JNK MAPK, markedly
promoted the expression of Bax and inhibited the Bcl-2/Bax ratio.
However, SP600125 had no significant effect on the PI. These
findings indicated that JNK MAPK may promote HMC proliferation by
downregulating the expression of Bax, thus inhibiting HMC
apoptosis. As SP600125 had no significant effect on the expression
levels of TGF-β1, Col-IV or Col-I, it was hypothesized that the
effects of JNK on HMCs may be secondary to its inhibitory effects
on cell apoptosis.
The present study also demonstrated that PD98059, a
specific ERK inhibitor, promoted HMC apoptosis, inhibited the
expression levels of TGF-β1, Col-IV, Col-I and Bcl-2, and
upregulated the expression of Bax, therefore, decreasing the
Bcl-2/Bax ratio. These findings coincided with the finding that MVA
promoted the phosphorylation of ERK. These results indicated that
MVA promoted the expression of TGF-β1 in an autocrine manner,
resulting in HMC proliferation and ECM accumulation. By contrast,
treatment with MVA inhibited HMC apoptosis by regulating the
expression levels of Bcl-2 and Bax.
P38 MAPK is another important component of the MAPK
family. In the present study SB203580, a specific inhibitor of p38
MAPK, exhibited no effects on HMC proliferation, cell apoptosis or
the expression levels of TGF-β1 Bcl-2 or Bax, indicating that the
effects of MVA on the HMCs were not mediated by the P38 MAPK
pathway.
Crosstalk among MAPKs is common in mammalian cells
(14). In the present study,
treatment with MVA activated JNK MAPK and ERK MAPK, but had no
effect on p38 MAPK. Despite this, whether any crosstalk occurs
between JNK MAPK and ERK MAPK remains to be elucidated (15,16).
The association between different MAPKs and their potential
crosstalk requires further investigation in HMCs.
In conclusion, the results of the present study
revealed the mechanism by which MVA stimulates MC proliferation,
and may provide a therapeutic strategy in the treatment of
proliferative glomerular disease.
Acknowledgments
The present study was supported by the Natural
Science Foundation of China (grant no. 81450033) and the Shanxi
International Cooperative Program (grant no. 2014081054).
References
|
1
|
Chiang CK, Sheu ML, Hung KY, Wu KD and Liu
SH: Honokiol, a small molecular weight natural product, alleviates
experimental mesangial proliferative glomerulonephritis. Kidney
Int. 70:682–689. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim SI, Na HJ, Ding Y, et al: Autophagy
promotes intracellular degradation of type I collagen induced by
transforming growth factor (TGF)-β1. J Biol Chem. 287:11677–11688.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zeng R, Xiong Y, Zhu F, et al: Fenofibrate
attenuated glucose-induced mesangial cells proliferation and
extracellular matrix synthesis via PI3K/AKT and ERK1/2. PLoS One.
8:e768362013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Owens TW, Foster FM, Valentijn A, Gilmore
AP and Streuli CH: Role for X-linked Inhibitor of apoptosis protein
upstream of mitochondrial permeabilization. J Biol Chem.
285:1081–1088. 2010. View Article : Google Scholar :
|
|
5
|
Kennedy DJ, Chen Y, Huang W, et al: CD36
and Na/K-ATPase-α1 form a proinflammatory signaling loop in kidney.
Hypertension. 61:216–224. 2013. View Article : Google Scholar
|
|
6
|
Jougasaki M, Ichiki T, Takenoshita Y and
Setoguchi M: Statins suppress interleukin-6-induced monocyte
chemoattractant protein-1 by inhibiting Janus kinase/signal
transducers and activators of transcription pathways in human
vascular endothelial cells. Br J Pharmacol. 159:1294–1303. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cassidy H, Radford R, Slyne J, et al: The
role of MAPK in drug induced kidney injury. J Signal Transduct.
2012:4636172012.
|
|
8
|
Feliers D and Kasinath BS: Erk in kidney
diseases. J Signal Transduct. 2011:7685122011.PubMed/NCBI
|
|
9
|
Huang K, Liu W, Lan T, et al: Berberine
reduces fibronectin expression by suppressing the S1P-S1P2 receptor
pathway in experimental diabetic nephropathy models. PLoS One.
7:e438742012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang W, He B, Shi W, Liang X, Ma J, et al:
Deletion of scavenger receptor A protects mice from progressive
nephropathy independent of lipid control during diet-induced
hyperlipidemia. Kidney Int. 81:1002–1014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang Y, Kong J, Deb DK, Chang A and Li
YC: Vitamin D receptor attenuates renal fibrosis by suppressing the
rennin-angiotensin system. J Am Soc Nephrol. 21:966–973. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiao HB, Liu RH, Ling GH, Xiao L, Xia YC,
et al: HSP47 regulates ECM accumulation in renal proximal tubular
cells induced by TGF-β1 through ERK1/2 and JNK MAPK pathways. Am J
Physiol Renal Physiol. 303:F757–765. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Linkermann A, Chen G, Dong G, Kunzendorf
U, Krautwald S and Dong Z: Regulated cell death in AKI. J Am Soc
Nephrol. 25:2689–2701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kondoh K, Sunadome K and Nishida E: Notch
signaling suppresses p38 MAPK activity via induction of MKP-1 in
myogenesis. J Biol Chem. 282:3058–3065. 2007. View Article : Google Scholar
|
|
15
|
Yang C, Patel K, Harding P, et al:
Regulation of TGF-beta1/MAPK-mediated PAI-1 gene expression by the
actin cytoskeleton in human mesangial cells. Exp Cell Res.
313:1240–1250. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim SI, Kwak JH, Na HJ, Kim JK, Ding Y and
Choi ME: Transforming growth factor-beta (TGF-beta1) activates TAK1
via TAB1-mediated autophosphorylation, independent of TGF-beta
receptor kinase activity in mesangial cells. J Biol Chem.
284:22285–22296. 2009. View Article : Google Scholar : PubMed/NCBI
|