Introduction
Sugars that contain aldehyde groups and are oxidized
to carboxylic acids are termed reducing sugars. Among reducing
sugars, 2-deoxy-D-ribose (dRib) has the highest reactivity to
induce protein glycation (1). Our
previous study reported that dRib causes cellular damage by
increasing the levels of oxidative stress and protein glycation in
pancreatic β-cells (2). Oxidative
stress is an important mechanism underlying β-cell failure in type
2 diabetes (3), and protein
glycation mediates chronic diabetic complications (4). High-glucose stimulation increases the
levels of intracellular reactive oxygen species (ROS) and protein
glycations in pancreatic β-cells, which is a lengthy process.
However, dRib promptly induces oxidative damage and protein
glycation in a short time (2).
Therefore, dRib may be used as a stimulant in place of glucose in
experimental studies of islet biology and diabetic
complications.
Captopril is an angiotensin-converting enzyme
inhibitor (ACEI) used in the treatment of hypertension. In large
randomized controlled trials, ACEIs exerted both
diabetes-preventive and blood pressure-lowering effects (5). These preventive effects may be
associated with the ACEI-induced preservation of β-cell function,
due to the fact that insulin secretion is an important mechanism
underlying the progression from glucose intolerance to diabetes
(6). Previous studies have
suggested that captopril decreases oxidative stress in cellular
(7) and animal (8) models. The ROS-scavenging activity of
the sulfhydryl group in the captopril structure is thought to be
responsible for its antioxidant properties (9).
The aim of the present study was to examine whether
captopril is able to prevent dRib-induced oxidative injury and
insulin suppression in pancreatic β-cells. In addition, the
mechanism underlying the antioxidative effects of captopril on
dRib-induced β-cell damage was investigated.
Materials and methods
Materials
Captopril, buthionine sulfoximine (BSO),
dihydrorhodamine 123 (DHR 123), Ficoll, sodium azide, Girard-T
reagent, 5,5-dimethyl-1-pyrroline-N-oxide, FeSO4, and
H2O2 were purchased from Sigma-Aldrich (St
Louis, MO, USA). dRib, HEPES, guanidine hydrochloride, potassium
phosphate monobasic, and potassium phosphate dibasic were purchased
from Amresco, LLC (Solon, OH, USA). Hydrochloric acid and ethanol
were purchased from Merck Millipore (Darmstadt, Germany).
RPMI-1640, Dulbecco's phosphate-buffered saline (DPBS), trypsin,
penicillin, streptomycin, and TRIzol® reagent were
purchased from Gibco Life Technologies (Carlsbad, CA, USA).
Collagenase P and Hank's balanced salt solution (HBSS) were
purchased from Roche Diagnostics GmbH (Mannheim, Germany). Fetal
bovine serum (FBS) was purchased from GE Healthcare Life Sciences
(Logan, UT, USA). All culture dishes and tubes were purchased from
BD Biosciences (Franklin Lakes, NJ, USA).
Cell culture
Syrian hamster insulinoma HIT-T15 cells were
obtained from the Korean Cell Line Bank (Seoul, Korea). The cells
were cultured at 37°C in an atmosphere containing 5% CO2
and 95% O2, in RPMI-1640 medium supplemented with 10%
FBS, 100 mg/ml penicillin, and 100 mg/ml streptomycin. The HIT-T15
cells were separated by trypsinization and subcultured until
reaching ~70% confluence. Two days following subculture, the
culture medium was discarded and replaced with RPMI-1640
supplemented with 10% FBS, and the cells were pretreated with 0.1,
0.5, or 1 mM captopril and/or 200 µM BSO for 30 min, prior
to the addition of 30 or 50 mM dRib to the medium. The cultures
were then further incubated for 6 or 24 h.
Islet isolation and culture
The method of islet isolation and culture was
carried out as previously described (10). Sprague Dawley rats (age, 6–8 weeks)
were provided by Orient Bio Inc. (Seongnam, Korea). The rats were
housed under a controlled temperature (23°C), 50% humidity and a 12
h light-dark cycle with ad libitum access to water and food.
A total of 10 rats were sacrificed by cervical dislocation, an
incision was made into the abdomen, and 9–10 ml collagenase P in 1
mg/ml HBSS was injected into the pancreas via the bile duct. The
distended pancreas was then removed for digestion at 37°C for ~12
min, and the tissue samples were separated using a Ficoll gradient.
Isolated islets were suspended in RPMI-1640 supplemented with 20%
FBS and 11.1 mM glucose for 24 h prior to experimentation.
Thereafter, the medium was replaced by fresh RPMI-1640 supplemented
with 10% FBS, and 10 mM dRib was added to the medium following
pretreatment with 0.1, 0.5, or 1 mM captopril for 30 min. The
cultures were incubated for 6 h. The present study was approved by
the Institutional Animal Care and Use Committee of Jeju National
University (Jeju, Korea).
Assessment of cell viability
The HIT-T15 cells were cultured in 24-well plates at
a density of 1×105 cells/well. The cells were then
incubated with 30 mM dRib for 24 h, with or without captopril. The
cells were subsequently harvested and stained with 0.4% trypan blue
(Sigma-Aldrich) for 5 min. The cells were transferred to a
hemocytometer (Marienfield-Superior, Baden-Württemberg, Germany),
and both the dead cells that did not exclude the dye, and the
viable cells that excluded the dye were counted. The dead and
viable cells were counted using an inverted fluorescence microscope
(Olympus IX50; Olympus, Tokyo, Japan). The results were expressed
as the percentage of viable cells in total cell population.
Flow cytometry to assess apoptosis
The HIT-T15 cells were stained with fluorescein
isothiocyanate (FITC)-conjugated Annexin V and propidium iodide
(PI) using a Vybrant Apoptosis Assay kit #2, according to the
manufacturer's instructions (Molecular Probes Life Technologies,
Carlsbad, CA, USA). Briefly, the HIT-T15 cells were cultured in
six-well plates at a density of 5×105 cells/well. The
cells were then treated with 50 mM dRib and captopril for 24 h.
Following gentle washing with PBS, the cells were collected by
trypsinization and centrifugation. The cell pellets were
resuspended in PBS and centrifuged (200 x g for 5 min at 4°C),
prior to being resuspended and incubated with FITC-conjugated
Annexin V and PI staining solution at room temperature for 15 min.
The stained cells were analyzed using a FACSCalibur
fluorescence-activated cell sorter (BD Bioscience, San Jose, CA,
USA), and the data were analyzed using CellQuest software (BD
Bioscience). A total of 1×104 cells were evaluated for
each sample.
Assessment of the intracellular levels of
ROS
Intracellular ROS levels were investigated using a
DHR 123 dye. Cellular fluorescence intensity is directly correlated
with intracellular ROS levels. The HIT-T15 cells were plated in
24-well culture plates at a density of 5×105 cells/well.
The cells were treated with 50 mM dRib and 1 mM captopril for ≤6 h,
due to the possibility of extensive cell death interfering with ROS
measurements. A total of 5 µM DHR 123 was added to the cell
cultures 30 min prior to harvesting. The cells were then washed
twice with PBS and harvested using 0.05% trypsin. The cells were
then centrifuged and resuspended in PBS, and the intracellular
levels of ROS were measured using a FACScan instrument (BD
Bioscience). A total of 1×104 cells were analyzed per
sample. The data were calculated as the mean fluorescence
intensity, and expressed as the fold difference, as compared with
control untreated cells.
Measurement of the intracellular levels
of glutathione
The intracellular levels of glutathione were
measured using a Glutathione Assay kit (Cayman Chemical, Ann Arbor,
MI, USA) according to an enzymatic recycling method using
glutathione reductase. The HIT-T15 cells were cultured in 6-well
plates at a density of 5×105 cells/well. The cells were
treated as described above prior to being washed with ice-cold DPBS
and sonicated using a Vibra Cell sonicator (Sonics & Materials,
Danbury, CT, USA) at 30% of the maximal amplitude, and were
subsequently centrifuged at 10,000 x g for 15 min. The resulting
supernatants were used to measure glutathiones. Exclusive
measurement of oxidized glutathione (GSSG) is accomplished by first
derivatizing reduced glutathione (GSH) as described by the
manufacturer of the Glutathione Assay kit. The amount of GSH can
also be determined by subtracting the quantity of GSSG from the
total amount of glutathiones. The glutathione concentrations were
expressed as µM/l in reference to a standard curve.
Assessment of reactive dicarbonyl and
advanced glycation end product (AGE) formation
The reaction mixture contained 10 mg/ml bovine serum
albumin (BSA; Gibco Life Technologies), 0.02% sodium azide, and 100
mM CuSO4 in 100 mM sodium phosphate buffer (pH 7.4),
with a final volume of 4 ml. The mixture was incubated with 10, 30,
or 50 mM dRib and 1 mM captopril at 37°C for three days in capped
polystyrene tubes. All incubations were carried out in
quadruplicate. The incubated sample mixtures were dialyzed against
100 mM sodium phosphate buffer (pH 7.4) for 48 h, in order to
remove unbound dRib and any other impurities. The sample mixtures
were then stored at ≤0°C prior to being assayed for dicarbonyl or
AGE fluorescence.
The levels of albumin-attached dicarbonyls were
measured using the Girard-T reagent as previously described
(11). Briefly, the reaction
mixture containing 60 ml sample or each standard solution, 20 ml
0.5 N sodium borate (pH 9.2), and 20 ml 0.1 N Girard-T reagent was
incubated at 30°C for 10 min. A total of 100 ml 0.1 N sodium borate
(pH 9.2) was added to the mixture, and the absorbance was measured
at 375 nm against a zero blank using a UVmini-1240
spectrophotometer (Shimadzu Corporation, Kyoto, Japan). The
concentration of the reactive dicarbonyl was calculated by
comparison with standard solutions, and was corrected with the
total protein concentration. The present study used glyoxal to
normalize the data.
AGE fluorescence of the incubation mixture was
measured at excitation and emission wavelengths of 350 and 450 nm,
respectively, against an unincubated blank containing dRib and
captopril using a DTX 880 Multimode Detector fluorescence reader
(Beckman Coulter, Inc., Fullerton, CA, USA). Any significant
difference observed between the sample and the control was used as
an indication of AGE formation.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Rat islets were pre-treated with captopril for 30
min at 0.1, 0.5, or 1.0 mM, and then cultured with 10 mM dRib for 6
h. Total RNA was isolated using TRIzol®, according to
the manufacturer's instructions. RNA isolation was carried out in
an RNase-free environment. A total of 4 mg aliquots of RNA were
reverse transcribed using MuLV reverse transcriptase (Promega
Corporation, Madison, WI, USA), oligo (dT)15 primer, dNTP (0.5 mM)
and 1 unit RNase inhibitor (Promega Corporation). RT-qPCR was
performed using a 2 SYBR Green PCR Master Mix (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) and an iQ™ 5 Multicolor
Real-Time PCR Detection system (Bio-Rad Laboratories, Inc.). The
PCR was performed to amplify the synthesized cDNA in the presence
of specific primers, for 40 cycles at 95°C for 20 sec, 60°C for 20
sec, and 72°C for 30 sec, with an initial cycle of 95°C for 5 min.
The PCR reactions were carried out using the following primers for
the cDNA sequences: Insulin forward,
5′-GAACGGCTACAATCTCCGGAGGCA-3′, and reverse,
5′-TTCCAGAGGCGGTCGTGGTCACAA-3′; pancreatic and duodenal homeobox 1
(PDX-1) forward, 5′-AGTTTGCAGGCTCGCTGGGAA-3′, and reverse,
5′-TTCCACGCGTGAGCTTTGGTGG-3′; and β-actin forward,
5′-TCCTGGCCTCACTGTCCAC-3′, and reverse, 5′-GGGCCGGACTCATCGTACT-3′.
All primers were synthesized by Bioneer Corporation (Daejeon,
Korea).
Measurement of insulin content
The isolated islets were pre-incubated with 0.1, 0.5
or 1 mM captopril for 30 min, and then stimulated with 10 mM dRib
for 6 h. The cells were subsequently harvested and washed twice
with cold DPBS. The cells were lysed using a lysis buffer
containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 2 mM EDTA, 1 mM
ethylene glycol tetraacetic acid, 1 mM
Na3VO4, 10 mM NaF, 1 mM dithiothreitol, 1 mM
phenylmethylsulfonylfluoride, 25 mg/ml aprotinin, 25 mg/ml
leupeptin, and 1% Nonidet P-40, in order to obtain whole cell
protein, which was kept on ice for 30 min. The cell lysates were
centrifuged at 15,000 x g for 15 min at 4°C. The supernatant was
used to determine the levels of insulin and protein contents. The
levels of insulin were determined using an ELISA kit (Mercodia,
Uppsala, Sweden) with rat insulin as a standard, according to the
manufacturer's instructions. Protein content was measured using a
BCA Protein Assay Reagent kit (Pierce Biotechnology, Inc.,
Rockford, IL, USA). The intracellular insulin content was corrected
for total protein content, and expressed as mg/mg protein.
Statistical analysis
All data are expressed as the mean ± standard error.
The statistical difference between the groups were analyzed by
one-way analysis of variance, followed by Duncan's post hoc and
Student's t-test. All analyses were performed using SPSS 14.0
software (SPSS Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Captopril prevents dRib-triggered
cytotoxicity and apoptosis
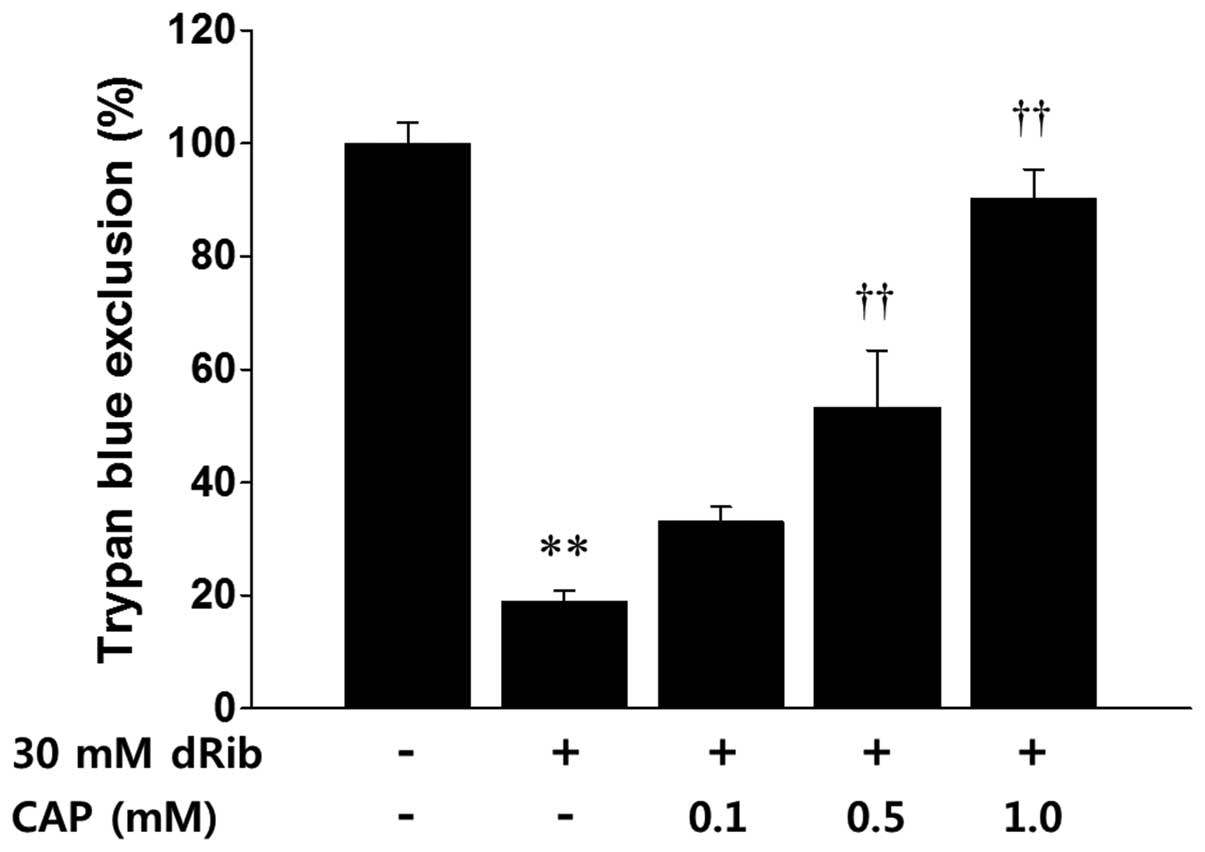
Trypan blue exclusion demonstrated that a 24 h
incubation of the HIT-T15 cells with 30 mM dRib led to cell death.
Conversely, treatment with 0.1, 0.5, or 1 mM captopril inhibited
the dRib-triggered cytotoxicity in a dose-dependent manner
(Fig. 1). Treatment with 50 mM
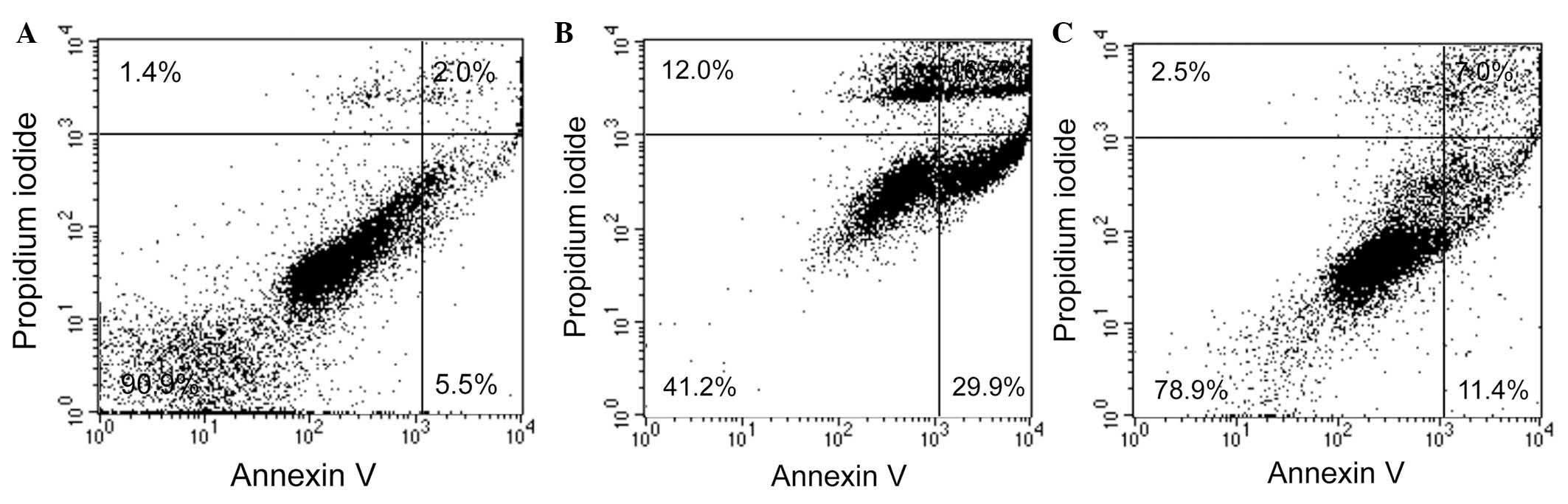
dRib increased the percentage of apoptotic cells as shown by
Annexin V and PI double staining; however, these changes were
reversed by pretreatment with 1 mM captopril (Table I, Fig.
2).
 | Table IPercentages of Annexin V-positive
cells in the control and dRib-treated Syrian hamster insulinoma
HIT-T15 cells, with or without CAP treatment. |
Table I
Percentages of Annexin V-positive
cells in the control and dRib-treated Syrian hamster insulinoma
HIT-T15 cells, with or without CAP treatment.
| Group | Annexin V-positive
cells (%) |
|---|
| Control | 8.3±2.3 |
| 50 mM dRib | 56.3±7.7a |
| 1 mM CAP + 50 mM
dRib | 20.1±1.3b |
Captopril reverses the dRib-induced
increase in the intracellular levels of ROS and glutathione
depletion
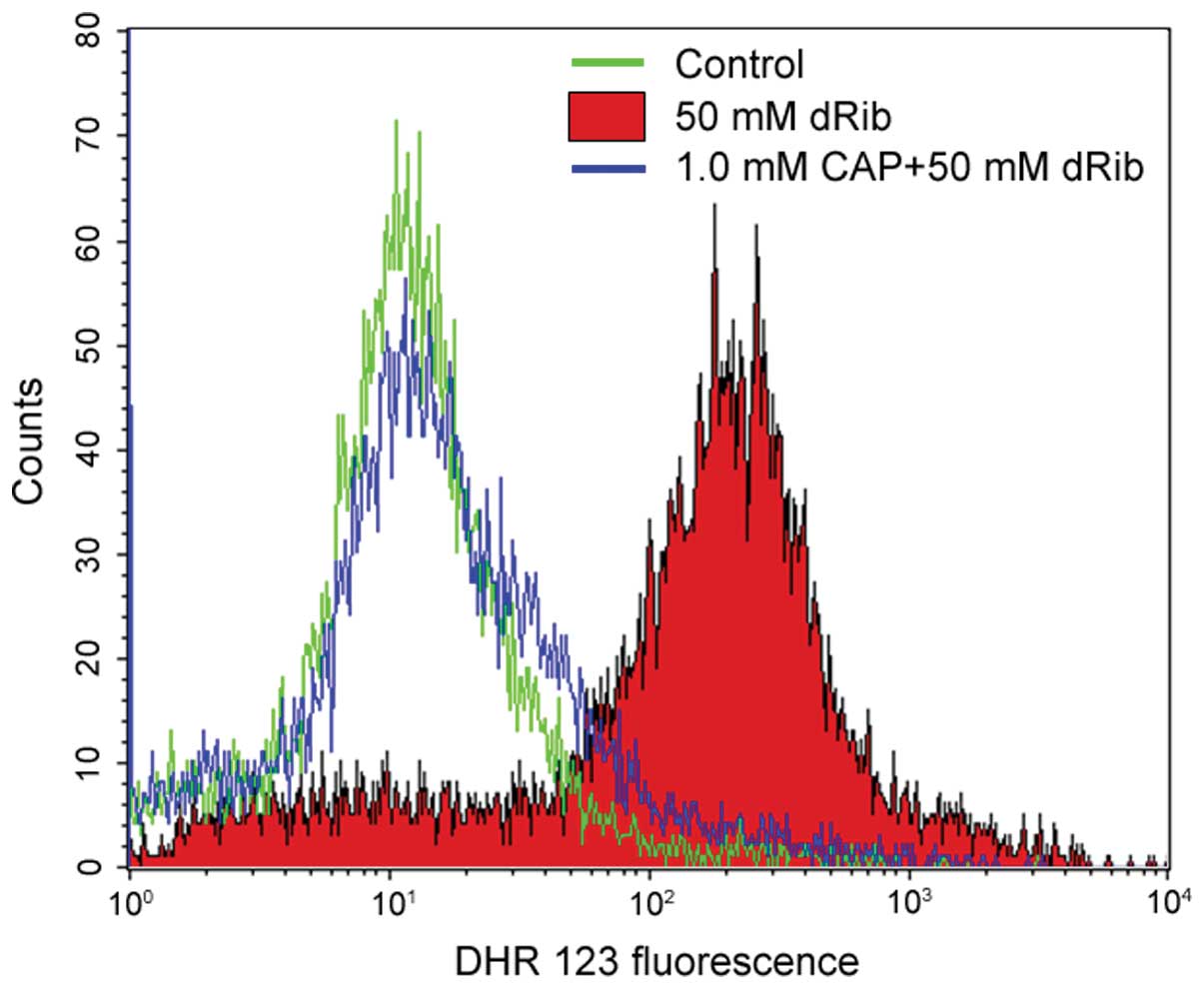
The flow cytometric analysis using DHR 123 dye
determined that 50 mM dRib stimulation for 6 h increased the levels
of ROS by ~12-fold in the HIT-T15 cells. The addition of 1 mM
captopril reversed the dRib-induced increase in intracellular
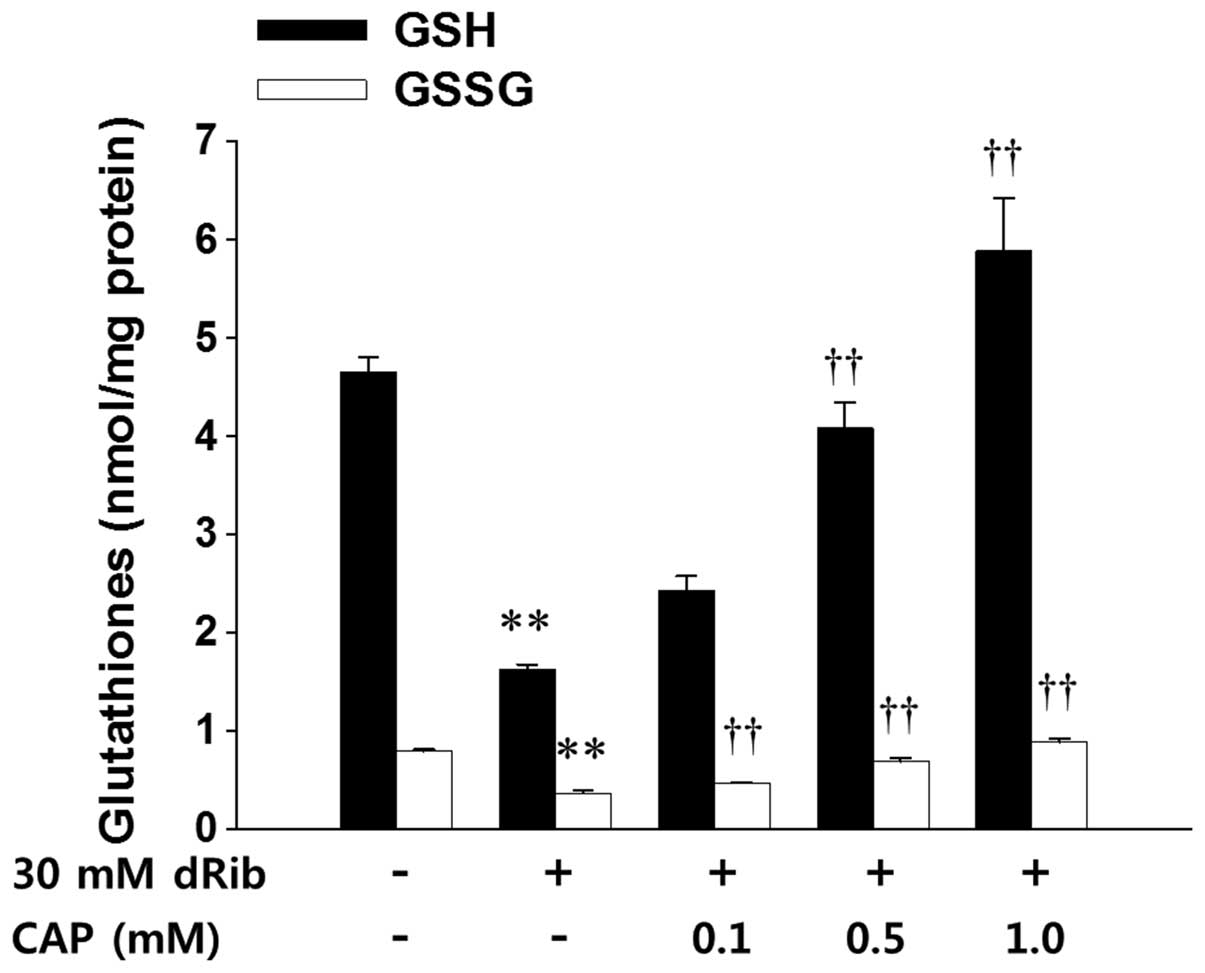
levels of ROS (Table II, Fig. 3). The present study measured the
intracellular levels of both GSH and GSSG in order to assess the
total levels of glutathione and intracellular redox potential in
HIT-T15 cells. A 6 h stimulation with 30 mM dRib decreased the
intracellular levels of GSH and GSSG, and preincubation with
various concentrations of captopril dose-dependently restored these
levels (Fig. 4). However, the
intracellular GSH/GSSG ratio did not differ between the treatment
conditions.
| Table IIRelative intracellular levels of
reactive oxygen species in the control and dRib-treated Syrian
hamster insulinoma HIT-T15 cells, with or without CAP
treatment. |
Table II
Relative intracellular levels of
reactive oxygen species in the control and dRib-treated Syrian
hamster insulinoma HIT-T15 cells, with or without CAP
treatment.
| Group | Relative fluorescence
(fold change from control) |
|---|
| Control | 1.0 |
| 50 mM dRib | 11.9±2.5a |
| 1 mM CAP + 50 mM
dRib | 1.8±0.4b |
Captopril restores the dRib-mediated
suppression of insulin transcription, and decreases the levels of
insulin in rat islets
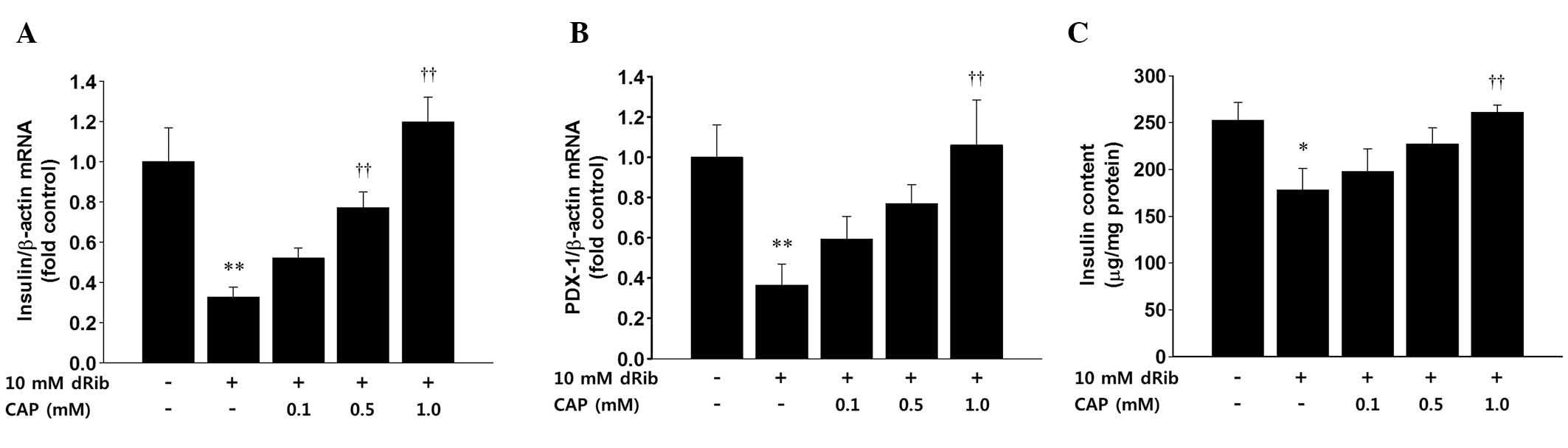
The present study examined the effects of dRib on
the mRNA expression levels of insulin and PDX-1, as well as the
insulin content in rat islets. A lower concentration of dRib and a
shorter treatment time were chosen to stimulate the islets due to
the fact that cell damage may interfere with mRNA and protein
quantification. RT-qPCR was used to quantify the mRNA expression
levels of insulin and PDX-1 in the rat islets. Stimulation of the
isolated rat islets with 10 mM dRib for 6 h significantly
suppressed the mRNA expression levels of insulin and PDX-1. This
suppression was reversed by pretreatment with captopril in a
dose-dependent manner (Fig. 5A and
B). The intracellular levels of insulin were measured by ELISA.
The 6 h stimulation of the islets with 10 mM dRib significantly
decreased the levels of insulin, and this insulin suppression was
dose-dependently attenuated by treatment with captopril (Fig. 5C).
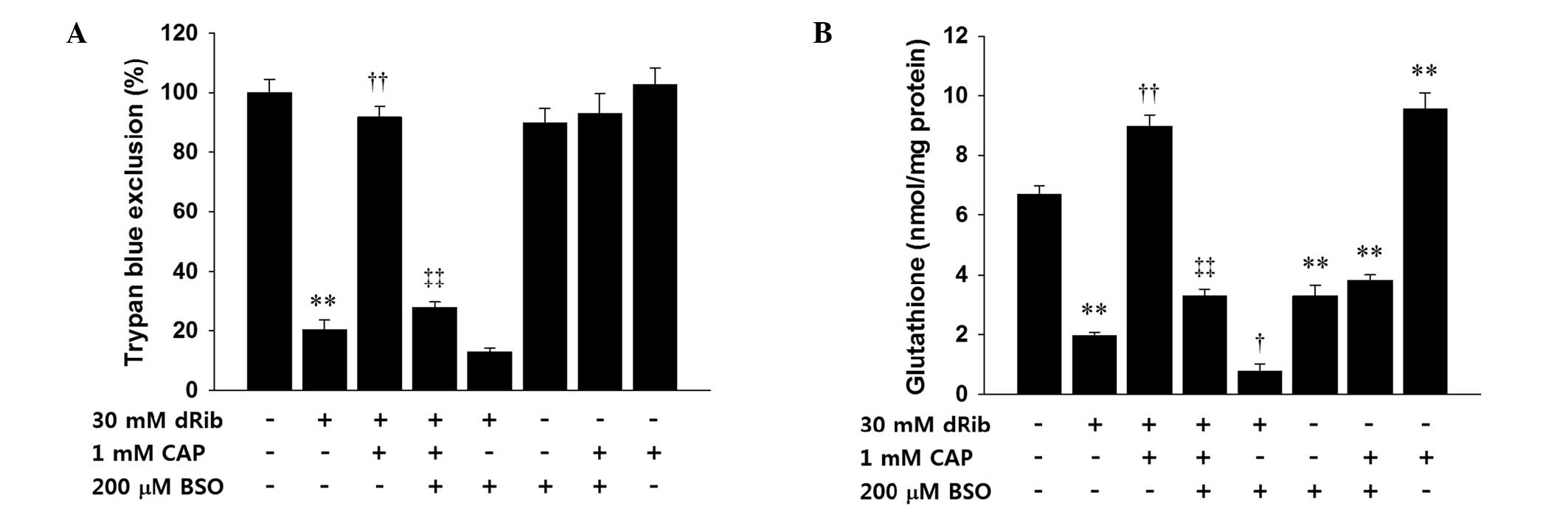
Treatment with BSO inhibits the
protective effects of captopril on dRib-induced glutathione
depletion and cytotoxicity
BSO is a specific and irreversible inhibitor of
glutamate cysteine ligase, which is the rate-limiting enzyme of
glutathione synthesis. The present study used BSO to investigate
whether the protective effects of captopril were related to an
increase in glutathione synthesis. Pre-treatment with BSO inhibited
the protective effects of captopril on dRib-induced cytotoxicity.
Co-administration of dRib and BSO appeared to increase cell death,
as compared with treatment with dRib alone; however, these apparent
results were not statistically significant. Treatment with
captopril significantly reduced the cytotoxicity caused by the
co-administration of dRib and BSO. BSO alone did not significantly
alter cell viability, as compared with the control (Fig. 6A). Treatment with BSO suppressed
the captopril-induced prevention of intracellular glutathione
depletion caused by dRib. Co-treatment with dRib and BSO increased
the depletion of glutathione, as compared with dRib alone. Addition
of captopril restored the glutathione depletion caused by
co-treatment with dRib and BSO. Unlike the Trypan blue exclusion,
the glutathione assay demonstrated that BSO alone significantly
decreased the intracellular levels of glutathione, as compared with
the control. These effects were not reversed by the addition of
captopril (Fig. 6B).
dRib-induced protein glycation is not
suppressed by treatment with captopril
The present study assessed the formation of
dicarbonyl and AGE in order to investigate whether captopril is
able to inhibit the dRib- induced protein glycation in
vitro. Protein-bound dicarbonyl and AGE formation were measured
using Girard-T reagent and fluorometry, respectively. Following 3
days incubation of albumin with 10, 20, or 30 mM dRib, dRib
dose-dependently increased dicarbonyl formation and AGE
fluorescence. Treatment with captopril did not suppress these
dRib-induced effects (Fig. 7A and
B).
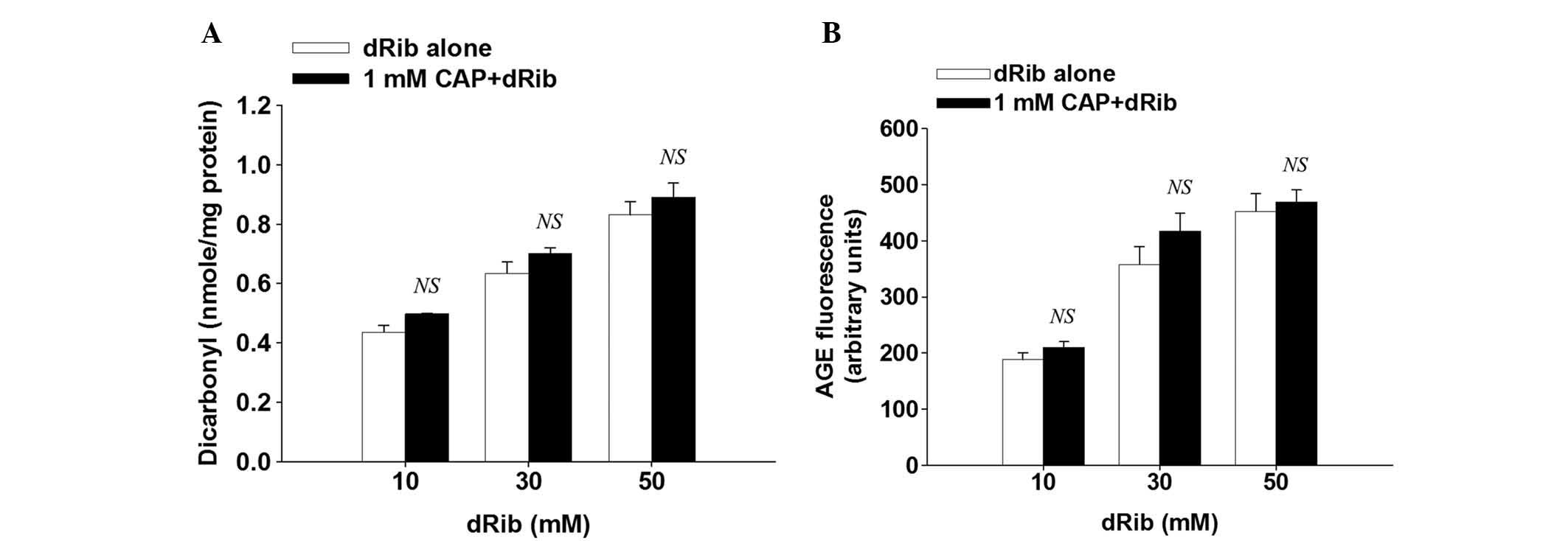 | Figure 7Inhibitory effects of captopril (CAP)
on the formation of (A) protein-bound dicarbonyl and (B) advanced
glycation end products. Bovine serum albumin (10 mg/ml) was
incubated with 10, 30, or 50 mM 2-deoxy-D-ribose (dRib) at 37°C for
3 days in the presence or absence of CAP. Dicarbonyl concentration
was estimated by adduct formation with the Girard-T reagent.
Advanced albumin glycation was determined by measuring the
fluorescence intensity (excitation and emission wavelength of 350
and 450 nm, respectively), as compared with a non-incubated blank
containing the protein, dRib, and CAP. The data are presented as
the mean ± standard error. This experiment was performed twice, in
quadruplicate. NS, no significant difference, as compared with the
dRib-alone group, as determined by Student's t test. |
Discussion
Our previous study investigated the mechanism
underlying dRib-induced damage in a pancreatic β-cell line
(2). In our previous study, the
metal chelator diethylenetriaminepentaacetic acid (DTPA) suppressed
dRib-induced increases in intracellular ROS in HIT-T15 cells;
however, DTPA did not reduce the formation of dRib-mediated
dicarbonyl and AGE in vitro. Pyridoxamine, which is a
post-Amadori inhibitor, was able to decrease the dRib-induced
formation of dicarbonyl and AGE; however, pyridoxamine did not
suppress the dRib-induced production of intracellular ROS. Both
DTPA and pyridoxamine protected the HIT-T15 cells against
dRib-induced cytotoxicity and apoptosis. These results suggested
that both oxidative stress and protein glycation are important
mechanisms underlying dRib-induced β-cell damage.
The present study demonstrated that captopril did
not suppress dRib-induced increases in protein-bound dicarbonyl
concentration and AGE fluorescence. Dicarbonyl is the
monosaccharide-derived precursor of AGE (12); however, captopril does not inhibit
protein glycation. In the present study, captopril decreased the
intracellular levels of oxidative stress via the replenishment of
depleted glutathione. The mechanism underlying the effects of
captopril, which is thought to protect against oxidative stress,
may involve an increase in the synthesis of intracellular GSH, due
to the fact that BSO, a specific inhibitor of GCL, suppressed the
protective effects of captopril on dRib-induced cytotoxicity and
glutathione depletion. Therefore, an increase in glutathione
synthesis, rather than an inhibition of protein glycation, may be
the mechanism underlying the protective effects of captopril in
dRib-induced β-cell damage.
Previous studies have reported that captopril
exhibits antioxidative effects (7,9,13,14).
The majority of these studies have suggested that captopril exerts
direct ROS-scavenging activity via its sulfhydryl group (7,9,13).
Using electron spin resonance spectroscopy, the present study also
investigated the ROS-scavenging effects of captopril in a cell-free
system (data not shown). However, these results did not support the
hypothesis that ROS-scavenging activity contributes to the
protective effects of captopril against dRib-induced oxidative
damage in β-cells. Pre-treatment with captopril increased the
intracellular concentrations of both GSH and GSSG. These results
indicated that captopril regulates the total intracellular levels
of glutathione rather than the intracellular glutathione redox
potential. Treatment with BSO inhibited the protective effects of
captopril on dRib-induced glutathione depletion and cytotoxicity.
These results demonstrated that intracellular glutathione synthesis
is an important mechanism underlying the protective effects of
captopril on dRib-induced β-cell damage. However, the results of
the present study did not identify a specific mechanism to explain
the captopril-induced increase in glutathione synthesis. The
possible mechanisms underlying glutathione synthesis are increased
activity and expression levels of GCL, and/or increased delivery of
GCL substrates. Further research is required in order to further
elucidate this mechanism.
Glucose toxicity is defined as the non-physiological
and potentially irreversible β-cell damage caused by chronic
exposure to supraphysiological glucose concentrations (3). Long-term stimulation via exposure to
high glucose concentrations causes ROS production in pancreatic
β-cells, and this oxidative stress suppresses the mRNA expression
levels of PDX-1, and PDX-1 binding to the insulin gene promoter. As
a result, insulin gene expression and secretion decrease (15,16).
In the present study, dRib stimulation decreased the mRNA
expression levels of PDX-1 and insulin in isolated islets, and
treatment with captopril reversed these dRib-induced effects. These
results suggested that dRib is able to induce β-cell damage, as
does high glucose concentrations in rat islets. It is also possible
that captopril may prevent glucose toxicity.
The present study had limitations. Captopril was
developed for the purpose of inhibiting ACE; however, the present
study did not evaluate the role of the inhibition of ACE in
preventing dRib-induced β-cell damage. The present study
investigated the extent of protein glycation using bovine serum
albumin in vitro. Technical issues disturbed the measurement
of the concentration levels of intracellular protein-bound
dicarbonyl and AGE in β-cells. The results of the present study did
not successfully elucidate the mechanism underlying dRib-induced
oxidative stress. Our previous study reported that intracellular
glutathione depletion was an important factor in dRib-mediated
oxidative damage (17). Both our
previous and present studies have prompted further investigation
into the mechanism underlying dRib depletion of intracellular
glutathione, in order to elucidate the mechanisms underlying for
the protective effects of captopril on dRib-induced β-cell
damage.
In conclusion, the results of the present study
demonstrated that captopril suppressed dRib-induced oxidative
damage by increasing the levels of glutathione synthesis in
β-cells. Protein glycation was not associated with the protective
effects of captopril. The ROS-scavenging activity of captopril did
not appear to be important for the prevention of dRib-triggered
β-cell damage. Further research is required in order to identify
the mechanism underlying the captopril-induced increase in
intracellular glutathione synthesis. Captopril may attenuate
oxidative stress in pancreatic β-cells and may prevent glucose
toxicity in type 2 diabetes. Captopril is used in the treatment of
high blood pressure, and the results of the present study suggest
that it may have potential as a novel treatment for preventing
β-cell failure in patients with type 2 diabetes.
Acknowledgments
The present study was supported by research grants
from Jeju National University Hospital and the Korean Diabetes
Association in 2010.
References
|
1
|
Monnier VM: Nonenzymatic glycosylation,
the Maillard reaction and the aging process. J Gerontol.
45:B105–B111. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Koh G, Lee DH and Woo JT: 2-Deoxy-D-ribose
induces cellular damage by increasing oxidative stress and protein
glycation in a pancreatic beta-cell line. Metabolism. 59:325–332.
2010. View Article : Google Scholar
|
|
3
|
Robertson RP, Harmon J, Tran PO, Tanaka Y
and Takahashi H: Glucose toxicity in beta-cells: Type 2 diabetes,
good radicals gone bad, and the glutathione connection. Diabetes.
52:581–587. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brownlee M: Biochemistry and molecular
cell biology of diabetic complications. Nature. 414:813–820. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Scheen AJ: Prevention of type 2 diabetes
mellitus through inhibition of the Renin-Angiotensin system. Drugs.
64:2537–2565. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
DeFronzo RA: Lilly lecture 1987. The
triumvirate: Beta-cell, muscle, liver A collusion responsible for
NIDDM. Diabetes. 37:667–687. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mak IT, Freedman AM, Dickens BF and
Weglicki WB: Protective effects of sulfhydryl-containing
angiotensin converting enzyme inhibitors against free radical
injury in endothelial cells. Biochem Pharmacol. 40:2169–2175. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hayek T, Attias J, Smith J, Breslow JL and
Keidar S: Antiatherosclerotic and antioxidative effects of
captopril in apolipoprotein E-deficient mice. J Cardiovasc
Pharmacol. 31:540–544. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Andreoli SP: Captopril scavenges hydrogen
peroxide and reduces, but does not eliminate, oxidant-induced cell
injury. Am J Physiol. 264:F120–F127. 1993.PubMed/NCBI
|
|
10
|
Lacy PE and Kostianovsky M: Method for the
isolation of intact islets of Langerhans from the rat pancreas.
Diabetes. 16:35–39. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mitchel RE and Birnboim HC: The use of
Girard-T reagent in a rapid and sensitive methods for measuring
glyoxal and certain other alpha-dicarbonyl compounds. Anal Biochem.
81:47–56. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Degenhardt TP, Thorpe SR and Baynes JW:
Chemical modification of proteins by methylglyoxal. Cell Mol Biol
(Noisy-le-grand). 44:1139–1145. 1998.
|
|
13
|
Bagchi D, Prasad R and Das DK: Direct
scavenging of free radicals by captopril, an angiotensin converting
enzyme inhibitor. Biochem Biophys Res Commun. 158:52–57. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen SX, Song T, Zhou SH, Liu YH, Wu SJ
and Liu LY: Protective effects of ACE inhibitors on vascular
endothelial dysfunction induced by exogenous advanced oxidation
protein products in rats. Eur J Pharmacol. 584:368–375. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Harmon JS, Gleason CE, Tanaka Y, Oseid EA,
Hunter-Berger KK and Robertson RP: In vivo prevention of
hyperglycemia also prevents glucotoxic effects on PDX-1 and insulin
gene expression. Diabetes. 48:1995–2000. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Robertson RP: Chronic oxidative stress as
a central mechanism for glucose toxicity in pancreatic islet beta
cells in diabetes. J Biol Chem. 279:42351–42354. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Koh G, Kim MK, Yang EJ and Lee DH:
Gliclazide does not fully prevent 2-deoxy-D-ribose-induced
oxidative damage because it does not restore glutathione content in
a pancreatic β-cell line. Oxid Med Cell Longev. 2012:3906782012.
View Article : Google Scholar
|