Introduction
Apoptosis of photoreceptors leads to retinal
dysfunction in retinal degenerative diseases, including retinitis
pigmentosa (RP), however, the exact mechanism remains to be
elucidated (1,2). Measurements using oxygen electrodes
in a RCS rat model of RP have revealed that oxidative stress may be
involved in the pathogenesis of photoreceptor degeneration,
whereas, oxidative damage in the outer retina in a transgenic pig
model results in gradual cone cell death, and administration of a
cocktail of antioxidants reduces markers of oxidative damage in
cones and reduces cone cell death in rd1 mice (3–5).
Oxidative stress also triggers the apoptosis of photoreceptors
in vitro, and antioxidants prevent oxidative stress-induced
apoptosis of photoreceptors (6).
Mitochondria are known to be a major source of
intracellular reactive oxygen species (ROS) and are particularly
vulnerable to oxidative stress. Increasing evidence suggests that
ROS are key in promoting the release of cytochrome c from
the mitochondria (7,8), and cytochrome c in the
cytoplasm triggers a series of apoptotic signal transduction
processes, resulting in apoptotic cell death (9,10).
It appears promising to target mitochondrial oxidative stress using
antioxidant therapy, however, there are several difficulties in
developing and using antioxidative drugs, in the delivery of drugs
to the mitochondria, minimization of adverse effects and delivering
drugs across the blood-retina barrier (11).
SS31 is a cell-permeable mitochondria-targeted
antioxidant peptide. Previous studies have demonstrated that SS31
selectively partitions to the inner mitochondrial membrane, where
it scavenges ROS generated by the electron transport chain. In
addition, studies have revealed that SS31 can prevent the
Ca2+-induced mitochondrial permeability transition (MPT)
and release of cytochrome c (11,12).
Several animal investigations have demonstrated that SS31 may be
beneficial in models of ischemia/reperfusion-induced myocardial
infarction (13), brain
infarction, Alzheimer's disease (AD) and amyotrophic lateral
sclerosis (ALS) (13–16). However, it whether SS31 has a
protective effect on retinal degenerative diseases by attenuating
oxidant injury to photoreceptor cells remains to be elucidated.
Therefore, in the present study, the effects of SS31 on
t-BHP-induced mitochondrial dysfunction and oxidative damage in
661W photoreceptor cells were investigated.
Materials and methods
Cell culture
The 661W cell line used in the present study was
provided by Dr Muayyad Al-Ubaidi (University of Oklahoma, Norman,
USA). These cells were cultured in Dulbecco's modified Eagle's
medium (DMEM; Gibco, NY, USA), supplemented with 10% fetal calf
serum (Sigma-Aldrich, St. Louis, MO, USA) at 37°C in a humidified
5% CO2 atmosphere. In all the following assays, 661W
cells were cultured at a density of 2×105 in growth
medium for 24 h at 37°C prior to the treatment. When grown to
75–80% confluence, the cells were incubated with different
concentrations of t-BHP (Sigma-Aldrich), either alone, or in the
presence of SS31 depending on the experimental requirements. In all
experiments, control cells were cultured without any treatment.
Cell viability assay
To determine the viability of the cells subsequent
to oxidative stress, the 661W cells were seeded into 96-well plates
and treated with t-BHP (25, 50, 100, 200 or 400 µM), with or
without SS31 (10, 100 nM or 1 µM) for 24 h, rinsed once in
PBS, then incubated with serum-free DMEM containing 0.25 mg/ml MTT
(Sigma-Aldrich). After 4 h, the MTT solution was aspirated,
dimethyl sulfoxide (0.1 ml/well) was added, and the plates were
shaken for 10 min at room temperature. The optical densities of the
supernatant were read at 490 nm using a microplate
spectrophotometer (Spectra Max 340; Molecular Devices, Sunnyvale,
CA, USA). The results were obtained from four independent
experiments. The optical density of the formazan formed in the
control cells was considered as 100% viability.
Detection of protein and DNA
peroxidation
To examine the antioxidative role of SS31,
nitrotyrosine and 8-hydroxy-deoxyguanosine (8-OHdG) were used as
markers of lipid and DNA peroxidation, respectively, to detect
oxidative damage. These markers were detected by immunofluorescence
using mouse anti-nitrotyrosine antibody (mouse monoclonal; 1:200;
cat. no. ab7048; Abcam, Cambridge, MA, USA) and goat anti-8-OHdG
antibody (goat polyclonal; 1:200; cat. no. AB5830; Chemicon,
Temecula, CA, USA). The 661W cells were seeded at a density of
2×105 and grown in culture dishes for 1 day. The cells
were subsequently treated with 100 mM t-BHP for 24 h in the absence
or presence of 100 nM SS31. The cells were fixed with freshly
prepared 4% formaldehyde in PBS for 10 min, washed with
phosphate-buffered saline (PBS), permeabilized with 0.1% Triton
X-100 in PBS for 2 min, and rinsed with PBS. Following blockade of
the non-specific binding sites by incubation with blocking buffer
(PBS and 5% BSA) for 60 min, the cells were incubated with the
primary antibodies overnight at 4°C. The cells were then rinsed
thoroughly with PBS and were incubated with anti-mouse
immunoglobulin (Ig)G-Alexa 555 and anti-goat IgG-Alexa 555
secondary antibodies (donkey polyclonal; 1:3,000; cat. no. A21292;
Invitrogen Life Technologies) for 1 h. Following repeated washing
twice with PBS, the slides were mounted and analyzed using a Zeiss
LSM 510 META confocal microscope (LSM510 META; Carl Zeiss,
Oberkochen, Germany). Fluorescence images were captured using
identical exposure settings. For the negative control, the sections
were stained without primary antibodies to produce no fluorescent
signals.
Determination of cell apoptosis using
Hoechst staining
Apoptosis was determined using Hoechst staining in
the 661w cells treated with 100 µM t-BHP, and without or
with 100 nM SS31 for 24 h. Nuclear integrity was evaluated
following staining of the cell nuclei using Hoechst, a fluorescent
dye that binds to DNA. Briefly, the cells were permeated using 0.1%
Triton X-100 in PBS, washed with PBS, and incubated with 0.05%
Hoechst for 10 min. The cells were considered to be apoptotic when
they exhibited either fragmented or condensed nuclei. To
quantitatively analyze the changes of nuclear morphology, the
percentage of cells with apoptotic nuclei was then calculated. The
results were obtained from 10 randomly-selected fields per sample
in four independent experiments with microscopy (Zeiss, Jena,
Germany).
Flow cytometry for the measurement of
apoptosis
The presence of apoptotic cells was evaluated by an
early change in membrane phospholipid asymmetry associated with
cells during the early phases of apoptosis. The loss of cell
membrane phospholipid asymmetry is accompanied by the exposure of
phosphatidylserine to the outer membrane (17). Apoptosis was assessed in the
present study using an Annexin V/Propidium Iodide (PI) kit,
according to the manufacturer's instructions (Bender Med Systems,
Vienna, Austria). The 661w cells were treated with 100 µM
t-BHP without or with 10 nM, 100 nM or 1 µM SS31 for 24 h.
Briefly, 4×105 cells were removed from the culture
dishes by 3 min incubation in 0.05% trypsin. The 661W cells were
washed twice with PBS and resuspended in 185 µl 1X binding
buffer. Subsequently, 5 µl annexin V and 10 µl l PI
were added to the cell suspension, which was then vortexed and
incubated for 15 min in the dark at room temperature. Finally, 200
µl 1X binding buffer was added, and the samples were
evaluated using flow cytometry (FASC Aria II SORP; BD Bioscience,
San Jose, CA, USA). In total ~20,000 cells were analyzed per
sample. Cells, which were negative for PI and annexin V staining
were live cells; PI-negative, annexin V-positive cells were early
apoptotic cells; and PI-positive, annexin V-positive cells were
primarily cells in the late stages of apoptosis.
Detection of ROS
Intra-mitochondrial production of ROS in the live
cells was estimated fluorimetrically using MitoSOX Red (Invitrogen
Life Technologies). Briefly, the cells were treated with 100
µM t-BHP for 1 h at 37°C, with or without 100 nM SS31.
Subsequently, the 661W cells were loaded with the MitoSOX Red
fluorogenic probe (5 µmol/l) for 20 min. Following removal
of MitoSOX Red and washing of the cells with Hanks' balanced salt
solution, fluorescent images were captured using a confocal
microscope. The mean fluorescence intensity per mm2 cell
area was calculated using Zeiss software (Carl Zeiss).
Measurement of mitochondrial membrane
potential (ΔΨm)
JC-1, a ΔΨm indicator, was used to demonstrate the
changes in ΔΨm in the 661W cells. JC-1 is a lipophilic and cationic
dye, which permeates plasma and mitochondrial membranes. The dye
fluoresces red when it aggregates in the matrix of healthy, high
ΔΨm mitochondria, whereas it exhibits green fluorescence in cells
with low ΔΨm (18). The JC-1
(Invitrogen Life Technologies) was freshly diluted in serum-free
DMEM to a final concentration of 1 µg/ml and was added to
the treated and non-treated cells at densities of 1×106
cells/ml. Following incubation for 20 min at 37°C in the dark, the
samples were rinsed twice in PBS and analyzed immediately using
flow cytometry. Data were collected at 525 nm emission for green
fluorescence and 590 nm for red fluorescence. The ratio of the
red:green (aggregate:monomer) fluorescence intensity values was
used to assess the ΔΨm.
Measurement of the release of cytochrome
c
The release of cytochrome c is a putative
event of the mitochondria apoptotic pathway following the loss of
ΔΨm. To evaluate whether cytochrome c (mouse polyclonal;
1:300; cat. no. sc4198; Santa Cruz Biotechnology, Inc., Santa Cruz,
CA, USA) was released from the mitochondria, immunocytochemical
labeling of cytochrome c was performed using confocal
microscopy. The 661W cells were treated with 100 mM t-BHP either
alone, or with 100 nM SS31 for 24 h. The cells were immunolabeled
with mouse monoclonal anti-cytochrome c and rabbit
anti-HSP60 antibodies (rabbit polyclonal; 1:500; cat. no. sc2714;
Santa Cruz Biotechnology, Inc.) at room temperature overnight,
followed by incubation with anti-mouse IgG-Alexa 555 (donkey
polyclonal; 1:3,000; cat. no. A21292; Invitrogen Life Technologies)
and anti-rabbit IgG-Alexa 488 secondary antibodies for 1 h
subsequent to thorough rinsing twice with PBS. Cells were then
washed and mounted in fluorescence mounting medium. For negative
control, sections stained without primary antibodies showed no
signals.
Statistical analysis
Statistical analysis was performed using SPSS 13.0
analytical software (SPSS, Inc., Chicago, MO, USA). All assays were
performed in at least three separate experiments. Data are
presented as the mean ± standard error of the mean and were
evaluated using one-way analysis of variance. P<0.05 was
considered to indicate a statistically significant difference.
Results
SS31 prevents the decrease in 661W cell
viability induced by oxidative damage
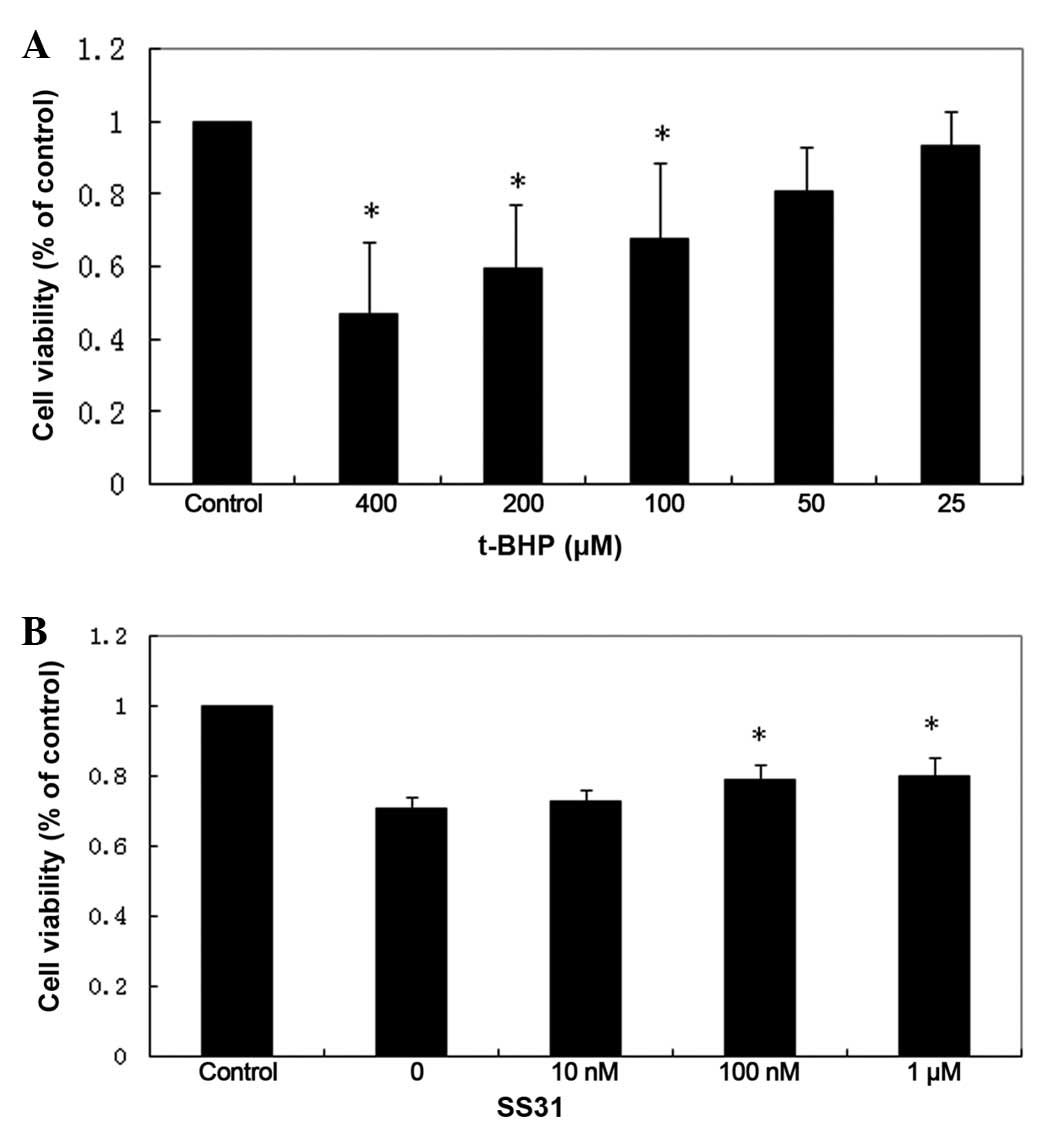
The viability of the 661W cells was reduced
following exposure to t-BHP for 24 h in a dose-dependent manner.
Marked cytotoxicity was observed at concentrations of ≥100
µM t-BHP, compared with the untreated control cells
(Fig. 1A). As shown in Fig. 1B, treatment of the 661W cells with
t-BHP at 100 µM for 24 h led to ~30% loss of cell viability
(P<0.01; n=4). By contrast, treatment with SS31 led to increased
cell survival (P<0.01; n-4), however, concentrations <10 nM
did not improve 661W cell survival following oxidant injury.
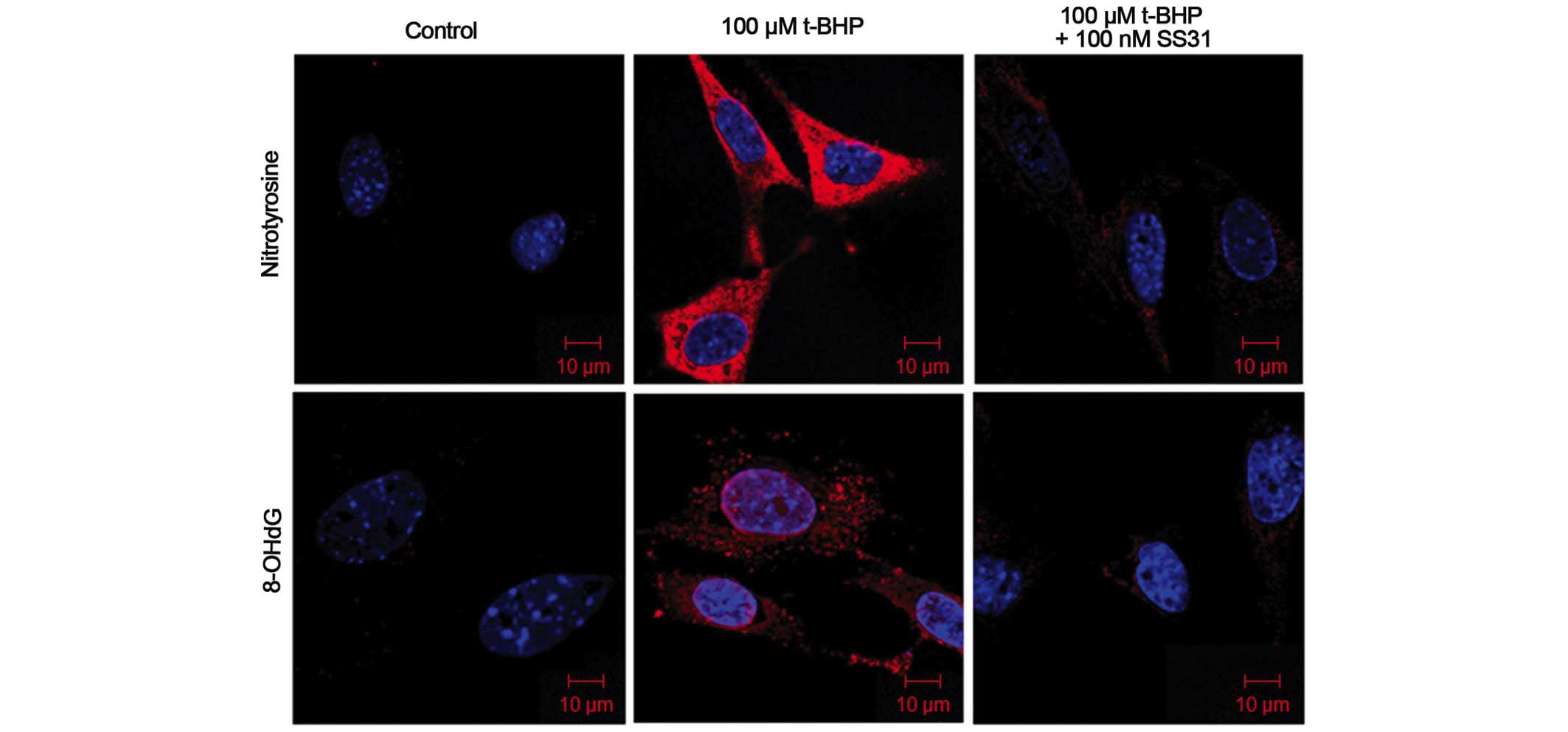
SS31 significantly attenuates
t-BHP-induced production of nitrotyrosine and 8-OHdG
When ROS interact with proteins, lipids or DNA, cell
dysfunction and death can occur. Certain sites of macromolecules
are particularly susceptible to particular ROS, resulting in
specific modifications that act as 'fingerprints', implicating
oxidative damage in disease pathogenesis (19). The occurrence of nitrotyrosine
residues in proteins is specific for peroxynitrite-induced protein
oxidative damage (20). Hydroxyl
radicals can also attack guanine at its C-8 position to yield
8-OHdG, which serves as another biomarker for DNA oxidative damage.
The confocal microscopic images in the present study indicated that
treatment of the 661W cells with 100 mM t-BHP for 24 h increased
the production of nitrotyrosine and 8-OHdG. Concurrent treatment
with 100 nM SS31 prevented the t-BHP-induced accumulation of
nitrotyrosine and 8-OHdG (Fig.
2).
SS31 provents t-BHP-induced cell death in
661W cells
Morphologically, a significant number of 661W cells
had detached from the culture dish following treatment with 100 mM
t-BHP for 24 h, and those that remained exhibited cell shrinkage
and shedding; whereas those co-cultured with 100 nM SS31 maintained
a fairly healthy appearance. Fewer cells were observed in these
cultures, partly due to cell death and partly due to cell
detachment (Fig. 3A). Following
Hoechst staining, the 661W cells treated with 100 mM t-BHP for 24 h
exhibited substantial fragmented or condensed nuclei,
characteristic of apoptotic cells, whereas few apoptotic nuclei
were identified in the control 661W cells. The 661W cells, which
were incubated with 100 nM SS31 had a markedly decreased number of
apoptotic nuclei, compared to the t-BHP group, indicating that SS31
protected the 661W cells from oxidant-induced 661W cell death
(Fig. 3B).
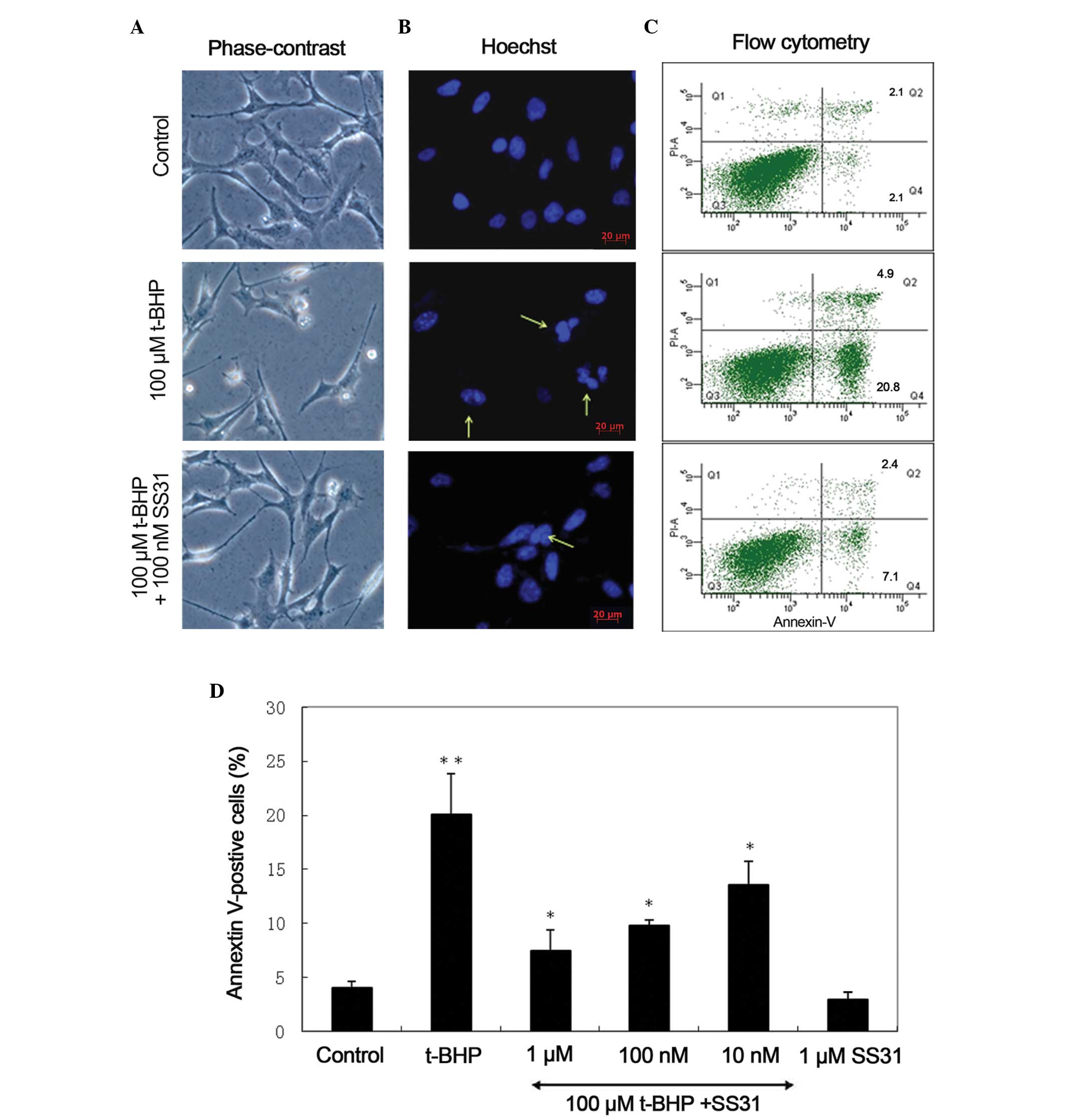 | Figure 3SS31 prevents apoptotic nuclei
condensation and externalization of membrane phosphatidylserine
residue in 661W cells. (A) Phase-contrast light micrographs
revealed cells exhibiting a generally unhealthy appearance
following treatment with 100 µM t-BHP for 24 h, whereas
those co-cultured with 100 nM SS31 maintained a relatively healthy
appearance. (magnification, ×1,000). (B) Representative images of
Hoechst-labeled nuclei, condensation of chromatin and apoptotic
bodies in the 661W cells following damage induced by 100 µM
t-BHP for 24 h (arrows). These changes were less visible in the
control cells and were markedly improved in the cells treated with
100 nM SS31. (C) Representative flow cytometric images indicated a
significant decrease of annexin V-positive cells in the 661W cells
treated with 100 nM SS31, compared with the cells treated with
t-BHP alone for 24 h. (D) Quantitative analysis of annexin
V-positive cells was measured in four independent experiments using
flow cytometry. The number of annexin V-positive cells decreased in
a dose-dependent manner to 13.6±2.6, 9.8±0.5 and 7.4±2.0% in
following treatment with 10 nM, 100 nM and 1 µM,
respectively (**P<0.001, vs. control;
*P<0.001, vs. t-BHP). Data are presented as the mean
± standard error of the mean. |
In addition, quantitative analysis of annexin
V-positive cells was performed in four independent experiments
using flow cytometry. In the flow cytometric images, cells negative
for PI and annexin V staining were live cells (bottom left);
PI-negative, annexin V-positive cells were early apoptotic cells
(bottom right); PI-positive, annexin V-positive cells were cells
primarily in the late stages of apoptosis (top right); and
PI-positive, annexin V-negative cells were necrotic cells (top
left). A significant increase in the number of annexin V-positive
cells was observed in the 661W cells following t-BHP treatment for
24 h (20.0±3.8%), compared with untreated control cultures
(4.0±0.6%; P<0.001). The number of annexin V-positive cells
decreased dose-dependently to 13.6±2.6, 9.8±0.5 and 7.4±2.0% in the
groups treated with 10 nm, 100 nm and 1 µM SS31,
respectively. SS31 at multiple doses demonstrated a significant
protective effect against t-BHP (P<0.001). No significant
toxicity was observed over a 24 h period in the cultures exposed to
1 µM SS31 alone (2.9±0.7%; P=0.429), compared with the
untreated control cultures (Fig. 3C
and D).
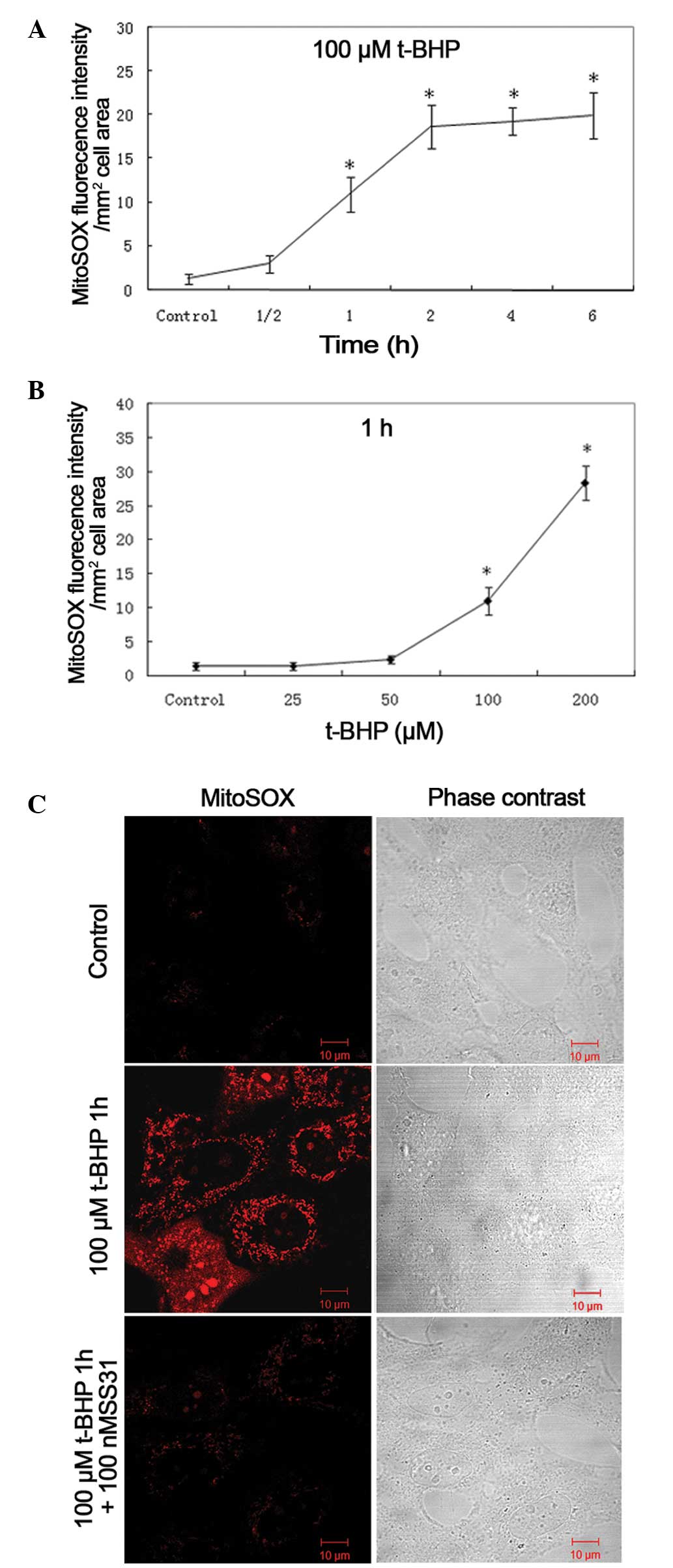
SS31 significantly reduces mitochondrial
ROS
To further examine the SS31 protective mechanisms,
the present study investigated its effect on mitochondrial ROS.
MitoSOX™, a specific dye for mitochondrial ROS, was used
to detect mitochondrial ROS. Confocal microscopy was used to
localize the oxidative stress-induced increase of ROS production in
the mitochondria of the 661W cells using MitoSOX™.
Quantitative measurements of the mean fluorescence intensities from
the samples demonstrated that t-BHP increased mitochondrial
superoxide generation in the 661W cells in a dose- and
time-dependent manner (Fig. 4A and
B). The MitoSOX™ fluorescence intensity per
mm2 cell area increased over time, beginning at 1 h
(11.0±2.0; P<0.001, vs. control). The mitochondrial ROS was
markedly increased following incubation with 100 µM t-BHP
for 1 h, whereas treatment with 100 nM SS31 significantly
ameliorated the oxidative stress-induced increase in mitochondrial
ROS (Fig. 4C).
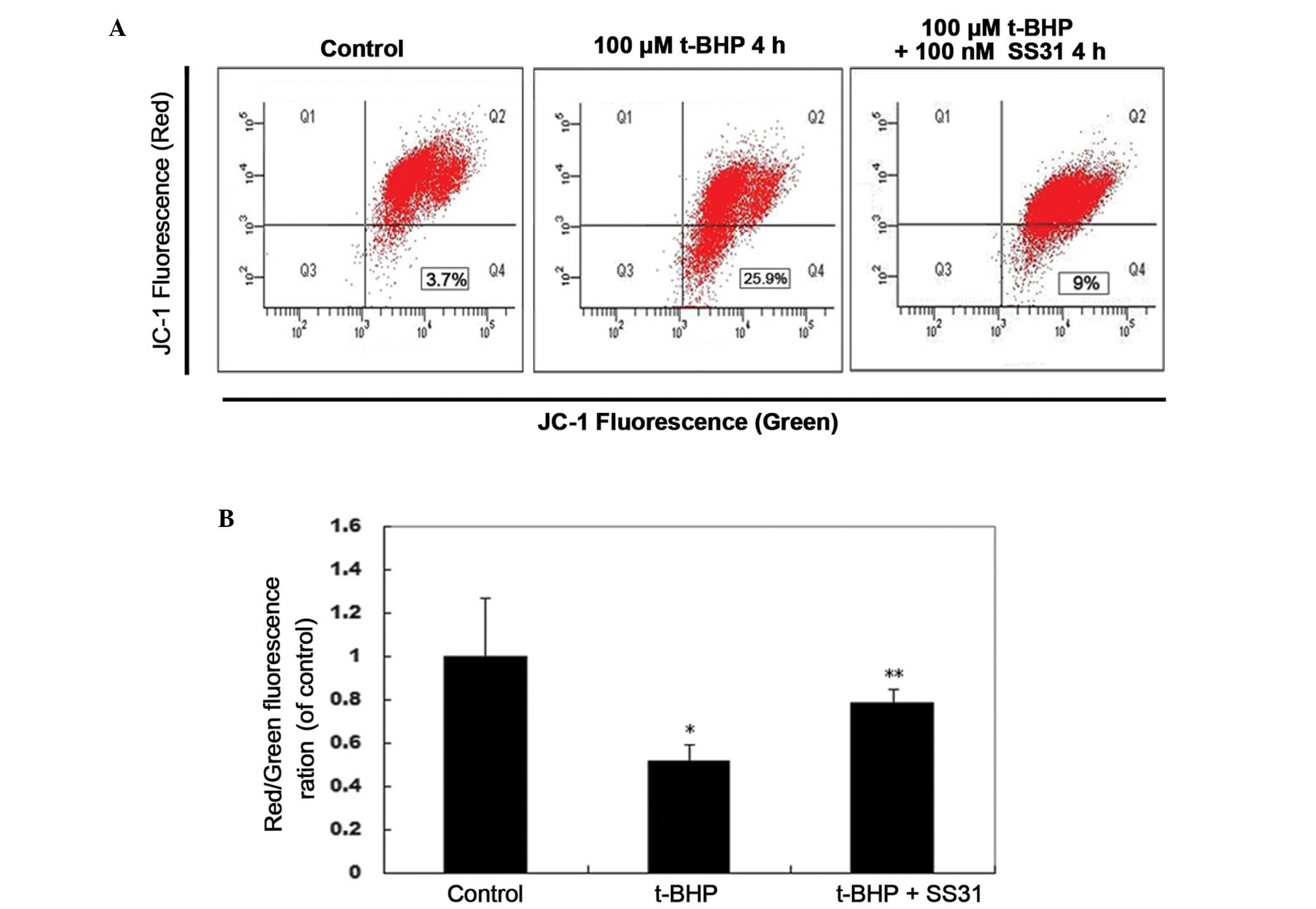
SS31 increases ΔΨm during oxidative
stress
To determine the involvement of the
mitochondrial-mediated pathway in oxidative stress induced cell
dysfunction, the present study measured ΔΨm using flow cytometry,
using the cationic membrane potential indicator JC-1. Treatment
with 100 nM SS31 for 4 h prevented the 100 µM t-BHP-induced
loss of ΔΨm in the 661W cells (Fig.
5A). Compared with the untreated control cultures, exposure to
100 µM t-BHP for 4 h resulted in a rapid decrease in the
red/green fluorescence intensity ratio to 51.49±7.59% of the
control (P<0.01; Fig. 5B).
Treatment with 100 nM SS31 significantly increased the red/green
fluorescence intensity of the 661W cells to 78.18±6.67% of the
control (P<0.05), which indicated that the ΔΨm was restored to
baseline.
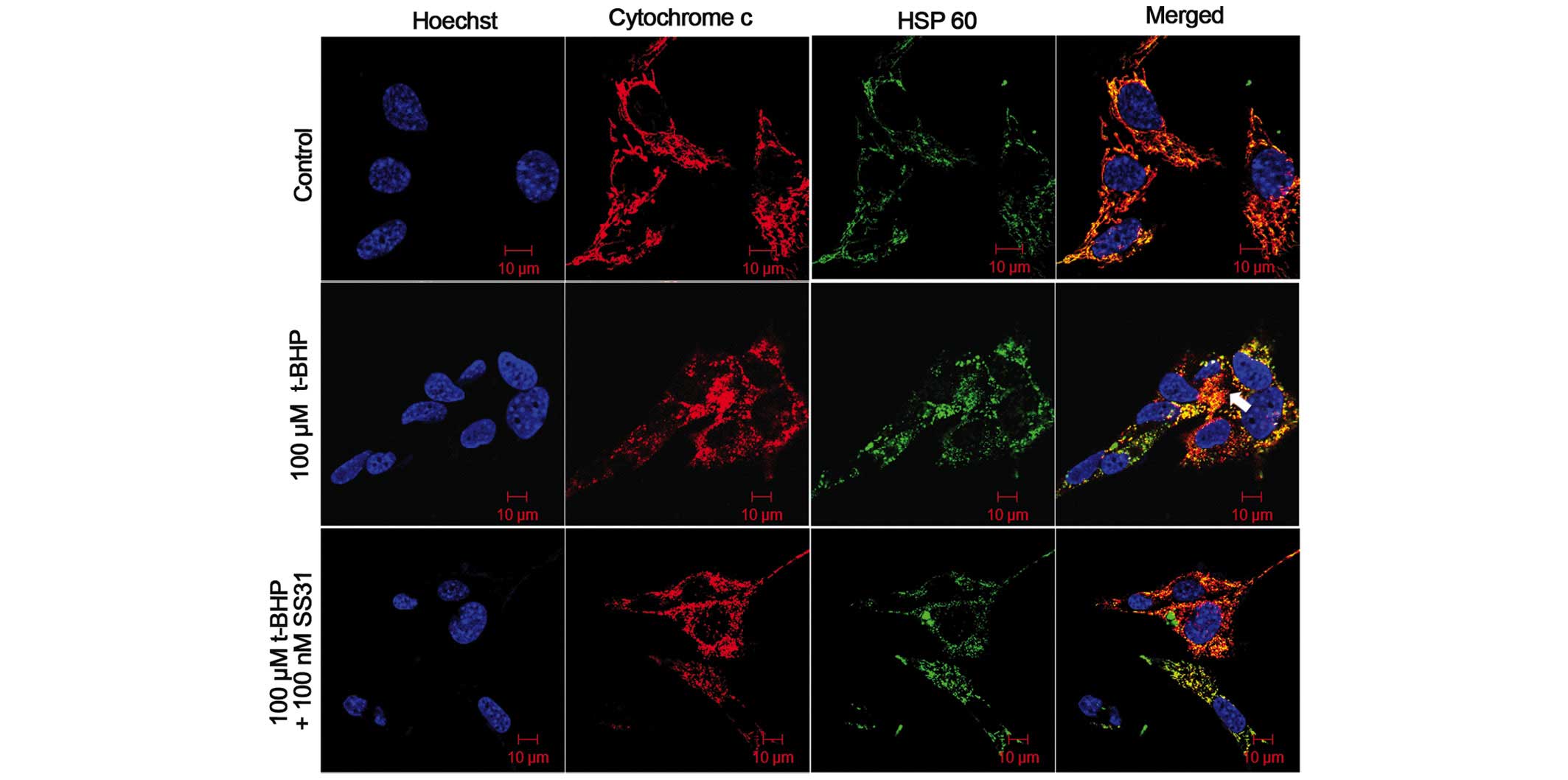
SS31 inhibits t-BHP-induced cytochrome c
release
As it is well known that excessive ROS and decreased
ΔΨm can induce apoptotic death, the present study examined the
release of mitochondrial cytochrome c, an important
signaling molecule in apoptosis. This included examining whether
oxidative stress induced the release of cytochrome c from
the mitochondria and whether the addition of SS31 prevented this
release. As shown in Fig. 6, the
661W cells in the control group exhibited an exact overlap of
anti-cytochrome c and HSP60-labeled mitochondrial
fluorescence in the confocal micrographs, indicating the
co-localization of cytochrome c and mitochondria in the
cells. No cytochrome c release was identified from the
mitochondria prior to treatment with t-BHP. Following treatment
with 100 µM t-BHP for 24 h, cytochrome c was observed
in the cytoplasm of the 661W cells, and this was not coincident
with the mitochondrial labeling, indicating that treatment with
t-BHP induced the release of cytochrome c from the
mitochondria in the 661W cells (Fig.
6, arrows). By contrast, treatment with 100 nM SS31 inhibited
the release of cytochrome c into the cytosol (Fig. 6).
Discussion
RP is a prevalent cause of blindness, caused by a
number of different mutations in several different genes (21). Why mutations in genes that are
exclusively expressed in rod photo-receptors can cause the death of
rod and cone cells remains to be elucidated, however, it is likely
to be multifactorial and it has been hypothesized that oxidative
stress-associated photoreceptor injury is important (3). The retina is particularly prone to
oxidative damage due to its relatively high oxygen consumption and
its constant exposure to light (5,22).
Cytotoxic levels of ROS have been found in ocular tissues in
vivo, and can be produced during photoreceptor outer segment
phagocytosis by the RPE cells (23). In an rd1 mouse model of RP,
oxidative damage in the outer retina results in gradual cone cell
death (24).
The 661W cell line, isolated from transgenic mice by
Al-Ubaidi MR, expresses photoreceptor cell markers, including
opsin, arrestin, phosphodiesterase, transducin, phosducin,
recoverin and IRBP (25,26). However, compared with in
vivo photoreceptor cells, 661W cells do not express outer
segment structural proteins, including RPE65, peripherin/rds and
ROM1, which support the cone origin of 661W cells (26). In addition, 661W cells have been
maintained in culture for >60 passages with no apparent slowing
of mitotic activity or loss of photoreceptor-specific markers
(27), and 661W cells grow to
confluence at ~2 days culture with a doubling rate of 1.1 days
(28). The patterns of expression
of cone opsin and arrestin are not modulated by treatment with
factors that stimulate differentiation, including retinoic acid and
hydrocortisone. In addition, 661W cells have more cytoplasm, a
characteristic usually used by pathologists to assess the
differentiation status of tumor cells (29,30).
Therefore, the 661W cell line has been demonstrated as a valuable
tool for in vitro investigations of photoreceptor cell
biology and function.
t-BHP is a membrane-permeant oxidant, which has been
used extensively as a model of oxidative stress in different
systems (31,32). Similar to previous studies, the
present study found that t-BHP causes apoptosis of 661W cells,
demonstrated by DNA fragmentation and condensation of nuclei at low
doses. When concentrations of t-BHP are ≥400 µM, the
percentage of cell necrosis is markedly increased, rather than
apoptosis (data not shown). In the present study, concentrations of
SS31 between 10 nM and 1 µM are necessary for protection
against oxidative damage. Although oxidant-induced cell death can
be prevented by a number antioxidants, none have been observed to
be effective at concentrations<1 µM. In the present
study, apoptosis of the 661W cells induced by t-BHP was largely
inhibited by SS31 treatment at a concentration of 100 nM.
Additionally, 1 µM SS31 had no cytotoxic effect in the
absence of t-BHP. Previous studies on different cell types also
demonstrated a therapeutic concentration range of 0.1–100 nM for
SS31 (12,14,33).
Natural antioxidants, including coenzyme Q (CoQ),
vitamin E and lipoic acid have protective effects in mouse models
of Parkinson's disease (PD), ALS, and AD (34–36).
It has been reported that patients with RP, who take natural
antioxidants, including vitamin A, vitamin E and docosahexaenoic
acid (DHA) exhibited slower declines in ERG amplitudes (37,38).
However, high doses of vitamin E may be harmful (39). Antioxidants generally require
concentrations of at least 100 mM to reduce oxidative cell death
(40). MitoQ has been observed to
inhibit H2O2-induced apoptosis at 1 mM,
however, concentrations >10 mM caused cytotoxicity (41). By contrast, SS31 are particularly
potent in reducing intracellular ROS and preventing cell death
following t-BHP treatment, with a half maximal effective
concentration in the nM range (12). Another possible reason for the lack
of efficacy of natural antioxidant is their difficulty in
penetrating the blood-brain barrier. It has been reported that
treatment with CoQ for 2 months failed to increase brain levels of
CoQ (42). Furthermore, certain
antioxidants may not reach the relevant sites of free radical
generation. Large proteins, including SOD and catalase, do not
penetrate cell membranes, and CoQ and Vitamin E are lipophilic,
tending to be retained in cell membranes and, therefore,
ineffective against intracellular ROS (11). SS31 represents a novel approach
with targeted delivery to the inner mitochondrial membrane,
containing an amino acid sequence that allows it to freely
penetrate cell membranes. Therefore, it has been observed to be
taken into cells in a potential-independent, non-saturable manner,
even if ΔΨm is compromised and does cause mitochondrial
depolarization, even at 1 mM (11,32).
In the present study, to investigate the effects of
oxidative stress, 661W cells were stimulated with various
concentrations of t-BHP. The results revealed that SS31 at
concentrations of 100 nM, which inhibited t-BHP-induced cell damage
and apoptosis, also prevented intracellular ROS production,
nitro-tyrosine formation and cytochrome c release. The
detailed mechanism by which SS31 protects against oxidative damage
remains to be elucidated. By reducing mitochondrial ROS, SS31 is
able to prevent opening of the mitochondrial permeability
transition pore, prevent mitochondrial swelling and reduce the
release of cytochrome c in response to high Ca2+
overload (12). SS31 also protects
against H2O2-induced mitochondrial
depolarization in immortalized human trabecular meshwork and
glaucomatous human trabecular meshwork cell lines by inhibiting the
activation of caspase 3 (33). Our
previous study demonstrated that SS31 attenuates the effects of
high glucose-induced injuries in human retinal endothelial cells by
decreasing the production of ROS, preventing the release of
cytochrome c from the mitochondria, decreasing the
expression of caspase-3 and increasing the expression of
thioredoxin 2 (Trx-2) (43). In
addition, SS31 can protect retinal structures and inhibit breakdown
of the inner blood retinal barrier by increasing the expression
levels of Trx-2 and B-cell lymphoma (Bcl)-2, and decreasing the
expression levels of p53, nuclear factor-κB, B-cell-associated X
protein and caspase-3 in the retina of diabetic rats (44).
In conclusion, the present study demonstrated that
SS31 prevented the intracellular production of ROS and revealed
significant antioxidant and anti-apoptotic effects on 661W cells.
Further investigations are required to elucidate the mechanism
underlying SS31 and to evaluate its protective effect in RP animal
models.
Acknowledgments
This study was supported by grants from National
Basic Research Development Program of China (973 program: no.
2013CB967000) and the National Natural Science Foundation of China
to Professor Yan Luo (no. 81371020) and Dr Xiaobo Zhu (no.
81271012).
References
|
1
|
Portera-Cailliau C, Sung CH, Nathans J and
Adler R: Apoptotic photoreceptor cell death in mouse models of
retinitis pigmentosa. Proc Natl Acad Sci USA. 91:974–978. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chang GQ, Hao Y and Wong F: Apoptosis:
final common pathway of photoreceptor death in rd, rds and
rhodopsin mutant mice. Neuron. 11:595–605. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shen J, Yang X, Dong A, et al: Oxidative
damage is a potential cause of cone cell death in retinitis
pigmentosa. J Cell Physiol. 203:457–464. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Komeima K, Rogers BS, Lu L and Campochiaro
PA: Antioxidants reduce cone cell death in a model of retinitis
pigmentosa. Proc Natl Acad Sci USA. 103:11300–11305. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yu DY, Cringle SJ, Su EN and Yu PK:
Intraretinal oxygen levels before and after photoreceptor loss in
the RCS rat. Invest Ophthalmol Vis Sci. 41:3999–4006.
2000.PubMed/NCBI
|
|
6
|
Rotstein NP, Politi LE, German OL and
Girotti R: Protective effect of docosahexaenoic acid on oxidative
stress-induced apoptosis of retina photoreceptors. Invest
Ophthalmol Vis Sci. 44:2252–2259. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar
|
|
8
|
Herrera B, Alvarez AM, Sanchez A, et al:
Reactive oxygen species (ROS) mediates the mitochondrial-dependent
apoptosis induced by transforming growth factor (beta) in fetal
hepatocytes. FASEB J. 15:741–751. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li N, Ragheb K, Lawler G, et al:
Mitochondrial complex I inhibitor rotenone induces apoptosis
through enhancing mitochondrial reactive oxygen species production.
J Biol Chem. 278:8516–8525. 2003. View Article : Google Scholar
|
|
10
|
Palanivel K, Kanimozhi V, Kadalmani B and
Akbarsha MA: Verrucarin A induces apoptosis through ROS-mediated
EGFR/MAPK/Akt signaling pathways in MDA-MB-231 breast cancer cells.
J Cell Biochem. 115:2022–2032. 2014.PubMed/NCBI
|
|
11
|
Szeto HH: Mitochondria-targeted peptide
antioxidants: novel neuroprotective agents. AAPS J. 8:E521–E531.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhao K, Zhao GM, Wu D, et al:
Cell-permeable peptide antioxidants targeted to inner mitochondrial
membrane inhibit mitochondrial swelling, oxidative cell death and
reperfusion injury. J Biol Chem. 279:34682–34690. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu D, Soong Y, Zhao GM and Szeto HH: A
highly potent peptide analgesic that protects against
ischemia-reperfusion-induced myocardial stunning. Am J Physiol
Heart Circ Physiol. 283:H783–H791. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT
and Pinto JT: A novel cell-permeable antioxidant peptide, SS31,
attenuates ischemic brain injury by down-regulating CD36. J Biol
Chem. 282:4634–4642. 2007. View Article : Google Scholar
|
|
15
|
Calkins MJ, Manczak M, Mao P, Shirendeb U
and Reddy PH: Impaired mitochondrial biogenesis, defective axonal
transport of mitochondria, abnormal mitochondrial dynamics and
synaptic degeneration in a mouse model of Alzheimer's disease. Hum
Mol Genet. 20:4515–4529. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Petri S, Kiaei M, Damiano M, et al:
Cell-permeable peptide antioxidants as a novel therapeutic approach
in a mouse model of amyotrophic lateral sclerosis. J Neurochem.
98:1141–1148. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pellicciari C, Bottone MG and Biggiogera
M: Detection of apoptotic cells by annexin V labeling at electron
microscopy. Eur J Histochem. 41:211–216. 1997.PubMed/NCBI
|
|
18
|
Chazotte B: Labeling mitochondria with
JC-1. Cold Spring Harb Protoc. 2011:2011.PubMed/NCBI
|
|
19
|
Soskic V, Groebe K and Schrattenholz A:
Nonenzymatic post-translational protein modifications in ageing.
Exp Gerontol. 43:247–257. 2008. View Article : Google Scholar
|
|
20
|
Schulz JB, Matthews RT, Muqit MM, Browne
SE and Beal MF: Inhibition of neuronal nitric oxide synthase by
7-nitroin-dazole protects against MPTP-induced neurotoxicity in
mice. J Neurochem. 64:936–939. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hartong DT, Berson EL and Dryja TP:
Retinitis pigmentosa. Lancet. 368:1795–1809. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Braun RD, Linsenmeier RA and Goldstick TK:
Oxygen consumption in the inner and outer retina of the cat. Invest
Ophthalmol Vis Sci. 36:542–554. 1995.PubMed/NCBI
|
|
23
|
Maslim J, Valter K, Egensperger R,
Holländer H and Stone J: Tissue oxygen during a critical
developmental period controls the death and survival of
photoreceptors. Invest Ophthalmol Vis Sci. 38:1667–1677.
1997.PubMed/NCBI
|
|
24
|
Sanz MM, Johnson LE, Ahuja S, Ekstrom PA,
Romero J and van Veen T: Significant photoreceptor rescue by
treatment with a combination of antioxidants in an animal model for
retinal degeneration. Neuroscience. 145:1120–1129. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
al-Ubaidi MR, Font RL, Quiambao AB, et al:
Bilateral retinal and brain tumors in transgenic mice expressing
simian virus 40 large T antigen under control of the human
interphotoreceptor retinoid-binding protein promoter. J Cell Biol.
119:1681–1687. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tan E, Ding XQ, Saadi A, Agarwal N, Naash
MI and Al-Ubaidi MR: Expression of cone-photoreceptor-specific
antigens in a cell line derived from retinal tumors in transgenic
mice. Invest Ophthalmol Vis Sci. 45:764–768. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sanvicens N and Cotter TG: Ceramide is the
key mediator of oxidative stress-induced apoptosis in retinal
photoreceptor cells. J Neurochem. 98:1432–1444. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Crawford MJ, Krishnamoorthy RR, Rudick VL,
et al: Bcl-2 overexpression protects photooxidative stress-induced
apoptosis of photoreceptor cells via NF-kappaB preservation.
Biochem Biophys Res Commun. 281:1304–1312. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li A, Zhu X and Craft CM: Retinoic acid
upregulates cone arrestin expression in retinoblastoma cells
through a Cis element in the distal promoter region. Invest
Ophthalmol Vis Sci. 43:1375–1383. 2002.PubMed/NCBI
|
|
30
|
Kyritsis A, Joseph G and Chader GJ:
Effects of butyrate, retinol and retinoic acid on human Y-79
retinoblastoma cells growing in monolayer cultures. J Natl Cancer
Inst. 73:649–654. 1984.PubMed/NCBI
|
|
31
|
Nieminen AL, Byrne AM, Herman B and
Lemasters JJ: Mitochondrial permeability transition in hepatocytes
induced by t-BuOOH: NAD(P)H and reactive oxygen species. Am J
Physiol. 272:C1286–C1294. 1997.PubMed/NCBI
|
|
32
|
Zhao K, Luo G, Giannelli S and Szeto HH:
Mitochondria-targeted peptide prevents mitochondrial depolarization
and apoptosis induced by tert-butyl hydroperoxide in neuronal cell
lines. Biochem Pharmacol. 70:1796–1806. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen M, Liu B, Gao Q, Zhuo Y and Ge J:
Mitochondria-targeted peptide MTP-131 alleviates mitochondrial
dysfunction and oxidative damage in human trabecular meshwork
cells. Invest Ophthalmol Vis Sci. 52:7027–7037. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Veldink JH, Kalmijn S, Groeneveld GJ, et
al: Intake of polyunsaturated fatty acids and vitamin E reduces the
risk of developing amyotrophic lateral sclerosis. J Neurol
Neurosurg Psychiatry. 78:367–371. 2007. View Article : Google Scholar
|
|
35
|
Sikorska M, Lanthier P, Miller H, et al:
Nanomicellar formulation of coenzyme Q10 (Ubisol-Q10) effectively
blocks ongoing neurodegeneration in the mouse
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model: potential use
as an adjuvant treatment in Parkinson's disease. Neurobiol Aging.
35:2329–2346. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Maczurek A, Hager K, Kenklies M, et al:
Lipoic acid as an anti-inflammatory and neuroprotective treatment
for Alzheimer's disease. Adv Drug Deliv Rev. 60:1463–1470. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fielder AR: A randomized trial of vitamin
A and vitamin E supplementation for retinitis pigmentosa. Arch
Ophthalmol. 111:1463Author Reply 1463–1466. 1993.PubMed/NCBI
|
|
38
|
Berson EL, Rosner B, Sandberg MA, et al:
Further evaluation of docosahexaenoic acid in patients with
retinitis pigmentosa receiving vitamin A treatment: subgroup
analyses. Arch Ophthalmol. 122:1306–1314. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Miller ER III, Pastor-Barriuso R, Dalal D,
Riemersma RA, Appel LJ and Guallar E: Meta-analysis: high-dosage
vitamin E supplementation may increase all-cause mortality. Ann
Intern Med. 142:37–46. 2005. View Article : Google Scholar
|
|
40
|
Jauslin ML, Meier T, Smith RA and Murphy
MP: Mitochondria-targeted antioxidants protect Friedreich Ataxia
fibroblasts from endogenous oxidative stress more effectively than
untargeted antioxidants. FASEB J. 17:1972–1974. 2003.PubMed/NCBI
|
|
41
|
Kelso GF, Porteous CM, Coulter CV, et al:
Selective targeting of a redox-active ubiquinone to mitochondria
within cells: antioxidant and antiapoptotic properties. J Biol
Chem. 276:4588–4596. 2001. View Article : Google Scholar
|
|
42
|
Beal MF and Matthews RT: Coenzyme Q10 in
the central nervous system and its potential usefulness in the
treatment of neurode-generative diseases. Mol Aspects Med.
18(Suppl): 169–179. 1997. View Article : Google Scholar
|
|
43
|
Li J, Chen X, Xiao W, et al:
Mitochondria-targeted antioxidant peptide SS31 attenuates high
glucose-induced injury on human retinal endothelial cells. Biochem
Biophys Res Commun. 404:349–356. 2011. View Article : Google Scholar
|
|
44
|
Huang J, Li X, Li M, et al:
Mitochondria-targeted antioxidant peptide SS31 protects the retinas
of diabetic rats. Curr Mol Med. 13:935–945. 2013. View Article : Google Scholar : PubMed/NCBI
|