Introduction
The incidence of hyperuricemia is steadily
increasing in the world population and has therefore become a focus
of recent studies (1,2). Hyperuricemia is not only a marker of
chronic kidney disease but is also an independent risk factor for
numerous types of kidney disease (3,4).
Experimental studies have demonstrated a variety of mechanisms by
which hyperuricemia causes the development of renal disease. One of
the key mechanisms of hyperuricemia-induced kidney injury is the
inflammation provoked by monosodium urate (MSU) crystals (5–7). MSU
crystals were first identified as the etiological agent of gout in
the eighteenth century and more recently as a danger signal
released from dying cells (8).
NOD-like receptor (NLR) family, pyrin domain
containing 3 (NALP3) inflammasome and interleukin (IL)-1β were
reported to be crucial molecules in MSU crystal-mediated
inflammation. NALP3 inflammasome is an innate immune complex that
contains NALP3, caspase-recruitment domain (CARD)-8, and
apoptosis-associated speck-like protein containing a CARD (ASC).
NALP3 is a member of the NLR family, which not only detects
microbial structure but also senses endogenous danger signals such
as uric acid released from injured cells (9). ASC is an essential component of the
NALP3 inflammasome, as it can recruit caspase-1 to the inflammasome
(10), while the protein CARD-8
normally inhibits activation of caspase-1 (11). NALP3 inflammasome controls IL-1β
production by recruiting caspase-1, which directly cleaves cytokine
IL-1β precursors into active forms (12). Martinon et al (9) reported that MSU crystals induced
inflammation, activation of NALP3 inflammasome, and production of
active IL-1β and IL-18. Furthermore, an impaired neutrophil influx
was found in inflammasome-deficient mice. Miao et al
(13) suggested that gene
mutations in NALP3 and CARD-8 may contribute to susceptibility to
gout.
IL-1β belongs to the IL-1 family of cytokines and is
pivotal to the regulation of innate and adaptive immunity (14). Chen et al (15) showed that Il-1β is important in MSU
crystal-induced inflammation based on findings of a markedly
decreased inflammatory response to MSU crystals in IL-1R-deficient
mice. The finding that IL-1β inhibition was efficacious in the
treatment of MSU crystal-induced inflammation suggested a crucial
role for IL-1β in this type of inflammation (16).
Peroxisome proliferator-activated receptor γ (PPARγ)
belongs to the nuclear hormone receptor superfamily and acts as a
transcriptional regulator of numerous target genes by forming
heterodimers with the retinoid X receptor (17). PPARγ is expressed not only in white
adipose tissue but also in proximal tubular cells (18). It has been evidenced that
activation of PPARγ attenuated the expression of pro-inflammatory
mediators (19,20). In addition, an increasing number of
studies suggested that PPARγ agonists, including pioglitazone and
troglitazone, have a protective effect on renal function in various
models of acute and chronic renal injury (21,22).
However, the effects of PPARγ ligand on NALP3 inflammasome and
IL-1β production in MSU crystal-stimulated HK-2 cells have remained
elusive. The present study was therefore performed to investigate
the expression of PPARγ in MSU crystal-stimulated HK-2 cells. The
PPARγ agonist pioglitazone was used to assess the regulatory
effects of PPARγ on NALP3 inflammasome and IL-1β expression
levels.
Materials and methods
Cell line and culture
The primary human proximal tubular cell line HK-2
was obtained from the American Type Culture Collection (Manassas,
VA, USA). HK-2 cells were cultured in Dulbecco's Modified Eagle's
Medium (DMEM)/F12 (Gibco-Brl, Grand Island, NY, USA) supplemented
with 10% fetal calf serum (FBS; Gibco-BRL) at 37°C in a humidified
5%-CO2 incubator.
Preparation of MSU crystals
MSU crystals were prepared according to the
following process: First, 0.8 g uric acid (Sigma-Aldrich, St.
Louis, MO, USA) was dissolved in 200 ml 0.1 M borate buffer (pH
8.5; Thermo Fisher Scientific, Waltham, MA, USA). Through the
addition of HCl, the pH of the solution was adjusted to 8.0. The
solution was then passed through a 0.22-µm filter
(Millipore, Billerica, MA, USA), and the supersaturated uric acid
solution was left at room temperature for seven days to allow the
formation of fine crystals. After two washes with absolute ethanol
and one wash with acetone, the crystals were allowed to air-dry and
were suspended in phosphate-buffered saline (PBS) at a
concentration of 8 mg/ml. All MSU crystals were verified to be
endotoxin-free by the Limulus amebocyte cell lysate assay (Xiamen
Limulus Reagent Company, Xiamen, China).
Cell treatments
Upon 80%-confluency, cells were divided into four
groups, which were incubated in serum-free medium for 24 h as
follows: (A) FBS-free medium only; (B) MSU crystals (200
µg/ml); (C) lipopolysaccharide (LPS) (100 µg/ml;
Sigma-Aldrich); (D) pre-treatment with pioglitazone for 12 h (5
µmol/l; Sigma-Aldrich) followed by incubation with MSU
crystals (200 µg/ml).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from HK-2 cells using TRIzol
reagent (Invitrogen Life Technologies, Carlsbad, CA, USA) according
to the manufacturer's instructions. cDNA was synthesized using the
Reverse Transcription system (PrimeScript™ RT master mix; Takara
Bio Inc., Shiga, Japan) consisting of 10 µl reaction mixture
(5 µl 5X PrimeScript buffer, 1 µl
PrimeScript® RT enzyme mix I, 1 µl oligo dT
Primer, 1.5 µl Random 6 mers and 1.5 µl Total RNA)
incubated at 37°C for 15 min followed by 85°C for 5 sec. PCR
amplification of cDNA was performed according to the instructions
in the Takara Taq™ HS PCR kit (Takara Bio Inc.) with 1 µl
cDNA in a final volume of 25 µl. The sequences of primers
for PCR were as follows: PPARγ (313 bp) forward,
5′-AGCCAACACTAAACCACA-3′ and reverse, 5′-AGAAACCCTTGCATCCT-3′;
GAPDH (638 bp) forward, 5′-AGTCCACTGGCGTCTTCAC-3′ and reverse,
5′-GCTTGACAAAGTGGTCGTTGAG-3′ (Sangon Biotech, Shanghai, China). PCR
was carried out in the GeneAmp®PCR System 9700 (Applied
Biosystems, Foster City, CA, USA) and PCR products were
electrophoresed on 1% agarose gel (Biowest, Barcelona, Spain) using
HE-120 Electrophoresis Cell (Shanghai Tanon Science and Technology
Ltd., Shanghai, China), and detected by ultraviolet
transillumination (Shanghai Furi Science & Technology Co. Ltd,
Shanghai, China). Gel-Pro Analyzer 4.0 (Media Cybernetics,
Bethesda, MD, USA) was used for quantification of the bands.
Western blot analysis
Protein concentrations were determined using the
Pierce BCA protein assay reagent kit (Thermo Fisher Scientific)
following the manufacturer's instructions. Proteins (20 µg
per lane) were separated by 12% SDS-PAGE and transferred onto a
polyvinylidene difluoride membrane (Millipore). The membranes were
blocked in 5% bovine serum albumin (Sigma-Aldrich), and incubated
with mouse monoclonal anti-human NALP3 antibody (1:1,000; cat. no.
ab17267; Abcam, Cambridge, UK), mouse monoclonal anti-human PPARγ
antibody (1:1,000; cat. no. ab70405; Abcam) and GAPDH antibody
(1:5,000, cat. no. ab8245; Abcam) overnight at 4°C, followed by
incubation with a horseradish peroxidase-labeled anti-mouse
antibody (1:5,000; cat. no. A0216; Beyotime Institute of
Biotechnology, Haimen, China) for 1 h at room temperature. Protein
was detected using an ECL western blotting kit (Thermo Fisher
Scientific) and X-OMAT BT film (Carestream, Xiamen, China) in an
X-ray film cassette (Shanghai Kunlei Medical Instrument Co., Ltd.,
Shanghai, China). The bands were quantified using Gel-Pro Analyzer
4.0. The results of protein expression were normalized to GAPDH in
all figures.
ELISA for detection of IL-1β
HK-2 cells were treated for 48 h as described above.
Supernatants were then collected, and the levels of IL-1β protein
were measured using an IL-1β (human) ELISA kit according to the
manufacturer's instructions (BioVision, San Francisco, CA, USA). OD
was determined by a microplate reader (Multiskan MK3; Thermo Fisher
Scientific) at 450 nm.
Statistical analysis
Statistical analysis was performed using SPSS 17.0
(SPSS, Inc., Chicago, IL, USA). Values are expressed as the mean ±
standard deviation. The standard error of the mean was shown for
all experiments. Comparisons between groups were evaluated by
analysis of variance. P<0.05 was considered to indicate a
statistically significant difference.
Results
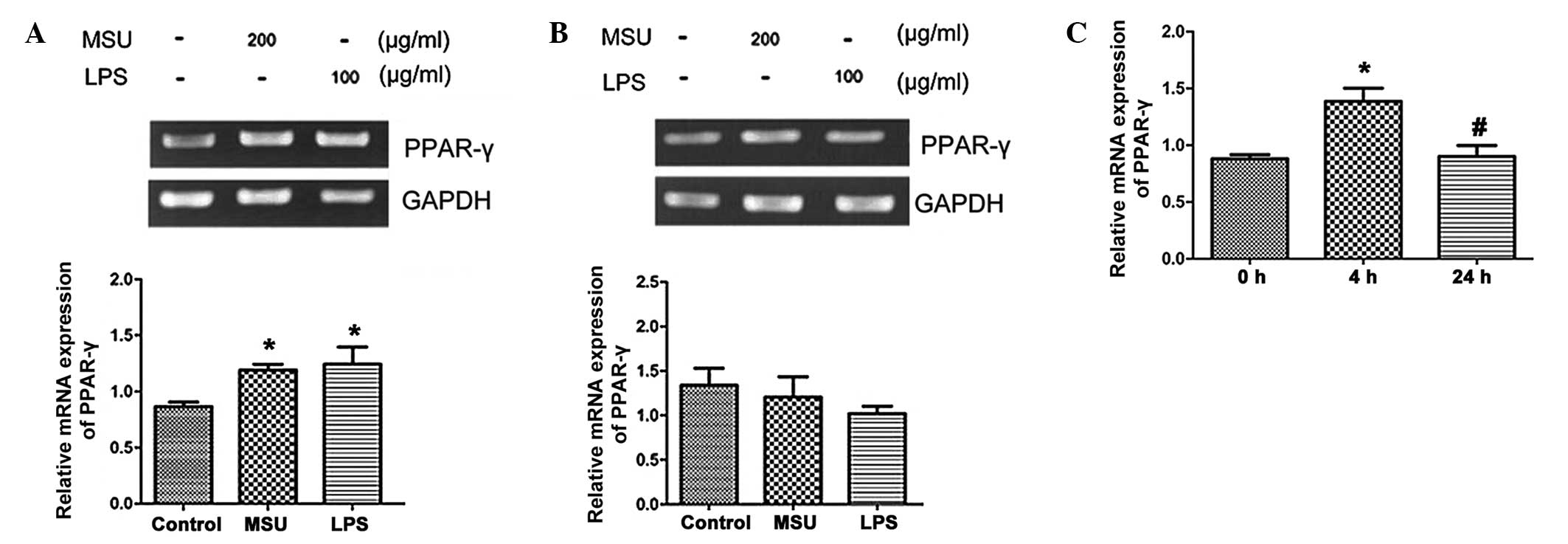
Effects of MSU crystals on PPARγ mRNA
expression in HK-2 cells
After 12-h stimulation, MSU crystals (200
µg/ml) and LPS (100 µg/ml) increased the levels of
PPARγ mRNA expression in HK-2 cells compared to that in the
untreated control group (P<0.05) (Fig. 1A). However, when HK-2 cells were
stimulated for 24 h, PPARγ expression levels in the MSU crystal-
and LPS-treated groups were slightly decreased compared with those
in the control group; however, the differences were not significant
(Fig. 1B). Since MSU crystals
regulated the expression of PPARγ in a time-dependent manner, the
present study further investigated the effects of MSU crystals on
the mRNA expression of PPARγ at 0, 4 and 24 h. As shown in Fig. 1C, MSU crystals (200 µg/ml)
significantly induced PPARγ mRNA expression at 4 h after
stimulation, while gene expression declined to basal levels at 24
h. Therefore, MSU crystals increased PPARγ mRNA expression at early
stages, while at later stages, PPARγ expression returned to basal
levels and declined eventually.
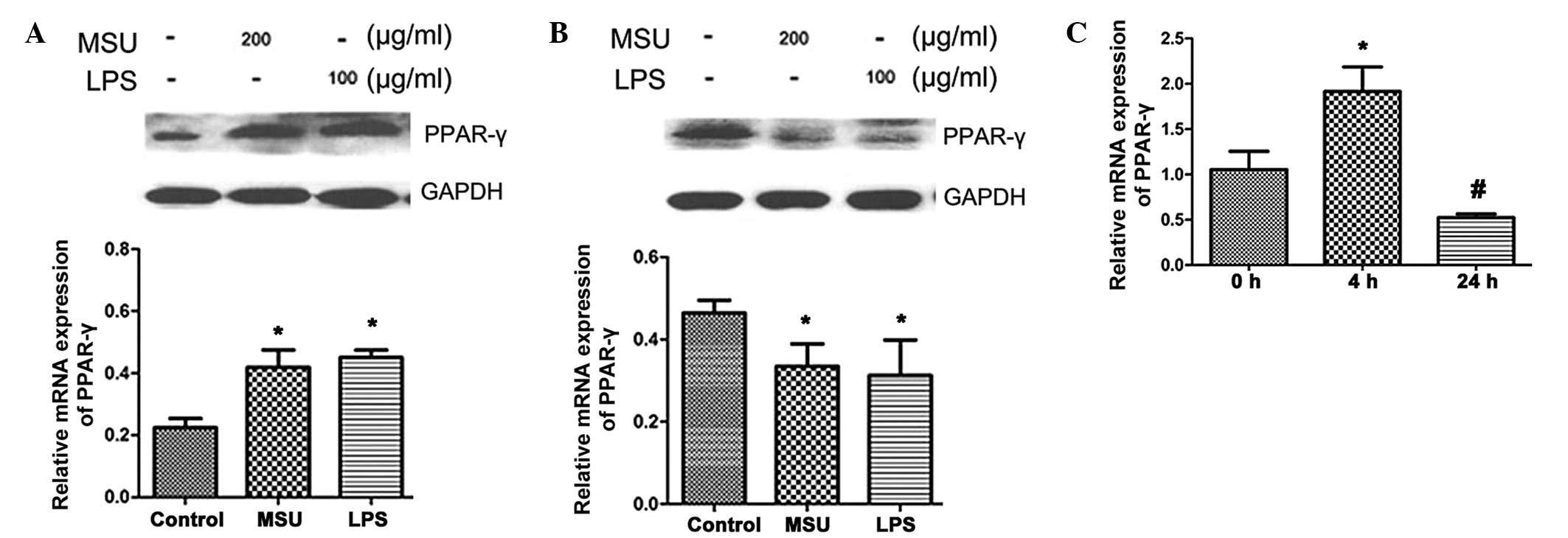
Effects of MSU crystals on PPARγ protein
expression in HK-2 cells
Next, the protein expression of PPARγ in HK-2 cells
was assessed using western blot analysis. As shown in Fig. 2A, after 24 h of treatment, PPARγ
protein levels were increased by MSU crystals (200 µg/ml) or
LPS (100 µg/ml) (P<0.05). However, after 48 h incubation,
the PPARγ protein expression levels in the MSU crystal- and
LPS-treated groups were decreased compared with those in the
control group (P<0.05) (Fig.
2B). Similarly to the effects of MSU crystals on mRNA levels,
PPARγ protein expression was affected in a time-dependent manner:
Increased PPARγ protein expression occurred at 4 h (P<0.05), but
at 24 h, expression was decreased compared with that at 0 h
(P<0.05) (Fig. 2C).
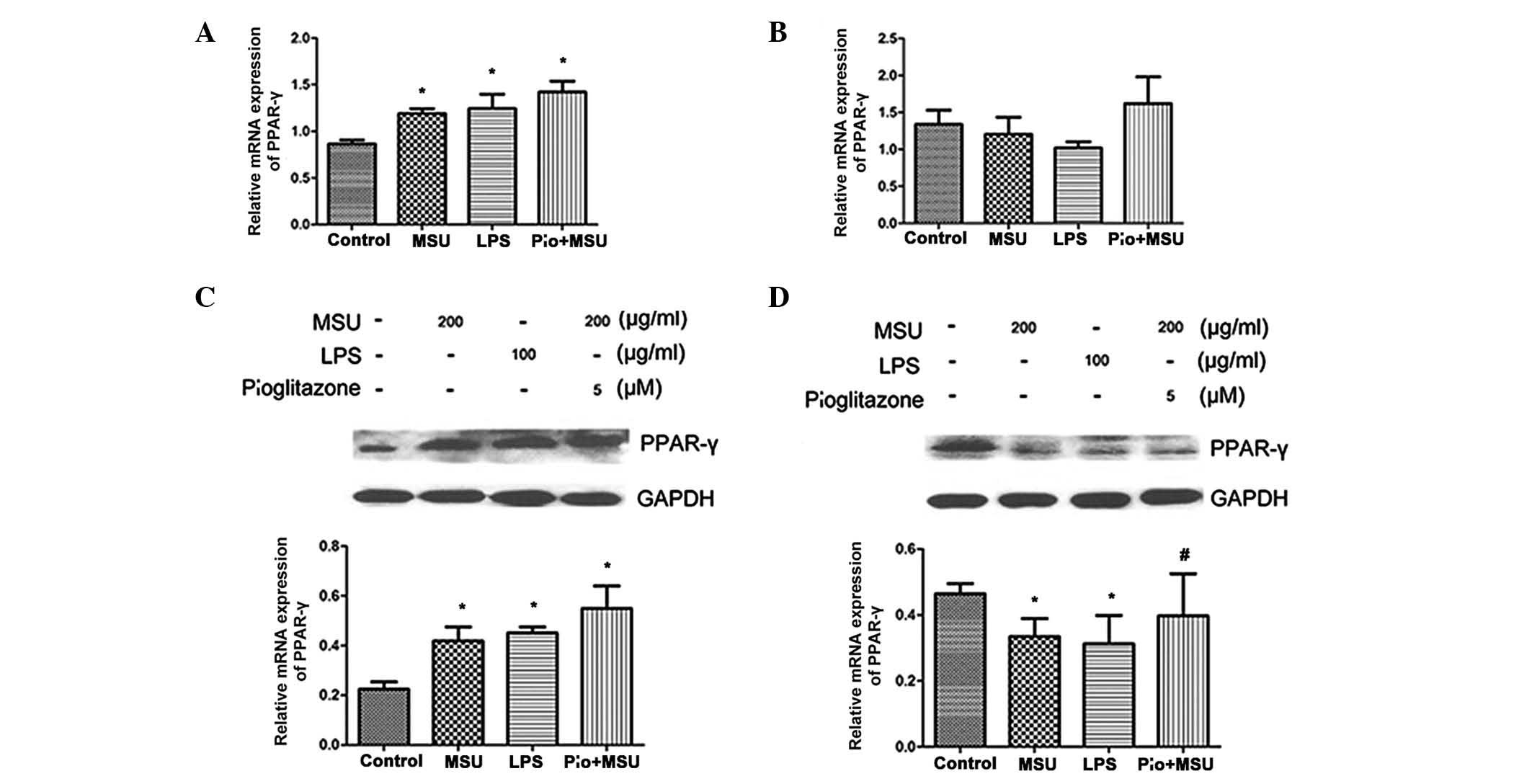
Effect of PPARγ agonist pioglitazone on
the expression of PPARγ in MSU crystal-stimulated HK-2 cells
To verify the role of PPARγ expression in HK-2 cells
stimulated by MSU crystals, the present study further investigated
the mRNA and protein levels of PPARγ in HK-2 cells which were
pre-treated with PPARγ agonist pioglitazone for 12 h and then
treated with 200 µg/ml MSU crystals. Pioglitazone
pre-treatment induced a further increase in PPARγ mRNA expression
at 12 h and partly restored basal PPARγ mRNA expression at 24 h;
however, the results were not significantly different from those of
the MSU crystals only-treated group (Fig. 3A and B). Similar results were
obtained by western blot analyses of protein levels of PPARγ
(Fig. 3C and D).
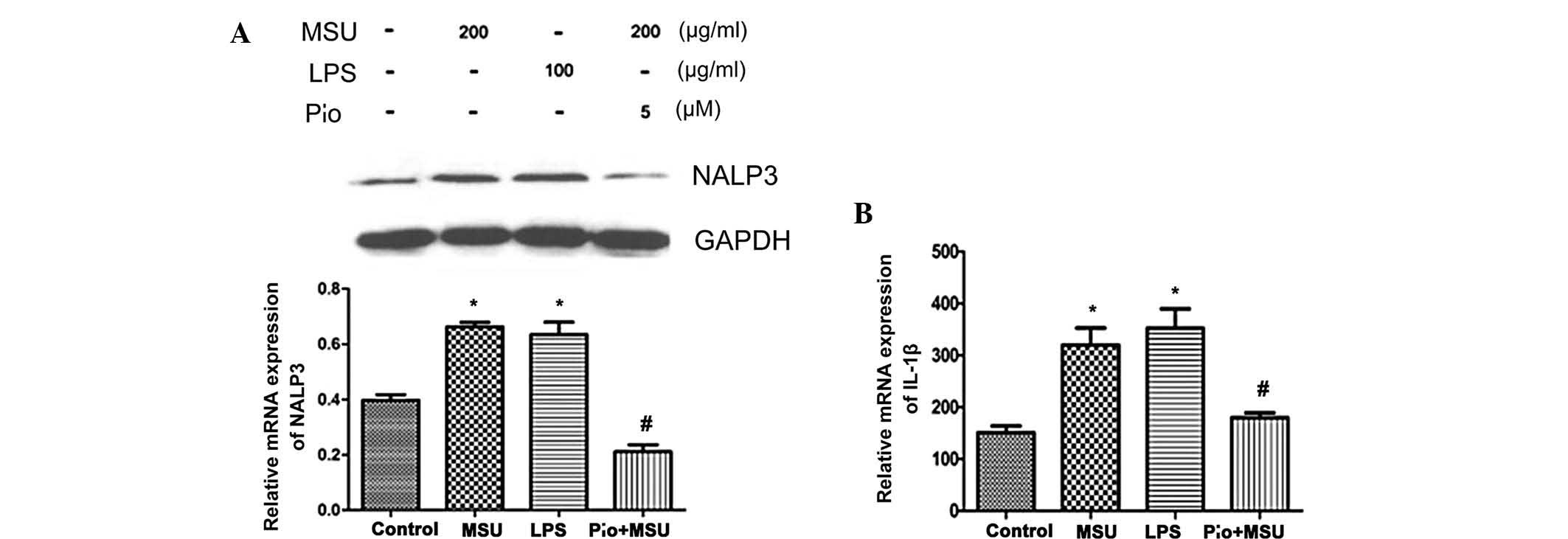
Effects of MSU crystals and PPARγ agonist
pioglitazone on NALP3 inflammasome expression and IL-1β
secretion
Studies have shown that the biological activity of
MSU crystals mainly depends on the activation of NALP3
inflammasome, while IL-1β mediates the release of cytokines in MSU
crystal-induced inflammation (23). Therefore, the present study
examined the effect of MSU crystals and pioglitazone on NALP3 and
IL-1β production in MSU crystal-induced HK-2 cells. NALP3 protein
levels were detected using western blot analysis and IL-1β levels
were examined by ELISA. As shown in Fig. 4A, MSU crystals elevated NALP3
protein expression in HK-2 cells compared to that in the untreated
control cells at 48 h (P<0.05), while pre-incubation with PPARγ
agonist pioglitazone resulted in a significant decrease of NALP3
protein expression (P<0.05). IL-1β secretion increased following
MSU-crystal treatment compared with that in the untreated control
group. Pioglitazone almost fully inhibited MSU crystal-induced
increases in IL-1β (P<0.05) (Fig.
4B).
Discussion
The present study demonstrated for the first time,
to the best of our knowledge, that MSU crystals affected PPARγ
expression in HK-2 cells. MSU crystals exerted a biphasic effect on
PPARγ expression, causing an increase during the first hours of
exposure, while later inhibiting PPARγ expression. In addition, it
was observed that the PPARγ agonist pioglitazone mildly, but not
significantly increased the MSU crystal-induced expression of PPARγ
in HK-2 cells at the mRNA and protein level. However, pioglitazone
significantly decreased the amount of NALP3 and IL-1β protein in
MSU crystal-stimulated HK-2 cells. Zou et al (24) reported that PPARγ ligand
troglitazone enhanced the activity of PPARγ in mesangial cells.
Therefore, it was hypothesized that pioglitazone inhibits NALP3
inflammasomes and IL-1β not by increasing the expression of the
transcriptional regulator PPARγ but, similarly to the effect of
troglitazone, by enhancing its activity.
The findings of the present study were consistent
with a number of studies focusing on the expression of PPARγ.
Akahoshi et al (25) have
shown that MSU crystals can induce PPARγ gene expression by
mononuclear cells in a time-dependent manner: mRNA expression was
rapidly increased and subsequently declined. Wang et al
(26) detected changes in PPARγ
activity using an electrophoretic mobility shift assay (EMSA),
which indicated that LPS upregulated PPARγ activity in HK-2 cells
at 6 h, which then decreased at 48 h. Similar findings were
reported by Bhatt et al (27), who examined the effect of
peptidoglycan (PGN) on PPARγ production. PGN induced a biphasic
effect on PPARγ expression in macrophages, leading to increases in
the early stage followed by suppression of PPARγ expression.
Further investigation of the mechanism of the late-phase inhibition
of PPARγ expression showed that the early increase is mediated by
extracellular signal-regulated kinase, while the late repression
occurs via c-Jun N-terminal kinase activation.
Since the results of the present study showed that
MSU crystals inhibited PPARγ expression at a later stage, MSU
crystal-stimulated HK-2 cells were pre-treated with PPARγ agonist
pioglitazone to investigate its effects on the expression of PPARγ,
NALP3 and IL-6. The results were consistent with previous studies
which explored the effect of PPARγ agonists. Jiang et al
(28) reported that troglitazone
inhibited LPS-induced IL-6, IL-8 and TNF-α secretion in
macrophages. Wang et al (26) demonstrated that rosiglitazone
inhibited LPS-induced IL-6 and IL-8 expression in HK-2 cells. In
cultured human proximal tubular epithelial cells (HPTECs),
rosiglitazone was reported to attenuate high-glucose-induced IL-6,
CCL-2 and transforming growth factor (TGF)-β expression (29). The same conclusions were inferred
from animal studies. Yang et al (30) showed that pioglitazone attenuated
podocyte injury-associated glomerulosclerosis by reducing
macrophage infiltration and inhibiting TGF-β and plasminogen
activator inhibitor-1 expression. Pioglitazone was also reported to
significantly decrease matrix metalloproteinase expression and
oxidative stress, and to reduce renal ischemia/re-perfusion injury
and acute inflammation in rats (31). In hyperoxaluric rats, Taguchi et
al (32) demonstrated that
pioglitazone suppressed kidney crystal formation through renal
tubular cell protection as well as anti-oxidative and
anti-inflammatory effects. All of these observations suggested that
PPARγ agonists have a protective effect on renal function through
the inhibition of inflammation.
However, in a number of studies, certain biochemical
stimuli significantly reduced PPARγ expression, which was contrary
to the results of the present study. Li et al (33) reported that the amount of PPARγ in
hypoxia-induced HPTECs was significantly decreased. PPARγ
expression in cyclosporine-treated rat kidneys was significantly
lower than that in the control groups (34). Matsuyama et al (35) also showed that PPARγ expression was
reduced in rats following ischemia/re-perfusion. The discrepancies
between these previous studies and the results of the present study
may be due to differences in treatment or stimulus intensity, or
due to differences between experimental in vitro and in
vivo models.
Based on the results of the present study, it is
hypothesized that the rapid induction of PPARγ may contribute to
the self-limiting nature of hyperuricemia-induced acute
inflammation in gouty patients, while the later suppression of
PPARγ may result in chronic renal injury in hyperuricemia patients.
The results of the present and other studies have shown that PPARγ
agonists downregulate MSU crystal-induced pro-inflammatory
cytokines. Since MSU crystal-induced inflammation in kidneys has an
important role in hyperuricemic nephropathy, PPARγ agonists have a
potential therapeutic value in preventing tubular injury associated
with hyperuricemia-associated renal disease.
Acknowledgments
This study was supported by the Shanghai Medical
Guide Science and Technology Project (no. 114119a6200).
References
|
1
|
Zhu Y, Pandya BJ and Choi HK: Prevalence
of gout and hyperuricemia in the US general population: The
national health and nutrition examination survey 2007–2008.
Arthritis Rheum. 63:3136–3141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
B L, T W, Hn Z, Ww Y, Hp Y, Cx L, J Y, Ry
J and Hw N: The prevalence of hyperuricemia in China: A
meta-analysis. BMC Public Health. 11:8322011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Iseki K, Ikemiya Y, Inoue T, Iseki C,
Kinjo K and Takishita S: Significance of hyperuricemia as a risk
factor for developing ESRD in a screened cohort. Am J Kidney Dis.
44:642–650. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Obermayr RP, Temml C, Gutjahr G,
Knechtelsdorfer M, Oberbauer R and Klauser-Braun R: Elevated uric
acid increases the risk for kidney disease. J Am Soc Nephrol.
19:2407–2413. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Khan SR: Crystal-induced inflammation of
the kidneys: Results from human studies, animal models and
tissue-culture studies. Clin Exp Nephrol. 8:75–88. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Akahoshi T, Murakami Y and Kitasato H:
Recent advances in crystal-induced acute inflammation. Curr Opin
Rheumatol. 19:146–150. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Maejima I, Takahashi A, Omori H, Kimura T,
Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y and
Yoshimori T: Autophagy sequesters damaged lysosomes to control
lysosomal biogenesis and kidney injury. EMBO J. 32:2336–2347. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shi Y, Evans JE and Rock KL: Molecular
identification of a danger signal that alerts the immune system to
dying cells. Nature. 425:516–521. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Martinon F, Pétrilli V, Mayor A, Tardivel
A and Tschopp J: Gout-associated uric acid crystals activate the
NALP3 inflammasome. Nature. 440:237–241. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mariathasan S, Newton K, Monack DM, Vucic
D, French DM, Lee WP, Roose-Girma M, Erickson S and Dixit VM:
Differential activation of the inflammasome by caspase-1 adaptors
ASC and Ipaf. Nature. 430:213–218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Razmara M, Srinivasula SM, Wang L, Poyet
JL, Geddes BJ, DiStefano PS, Bertin J and Alnemri ES: CARD-8
protein, a new CARD family member that regulates caspase-1
activation and apoptosis. J Biol Chem. 277:13952–13958. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Martinon F, Mayor A and Tschopp J: The
inflammasomes: Guardians of the body. Annu Rev Immunol. 27:229–265.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miao ZM, Zhao SH, Yan SL, Li CG, Wang YG,
Meng DM, Zhou L and Mi QS: NALP3 inflammasome functional
polymorphisms and gout susceptibility. Cell Cycle. 8:27–30. 2009.
View Article : Google Scholar
|
|
14
|
Sims JE and Smith DE: The IL-1 family:
Regulators of immunity. Nat Rev Immunol. 10:89–102. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen CJ, Shi Y, Hearn A, Fitzgerald K,
Golenbock D, Reed G, Akira S and Rock KL: MyD88-dependent IL-1
receptor signaling is essential for gouty inflammation stimulated
by monosodium urate crystals. J Clin Invest. 116:2262–2271. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
So A, De Smedt T, Revaz S and Tschopp J: A
pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis
Res Ther. 9:R282007. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kersten S, Desvergne B and Wahli W: Roles
of PPARs in health and disease. Nature. 405:421–424. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chana RS, Lewington AJ and Brunskill NJ:
Differential effects of peroxisome proliferator activated
receptor-gamma (PPAR gamma) ligands in proximal tubular cells:
Thiazolidinediones are partial PPAR gamma agonists. Kidney Int.
65:2081–2090. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ricote M, Li AC, Willson TM, Kelly CJ and
Glass CK: The peroxisome proliferator-activated receptor-gamma is a
negative regulator of macrophage activation. Nature. 391:79–82.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Su CG, Wen X, Bailey ST, Jiang W, Rangwala
SM, Keilbaugh SA, Flanigan A, Murthy S, Lazar MA and Wu GD: A novel
therapy for colitis utilizing PPAR-gamma ligands to inhibit the
epithelial inflammatory response. J Clin Invest. 104:383–389. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Matsuyama M, Yoshimura R, Hase T, Uchida
J, Tsuchida K, Takemoto Y, Kawahito Y, Sano H and Nakatani T:
Expression of peroxisome proliferator-activated receptor-gamma in
renal ischemia-reperfusion injury. Transplant Proc. 37:1684–1685.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma LJ, Marcantoni C, Linton MF, Fazio S
and Fogo AB: Peroxisome proliferator-activated receptor-gamma
agonist troglitazone protects against nondiabetic
glomerulosclerosis in rats. Kidney Int. 59:1899–1910. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Busso N and Ea HK: The mechanisms of
inflammation in gout and pseudogout (CPP-induced arthritis).
Reumatismo. 19:230–237. 2012.
|
|
24
|
Zou R, Xu G, Liu XC, Han M, Jiang JJ,
Huang Q, He Y and Yao Y: PPARgamma agonists inhibit TGF-beta-PKA
signaling in glomerulosclerosis. Acta Pharmacol Sin. 31:43–50.
2010. View Article : Google Scholar
|
|
25
|
Akahoshi T, Namai R, Murakami Y, Watanabe
M, Matsui T, Nishimura A, Kitasato H, Kameya T and Kondo H: Rapid
induction of peroxisome proliferator-activated receptor gamma
expression in human monocytes by monosodium urate mono-hydrate
crystals. Arthritis Rheum. 48:231–239. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang WM, Chen H, Zhong F, Lu Y, Han L and
Chen N: Inhibitory effects of rosiglitazone on
lipopolysaccharide-induced inflammation in a murine model and HK-2
cells. Am J Nephrol. 34:152–162. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bhatt KH, Sodhi A and Chakraborty R:
Peptidoglycan induced expression of peroxisome
proliferator-activated receptor γ in mouse peritoneal macrophages:
Role of ERK and JNK MAP kinases. Cytokine. 60:778–786. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jiang C, Ting AT and Seed B: PPAR-gamma
agonists inhibit production of monocyte inflammatory cytokines.
Nature. 391:82–86. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tang SC, Chan LY, Leung JC, Cheng AS, Chan
KW, Lan HY and Lai KN: Bradykinin and high glucose promote renal
tubular inflammation. Nephrol Dial Transplant. 25:698–710. 2010.
View Article : Google Scholar
|
|
30
|
Yang HC, Ma LJ, Ma J and Fogo AB:
Peroxisome proliferator-activated receptor-gamma agonist is
protective in podocyte injury-associated sclerosis. Kidney Int.
69:1756–1764. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Reel B, Guzeloglu M, Bagriyanik A, Atmaca
S, Aykut K, Albayrak G and Hazan E: The effects of PPAR-γ agonist
pioglitazone on renal ischemia/reperfusion injury in rats. J Surg
Res. 182:176–184. 2013. View Article : Google Scholar
|
|
32
|
Taguchi K, Okada A, Yasui T, Kobayashi T,
Ando R, Tozawa K and Kohri K: Pioglitazone, a peroxisome
proliferator activated receptor γ agonist, decreases renal crystal
deposition, oxidative stress and inflammation in hyperoxaluric
rats. J Urol. 188:1002–1011. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li X, Kimura H, Hirota K, Sugimoto H,
Kimura N, Takahashi N, Fujii H and Yoshida H: Hypoxia reduces the
expression and anti-inflammatory effects of peroxisome
proliferator-activated receptor-gamma in human proximal renal
tubular cells. Nephrol Dial Transplant. 22:1041–1051. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ahn KO, Lim SW, Yang HJ, Li C, Sugawara A,
Ito S, Choi BS, Kim YS, Kim J and Yang CW: Induction of PPAR gamma
mRNA and protein expression by rosiglitazone in chronic
cyclosporine nephropathy in the rat. Yonsei Med J. 48:308–316.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Matsuyama M, Yoshimura R, Kawahito Y, Sano
H, Chargui J, Touraine JL and Nakatani T: Relationship between
peroxisome proliferator-activated receptor-γ and renal
ischemia-reperfusion injury. Mol Med Rep. 1:499–503.
2008.PubMed/NCBI
|