Introduction
Accumulating evidence supports that various types of
cancer are triggered by infection and chronic inflammatory disease
(1), and gastric cancer is a
classical model of inflammation-associated cancer. Chronic
inflammation actuates cellular events, which prompt malignant
transformation of cells and even carcinogenesis (2). Previous studies have confirmed that
inflammatory cytokines are significant in the oncogenesis of
gastric cancer (3,4). In addition, a number of previous
studies have suggested that polymorphisms in pro-inflammatory
cytokine genes, including IL-1β, IL-6, CC chemokine ligand (CCL)2,
IL-8, CCL5 and PDGF, are associated with cancer (5–8).
CCL2 is a member of the CC chemokine family, which
predominantly binds with high affinity to the CC-chemokine receptor
2 (CCR2). CCL2 is a critical modulator of inflammation and is
produced by a variety of cell types, including fibroblast cells,
epithelial cells, mononuclear cells, astrocytes and certain tumor
cells (9). Previous studies have
reported that CCL2 is widely detected in several types of cancer
and is important in cancer progression. In prostate cancer, CCL2
mediates the interactions between normal and malignant cells in the
tumor microenvironment and promotes prostate cancer tumorigenesis
and metastasis (10). A
CCL2/reactive oxygen species autoregulation loop increases the
growth of oral squamous cell carcinoma (11). In addition, in colon and breast
cancers, CCL2 enhances endothelial cell activities in angiogenesis
and metastasis (12). Rokavec
et al (13) revealed that
recombinant CCL2 (MCP-1) alone was sufficient to transform mammary
epithelial cells and develop tumors. Notably, the expression of
CCL2 has been demonstrated to be high in gastric cancer tissue and
the peripheral blood of patients with gastric cancer (14,15).
Whether CCL2 is involved in gastric carcinogenesis remains to be
elucidated.
A number of factors contribute to the initiation and
development of gastric cancer. Diet, smoking, obesity and chronic
infections are all major factors, which are involved in the
occurrence and development of cancer (16,17).
The intake of salted foods, containing high levels of N-nitroso
compounds (NOCs), which are powerful carcinogens, is a critical
component of the carcinogenesis of gastric cancer (18). N-methyl-N-nitro-N′-nitrosoguanidine
(MNNG) is one of the most active carcinogenic NOCs and has been
used to establish stomach carcinomas successfully in animal models
(19). Human GES-1 gastric mucosa
epithelial cells can be transformed by MNNG into the precancerous
cell model, termed MC cells, which are widely used to investigate
the mechanism underlying gastric carcinogenesis (20).
In the present study, parental GES-1 cells and
MNNG-pretreated GES-1 or MC cells were stimulated with CCL2. It was
demonstrated that the expression of CCR2 was markedly low and CCL2
revealed no effect on the parental GES-1 cells. However, following
pretreatment or transformation into MC cells by MNNG, the
expression of CCR2 in the GES-1 cells was significantly increased.
CCL2 promoted the migration of MNNG-pretreated GES-1 cells and MC
cells through the induction of the epithelial-mesenchymal
transition (EMT).
Materials and methods
Cell culture
The U937 and human gastric epithelial GES-1 cell
lines were purchased from the Institute of Biochemistry and Cell
Biology at the Chinese Academy of Sciences (Shanghai, China). The
GES-1 cell line was transformed into an MC cell line using MNNG, as
follows: MNNG (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in
dimethyl sulfoxide (Sigma-Aldrich) at a concentration of 0.2 mol/l
and the GES-1 cells were induced with 2×10−5 mol/l MNNG
for 24 h in the dark. Following the removal of MNNG, normal
RPMI-1640 medium (Invitrogen Life Technologies, Carlsbad, CA, USA)
was used to culture the cells and was changed every 3 days. During
the following week, some of the cells initially died and
subsequently the surviving cells grew out. The cells were cultured
in RPMI-1640 medium, supplemented with 10% fetal bovine serum (FBS;
Invitrogen Life Technologies) and were maintained at 37°C in a
humidified chamber with 5% CO2.
Cell colony formation assay
The cells were collected and seeded into 6-well
plates (1,000 cells/well) in growth medium in the presence or
absence of CCL2 (10 ng/ml; R&D systems, Minneapolis, MN, USA).
The cultures were grown for 10 days at 37°C in a humidified
incubator and the growth medium was changed every 3 days. The
colonies were fixed with 99.5% methanol for 30 min at room
temperature and visualized by staining with 1% crystal violet
solution (Beijing Airan Bio-Engineering Co., Ltd., Beijing, China)
for 15 min on the indicated day. The cell colonies were counted for
statistical analysis following images being captured (SX240 HS;
Canon, Tokyo, Japan).
Transwell migration assay
The control cells or pretreated cells
(1×105 in 200 µl) suspended in serum-free medium
were seeded into the upper compartment of transwell chambers with 8
µm pore filters (Corning, Inc., Corning, NY, USA). Complete
medium, containing 10% FBS (600 µl), with or without CCL2
(10 ng/ml) was added into the lower chamber as a chemoattractant.
Following 10 h of incubation at 37°C, the insert was washed with
phosphate-buffered saline (PBS) and the non-migrated cells in the
top chamber were mechanically removed with cotton swabs. The cells
migrating through the membrane were fixed with 99.5% methanol and
stained with 1% crystal violet. The number of migrated cells were
quantified manually under an inverted microscope (TI-TS100W; Nikon,
Tokyo, Japan).
Western blotting
The cells were harvested and lysed with
radioimmunoprecipitation buffer [Vazyme Biotech (Nanjing) Co.,
Ltd., Nanjing, China], supplemented with protease inhibitors. Equal
quantities (150 µg) of individual proteins were prepared for
electrophoresis on 8–12% SDS-PAGE gels (Biolink, Beijing, China)
and were subsequently transferred onto polyvinylidene fluoride
membranes (EMD Millipore, Billerica, MA, USA). Following blocking
with 5% non-fat milk in Tris-buffered saline, containing 0.05%
Tween-20 (TBST), for 1 h, the membranes were incubated with the
appropriate dilutions of specific monoclonal antibodies overnight
at 4°C. The following monoclonal antibodies were used: Rabbit
monoclonal anti-E-cadherin (1:500; sc-7870; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), rabbit monoclonal
anti-N-cadherin (1:1,000; BS2224; Bioworld Technology, Louis Park,
MN, USA), rabbit monoclonal anti-CCR2 (1:500; 2068–1; Wuhan Boster
Bio-Engineering Co., Ltd., Wuhan, China) and mouse monoclonal
anti-GAPDH (1:2,000; CW0100A; Kangcheng, Shanghai, China).
Following washing with TBST, the membranes were incubated with the
following secondary antibodies for 1 h: Horseradish
peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG;
1:2,000; sc-2301) and anti-mouse IgG (1:2000; sc-358923) (Santa
Cruz Biotechnology, Inc.). The protein bands were visualized by
enhanced chemiluminescence, according to the manufacturer's
instructions (EMD Millipore). GAPDH was used as a loading
control.
Immunofluorescence staining
The cells (2×104) were seeded into a
24-well dish for 24 h and were subsequently washed with PBS twice,
fixed with 4% paraformaldehyde for 2 h, permeabilized with 0.1%
Triton X-100 for 10 min, blocked with 5% bovine serum albumin for
20 min (Wuhan Boster Bio-Engineering Co., Ltd.) and incubated with
the primary antibodies overnight at 4°C. The primary antibodies
were rabbit monoclonal anti-CCR2 (1:100; 2068–1; Wuhan Boster
Bio-Engineering Co., Ltd.), rabbit monoclonal anti-E-cadherin
(1:100; 21473; Signalway Antibody, College Park, MD, USA) and
rabbit monoclonal anti-N-cadherin (1:200; BS2224; Bioworld
Technology, Minneapolis, MN, USA). Following washing with PBS, the
cells were incubated with fluorescein isothiocyanate-labeled donkey
anti-rabbit IgG secondary antibody (1:500; A31572; Invitrogen Life
Technologies) at 37°C. The nuclei were subsequently counterstained
with Hoechst (1:200; Invitrogen Life Technologies) and all samples
were imaged using a confocal immunofluorescence microscope
(TI-TS100W; Nikon) at the same exposure times.
Statistical analysis
All statistical analyses were performed using of
SPSS 16.0 software (SPSS Inc., Chicago, IL, USA). The data are
presented as the mean ± standard deviation. Student's t-test was
used for two-group comparisons. Each experiment was performed
independently at least three times with similar results. P<0.05
was considered to indicate a statistically significant
difference.
Results
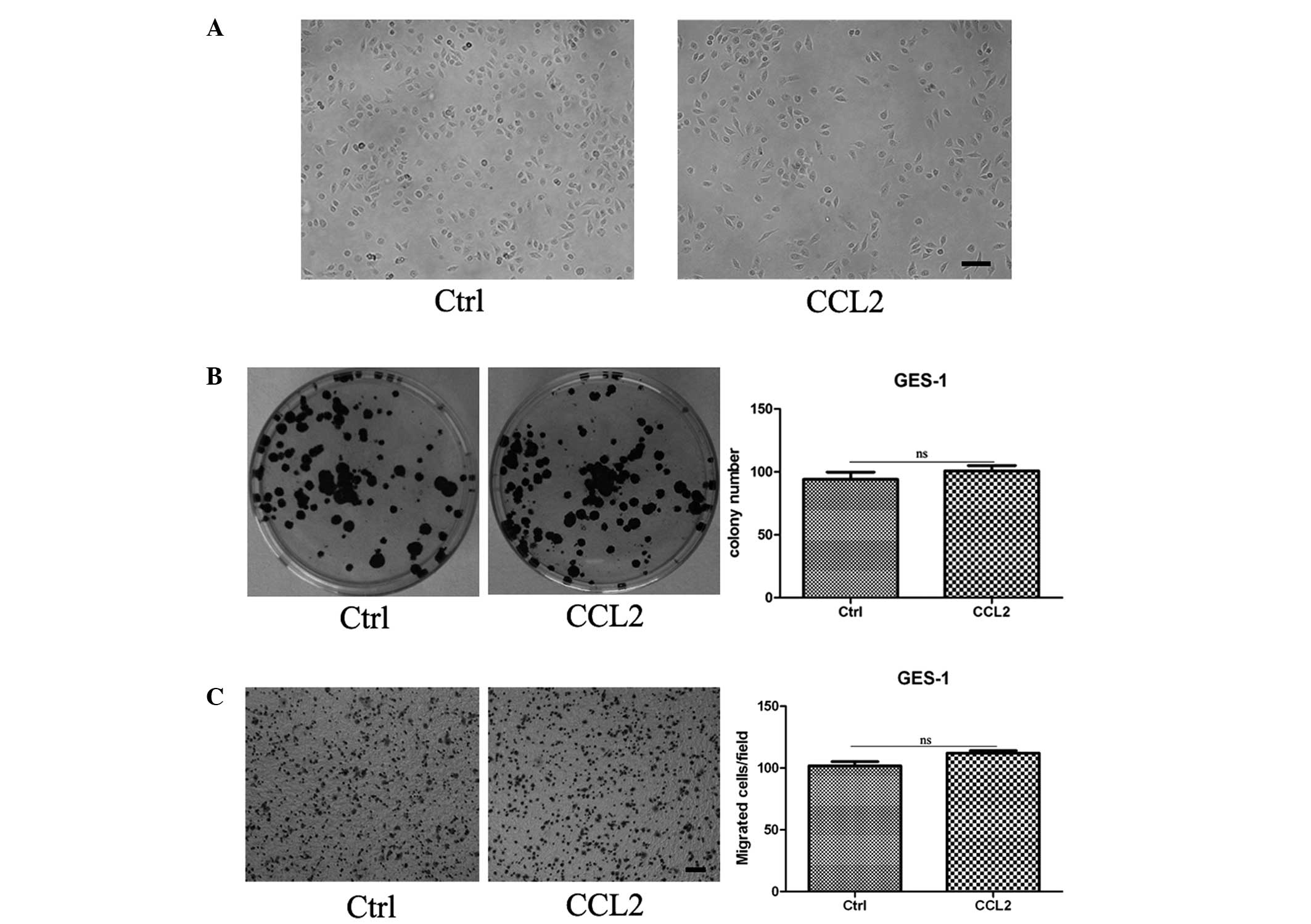
CCL2 has no effect on the morphology,
proliferation and migration of GES-1 cells
To investigate the role of CCL2 in gastric
carcinogenesis, GES-1, a human gastric epithelial cell line, was
continuously pretreated with human recombinant CCL2 (10 ng/ml) for
9 days and collected for cell biology analysis. It was revealed
that the cell morphology of the GES-1 cells pretreated with CCL2
was similar to the control group (Fig.
1A). A cell colony formation assay revealed no difference in
the number and size of the cell colonies between the control group
and the treated group (Fig. 1B).
Transwell migration analysis revealed that the migration capacity
of the GES-1 cells in the treated group was no different compared
with the control group (Fig.
1C).
To further investigate the direct effects of CCL2 on
GES-1, a cell colony formation assay was performed for GES-1 cells
in the presence or absence of CCL2. The results revealed that CCL2
had no effect on GES-1 (data not shown). The chemokine, CCL2, is
best known for its chemotactic functions, whereas no differences
were observed in the motility of gastric epithelial cells incubated
with or without CCL2 in the lower chamber (data not shown).
Therefore, CCL2 stimulation had no effect on the morphology,
proliferation and migration of gastric epithelial cells.
MNNG induces the expression of CCR2 in
gastric epithelial cells
CCL2 signals primarily through the chemokine
receptor, CCR2. It was hypothesized that GES-1 cells failed to
respond to CCL2 as a result of a lack of CCR2. To confirm this
hypothesis, the protein expression of CCR2 was determined in GES-1
cells by western blotting, using human U937 monocytes as the
positive control. As shown in Fig.
2A, GES-1 demonstrated a considerably low expression of
CCR2.
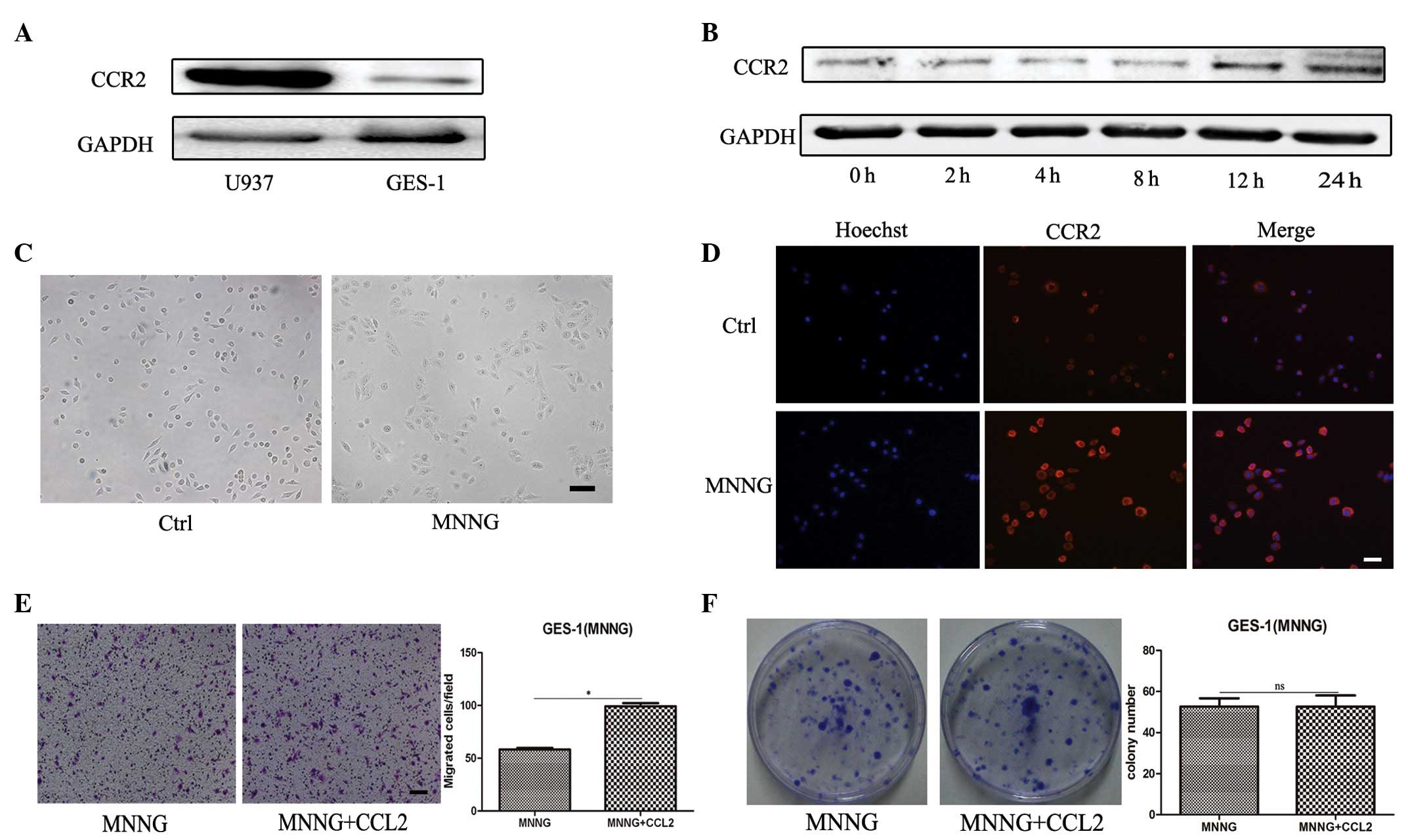 | Figure 2Pretreatment with MNNG induces the
expression of CCR2 in GES-1 cells. (A) Western blotting was
performed to determine the protein expression of CCR2 in U937 and
GES-1 cells. (B) Western blotting was performed to determine the
protein expression of CCR2 in GES-1 cells treated with MNNG for 0,
2, 4, 8, 12 and 24 h. (C) Representative images of cells
(magnification, ×100; scale bar, 100 µm). (D)
Immunofluorescence staining of CCR2 was performed to determine the
expression levels of CCR2 (magnification, ×200; scale bar, 50
µm). (E) A Transwell assay was performed to determine the
migration capability of the cells. The cells were seeded onto the
top of the upper chamber and the number of cells that migrated
through the uncoated filter in response to CCL2 in the lower
chamber was quantified (magnification, ×100; scale bar, 100
µm; *P<0.05). (F) A cell colony formation
assay of the MNNG-pretreated GES-1 cells cultured alone or in the
presence of CCL2 (*P<0.05). Ctrl, GES-1 cells; CCL2,
CC chemokine ligand 2; ns, non-significant; MNNG, cells treated for
12 h with N-methyl-N-nitro-N′-nitrosoguanidine. |
The cell model was changed and 1×10−5
mol/l of MNNG was used to pretreat the GES-1 cells for varying
durations. To determine whether the expression of CCR2 in GES-1
cells was altered following exposure to MNNG, the
duration-dependent effects of MNNG on the expression of CCR2 in
GES-1 cells were investigated. Western blotting revealed that the
expression of CCR2 was significantly increased in the GES-1 cells
following treatment with MNNG for 12 and 24 h (Fig. 2B). Therefore, the present study
focused on GES-1 pretreatment with MNNG for 12 h. The morphology of
the GES-1 cells treated with MNNG exhibited more unclear cell edges
and were flatter and bigger compared with the parental GES-1 cells
(Fig. 2C). Immunofluorescence
staining revealed that MNNG treatment significantly increased the
fluorescence intensity of CCR2, which was consistent with the
upregulation of CCR2 observed by immunoblotting (Fig. 2D). A Transwell migration assay
demonstrated that CCL2 significantly induced the chemotaxis of the
GES-1 cells pretreated with MNNG in vitro (Fig. 2E). However, the cell colony
formation ability of the GES-1 cells was not affected by MNNG
(Fig. 2F). These results indicated
that MNNG induced the expression of CCR2 in GES-1 cells and the
migration of GES-1 cells in response to CCL2.
CCL2 induces the EMT of MNNG-pretreated
GES-1 cells
Since treatment with MNNG induced the expression of
CCR2 in GES-1 cells, whether MNNG treatment enhanced the role of
CCL2 in GES-1 cells was assessed. Three experimental groups,
including the parental GES-1 group, the MNNG-pretreated GES-1 group
and the MNNG-pretreated GES-1 stimulated by CCL2 group, were used.
As shown in Fig. 3A, the cells in
the CCL2 treatment group exhibited a fibroblastic spindle-shaped
morphology and were marginally more slender and longer compared
with the cells in the MNNG-pretreated group. Treatment of
MNNG-pretreated GES-1 cells with CCL2 for 9 days resulted in
significant cell migration over 12 h, as determined by a Transwell
migration assay (Fig. 3B). Western
blotting revealed that CCL2 treatment notably decreased the
expression of the epithelial cell marker, E-cadherin, and increased
the expression of the mesenchymal cell marker, N-cadherin, in the
MNNG-pretreated GES-1 cells (Fig.
3C). To confirm these results, immunofluorescence staining was
performed to detect the expression of N-cadherin and E-cadherin.
Consistent with the western blot analysis, CCL2 downregulated the
expression of E-cadherin and upregulated the expression of
N-cadherin in the MNNG-pretreated GES-1 cells (Fig. 3D and E). Taken together, CCL2
enhanced the migration capacity of the MNNG-pretreated GES-1 cells
and prompted their EMT.
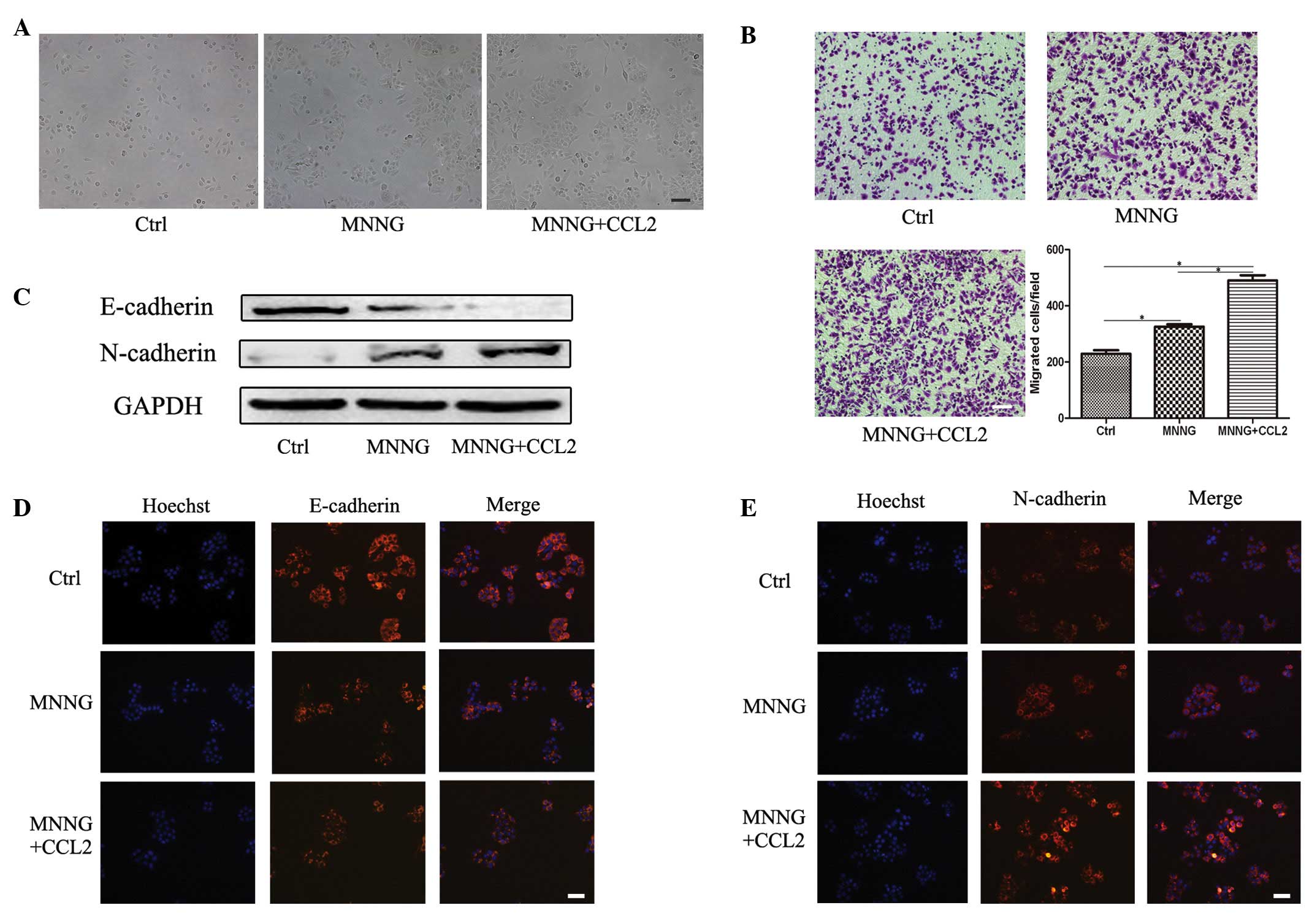 | Figure 3CCL2 promotes the EMT in
MNNG-pretreated GES-1 cells. The GES-1 cells were pretreated with
MNNG and subsequently induced with CCL2 for 9 days. (A)
Representative images of the cells (magnification, ×100; scale bar,
100 µm). (B) A Transwell migration assay was performed to
assess the migration capacity of the cells (magnification, ×100;
scale bar, 100 µm; *P<0.05). (C) Western
blotting was performed to determine the protein expression levels
of E-cadherin and N-cadherin. Immunofluorescence staining was
performed to determine the protein expression levels of (D)
E-cadherin and (E) N-cadherin (magnification, ×200; scale bar, 50
µm). Ctrl, GES-1 cells; MNNG,
N-methyl-N-nitro-N′-nitrosoguanidine-pretreated GES-1 cells; CCL2,
CC chemokine ligand 2; MNNG+CCL2, MNNG-pretreated GES-1 cells
induced with CCL2 for 9 days. |
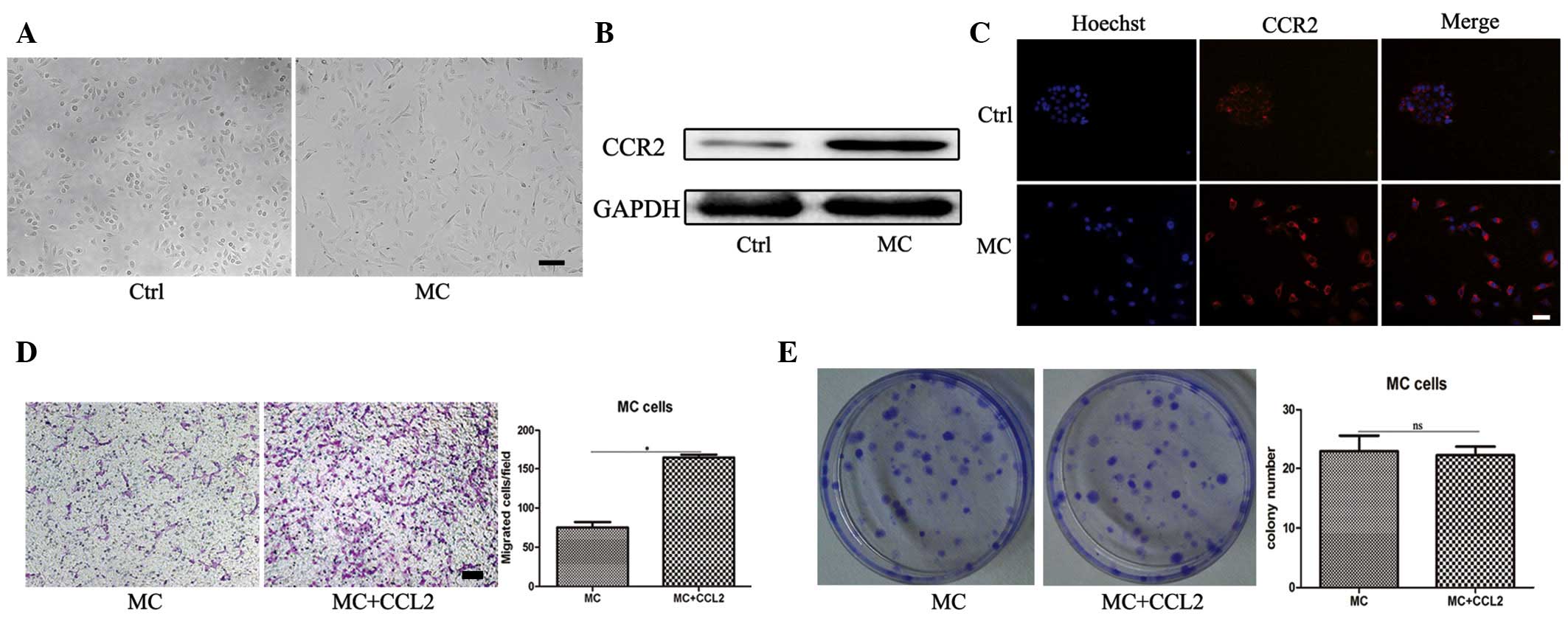
CCR2 upregulation in the MNNG transformed
GES-1 cell line
The GES-1 cells transformed by MNNG (termed MC
cells) have been regarded as a precancerous cell model. The present
study established MC cells using MNNG (2×10−5 mol/l) for
24 h, as described previously (20) (Fig.
4A). To investigate whether the expression of CCR2 in MC cells
was also increased, western blot analysis was performed and
revealed CCR2 was significantly upregulated in the MC cells
(Fig. 4B). This was further
confirmed by an immunofluorescence staining assay (Fig. 4C). The results of the Transwell
migration assay revealed that the migration capacity of the MC
cells was significantly increased in response to CCL2 (Fig. 4D). However, CCL2 still revealed no
effect on the MC cell proliferation, as determined by a cell colony
formation assay (Fig. 4E). These
data indicated that following transformation by MNNG, the
expression of CCR2 in MC cells was markedly upregulated.
CCL2 promotes the EMT in MNNG transformed
GES-1 cells
The MC cells were continuously stimulated with CCL2
for 9 days and collected for analysis. Following treatment with
CCL2, the mesenchymal-like morphology of the MC cells revealed no
change (Fig. 5A). A Transwell
migration assay revealed that the migration ability of the MC cells
stimulated by CCL2 was the most marked (Fig. 5B). Western blot analysis revealed
that the MC cells expressed a lower level of E-cadherin and a
higher level of N-cadherin following CCL2 treatment (Fig. 5C). Immunofluorescence staining
further supported the results of the western blotting (Fig. 5D and E), demonstrating that the
levels of E-cadherin were downregulated and the levels of
N-cadherin were upregulated. These results suggested that CCL2
promoted the migration capacity and the EMT of the MNNG transformed
GES-1 cell line.
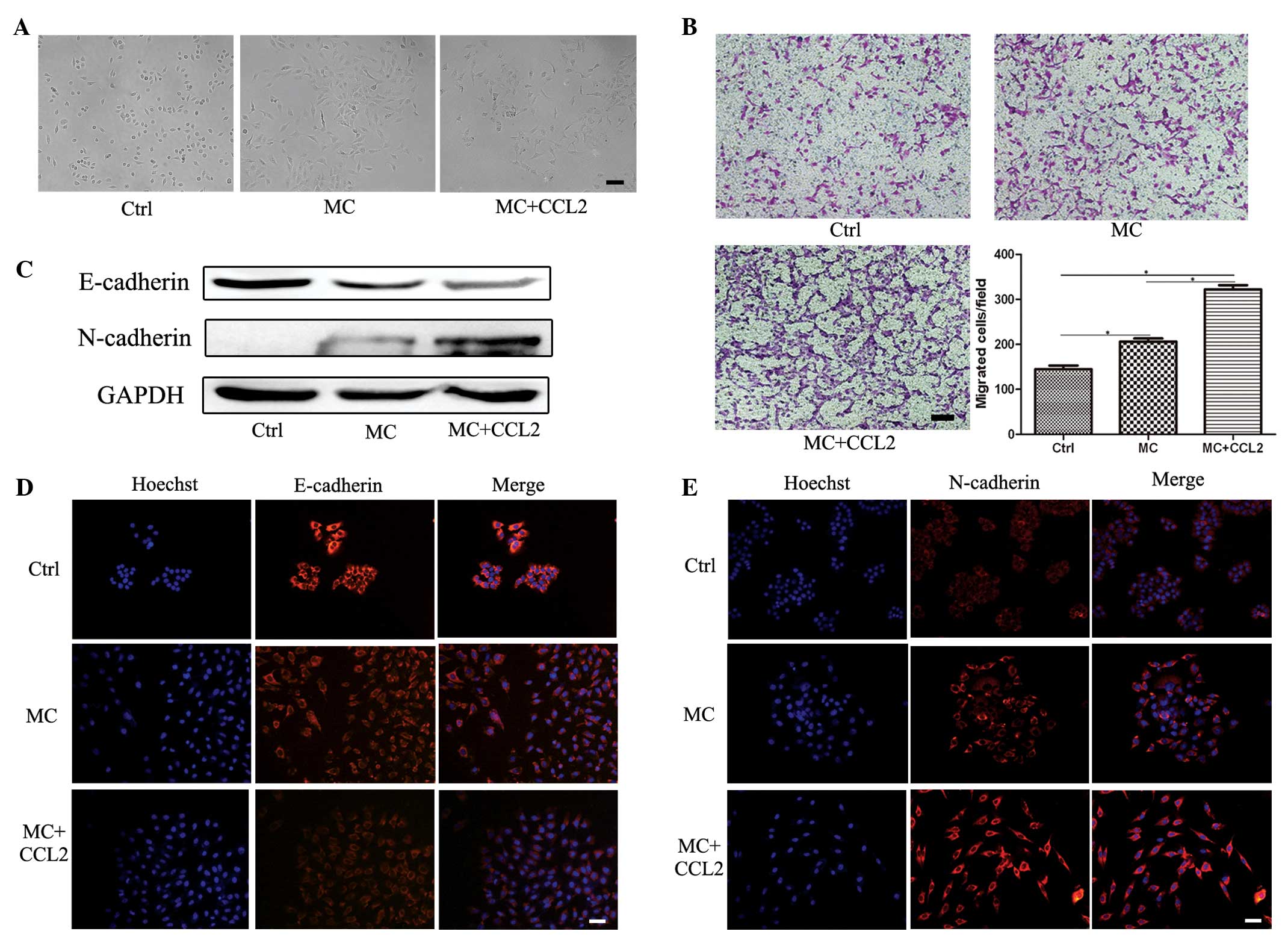 | Figure 5CCL2 promotes the EMT in the MNNG
transformed GES-1 cell line. The MNNG transformed GES-1 cells (MC
cells) were stimulated with CCL2 for 9 days. (A) Representative
images of the cells (magnification, ×100; scale bar, 100
µm). (B) A Transwell migration assay was performed to
determine the migration ability of the cell line (magnification,
×100; scale bar, 100 µm; *P<0.05). (C) Western
blotting was performed to determine the protein expression levels
of E-cadherin and N-cadherin. Immunofluorescence staining was
performed to asses the protein expression of (D) E-cadherin and (E)
N-cadherin (magnification, ×200; scale bar, 50 µm). Ctrl,
GES-1 cells; MC, MC cells; CCL2, CC chemokine ligand 2; MC + CCL2,
MC cells were stimulated with CCL2 for 9 days. |
Discussion
A number of types of human cancer appear to be
initiated by chronic inflammation (21). Epithelial cells are exposed to an
inflammatory microenvironment in the long-term, leading to the EMT
and malignant transformation (22,23).
Significantly high expression of CCL2 is detectable in the
epithelial region of certain tumor types, including breast cancer
(24). There is evidence to
support that CCL2 is expressed predominantly in the epithelial
regions of prostate cancer tissues (25). Therefore, the present study
hypothesized that CCL2 triggered the EMT and the malignant
transformation of gastric epithelial cells.
However, the morphology, proliferation and migration
capacity of GES-1 cells treated with CCL2 exhibited no changes.
Since CCL2 exhibits a particular affinity for the receptor, CCR2,
abundant evidence supports that the CCL2/CCR2 signaling axis may be
important in carcinogenesis and cancer progression (26–28).
Previous data demonstrates that cancer cell derived CCL2 can
activate CCR2 on endothelial cells, leading to efficient tumor cell
extravasation (29). Data from the
present study suggested that the expression of CCR2 was very low in
GES-1 cells. Gao et al (30) treated PC-3M cells with CCR2-small
interfering RNA and revealed that the cell proliferation rate of
the prostate cancer cells was significantly slow and the apoptotic
rate was increased. CCR2 knockdown also reduced the migration and
invasion ability of PC-3M cells (30). In addition, RNA interference of
CCR2 in breast cancer cells markedly inhibits CCL2-induced
migration (27). Therefore, these
results may provide an explanation as to why CCL2 has no direct
effect on the proliferation and migration of GES-1 cells.
CCL2 mediates its effects through the receptor,
CCR2, the expression of which is relatively restricted to certain
types of cell (9). The expression
of the G protein-coupled receptor, CCR2, was increased in breast
cancer (27), prostate cancer
(31) and ovarian cancer (32), correlating with the expression of
CCL2. It is generally accepted that the etiology of gastric cancer
is multifactorial and predominantly a dietary habit. NOCs,
including MNNG, have been implicated as one of major risk factors
for gastric cancer. Notably, GES-1 cells pretreated or transformed
with MNNG upregulated the expression of CCR2. Based on these cell
models, CCL2 promoted the migration of MNNG-pretreated GES-1 cells
and MC cells. In short, these findings suggested that MNNG induced
the expression of CCR2, which is crucial for the CCL2-induced
migration of GES-1 cells.
A crucial step in reinforcing cell migration may be
the acquisition of the EMT phenotype, which reduces cell-cell
adhesion and destabilizes the epithelial architecture. The EMT is
well-characterized in the initiation and development of cancer and
even the upregulation of cancer stemness (33). During the EMT, E-cadherin, which is
involved in maintaining cell polarity and organizing the epithelium
by strengthening intercellular adhesion, is downregulated and
mesenchymal proteins, including N-cadherin, are upregulated
(34). A crucial step in
reinforcing GES-1 cell migration may be associated with the
acquisition of the EMT phenotype. Following treatment with CCL2,
the expression of the epithelial marker, E-cadherin, was markedly
attenuated, while the expression of the mesenchymal marker,
N-cadherin, was notably intensified. As an expectation, the EMT
occurred in the MNNG pretreated GES-1 cells and MC cells following
CCL2 stimulation. During the EMT process, epithelial cells acquire
mesenchymal and stem cell phenotypes, and the EMT-induced cancer
stem cell phenotypes may contribute to carcinogenesis in the
stomach (35). These results
suggested that CCL2 may be involved in the transformation
progression of gastric epithelial cells during the chronic
inflammatory microenvironment.
Although CCL2 induced the migration and the EMT of
the GES-1 cells following treatment with MNNG, no confirmation as
to whether GES-1 induced carcinogenesis following long-term
exposure to CCL2 was demonstrated. Therefore, future work is
required to assess the role of CCL2 in gastric carcinogenesis.
The present study suggested that although normal
GES-1 cells lack the expression of CCR2, the chemical carcinogen,
MNNG, significantly induced the expression of CCR2 in gastric
epithelial cells, which is beneficial for CCL2 prompting the
migration of gastric epithelial cells by facilitating the EMT. In
conclusion, these findings suggested that the inflammatory
cytokine, CCL2, and chemical carcinogens have a synergistic effect
on the development of gastric carcinogenesis.
Acknowledgments
This study was supported by the Major Research Plan
of the National Natural Science Foundation of China (no. 91129718),
the National Natural Science Foundation of China (no. 81302119),
the Natural Science Foundation of the Jiangsu Province (nos.
BK2012709 and BK20130540), the Doctoral Program Foundation of State
Education Ministry (no. 20113227110011), the Jiangsu Province for
Natural Science Research in Colleges and Universities (no.
13KJB320001), the Jiangsu University Excellent Young Teacher
Project, the Scientific Research Foundation of Jiangsu University
for Senior Professional Talents (no. 13JDG088), the Jiangsu
Province for Outstanding Sci-tech Innovation Team in Colleges and
Universities (no. SJK2013–10) and a Project Funded by the Priority
Academic Program Development of Jiangsu Higher Education
Institutions (no. LJ201117).
References
|
1
|
Hussain SP and Harris CC: Inflammation and
cancer: An ancient link with novel potentials. Int J Cancer.
121:2373–2380. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Landskron G, De la Fuente M, Thuwajit P,
Thuwajit C and Hermoso MA: Chronic inflammation and cytokines in
the tumor microenvironment. J Immunol Res. 2014(149185)2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grivennikov SI, Greten FR and Karin M:
Immunity, inflammation and cancer. Cell. 140:883–899. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schetter AJ, Heegaard NH and Harris CC:
Inflammation and cancer: Interweaving microRNA, free radical,
cytokine and p53 pathways. Carcinogenesis. 31:37–49. 2010.
View Article : Google Scholar :
|
|
5
|
Schauer IG, Zhang J, Xing Z, Guo X,
Mercado-Uribe I, Sood AK, Huang P and Liu J: Interleukin-1β
promotes ovarian tumorigenesis through a p53/NF-κB-mediated
inflammatory response in stromal fibroblasts. Neoplasia.
15:409–420. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin JT, Wang JY, Chen MK, Chen HC, Chang
TH, Su BW and Chang PJ: Colon cancer mesenchymal stem cells
modulate the tumorigenicity of colon cancer through interleukin 6.
Exp Cell Res. 319:2216–2229. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Anton K, Banerjee D and Glod J:
Macrophage-associated mesenchymal stem cells assume an activated,
migratory, pro-Inflammatory phenotype with increased IL-6 and
CXCL10 secretion. PLoS One. 7:e350362012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huang F, Wang M, Yang T, Cai J, Zhang Q,
Sun Z, Wu X, Zhang X, Zhu W, Qian H and Xu W: Gastric
cancer-derived MSC-secreted PDGF-DD promotes gastric cancer
progression. J Cancer Res Clin Oncol. 140:1835–1848. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deshmane SL, Kremlev S, Amini S and Sawaya
BE: Monocyte chemoattractant protein-1 (MCP-1): An overview. J
Interferon Cytokine Res. 29:313–326. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang J, Patel L and Pienta KJ: CC
chemokine ligand 2 (CCL2) promotes prostate cancer tumorigenesis
and metastasis. Cytokine Growth Factor Rev. 21:41–48. 2010.
View Article : Google Scholar :
|
|
11
|
Li X, Xu Q, Wu Y, Li J, Tang D, Han L and
Fan Q: A CCL2/ROS autoregulation loop is critical for
cancer-associated fibroblasts-enhanced tumor growth of oral
squamous cell carcinoma. Carcinogenesis. 35:1362–1370. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen C, Duckworth CA, Fu B, Pritchard DM,
Rhodes JM and Yu LG: Circulating galectins -2, -4 and -8 in cancer
patients make important contributions to the increased circulation
of several cytokines and chemokines that promote angiogenesis and
metastasis. Br J Cancer. 110:741–752. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rokavec M, Wu W and Luo JL: IL6-mediated
suppression of miR-200c directs constitutive activation of
inflammatory signaling circuit driving transformation and
tumorigenesis. Mol Cell. 45:777–789. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu J, Liu X and Wang Y: Predictive value
of preoperative serum CCL2, CCL18 and VEGF for the patients with
gastric cancer. BMC Clin Pathol. 13(15)2013. View Article : Google Scholar
|
|
15
|
Tao LL, Shi SJ, Chen LB and Huang GC:
Expression of Monocyte chemotactic protein-1/CCL2 in gastric cancer
and its relationship with tumor hypoxia. World J Gastroenterol.
20:4421–4427. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Crew KD and Neugut AI: Epidemiology of
gastric cancer. World J Gastroenterol. 12:354–362. 2006.PubMed/NCBI
|
|
17
|
Aggarwal BB and Gehlot P: Inflammation and
cancer: How friendly is the relationship for cancer patients? Curr
Opin Pharmacol. 9:351–369. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Abe M, Yamashita S, Kuramoto T, Hirayama
Y, Tsukamoto T, Ohta T, Tatematsu M, Ohki M, Takato T, Sugimura T
and Ushijima T: Global expression analysis of
N-methyl-N′-nitro-N-nitrosoguanidine-induced rat stomach carcinomas
using oligonucleotide microarrays. Carcinogenesis. 24:861–867.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Raphael KR, Sabu M, Kumar KH and Kuttan R:
Inhibition of N-Methyl-N′-nitro-N-nitrosoguanidine (MNNG) induced
gastric carcinogenesis by Phyllanthus amarus extract. Asian Pac J
Cancer Prev. 7:299–302. 2006.PubMed/NCBI
|
|
20
|
Li JX, Wang ZB, Zhu LQ, Niu FL and Cui W:
Effects of Radix notoginseng extracts drug-containing serum on
expressions of bcl-2, Bax and p21WAF1 proteins in MNNG transformed
GES-1 cells. Zhong Xi Yi Jie He Xue Bao. 6:817–820. 2008.PubMed/NCBI
|
|
21
|
Kundu JK and Surh YJ: Inflammation:
Gearing the journey to cancer. Mutat Res. 659:15–30. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu Y, Zhao Y, Xu W, Luo F, Wang B, Li Y,
Pang Y and Liu Q: Involvement of HIF-2α-mediated inflammation in
arsenite-induced transformation of human bronchial epithelial
cells. Toxicol Appl Pharmacol. 272:542–550. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Luo F, Xu Y, Ling M, Zhao Y, Xu W, Liang
X, Jiang R, Wang B, Bian Q and Liu Q: Arsenite evokes IL-6
secretion, autocrine regulation of STAT3 signaling and miR-21
expression, processes involved in the EMT and malignant
transformation of human bronchial epithelial cells. Toxicol Appl
Pharmacol. 273:27–34. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fujimoto H, Sangai T, Ishii G, Ikehara A,
Nagashima T, Miyazaki M and Ochiai A: Stromal MCP-1 in mammary
tumors induces tumor-associated macrophage infiltration and
contributes to tumor progression. Int J Cancer. 125:1276–1284.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lu Y, Cai Z, Galson DL, Xiao G, Liu Y,
George DE, Melhem MF, Yao Z and Zhang J: Monocyte chemotactic
protein-1 (MCP-1) acts as a paracrine and autocrine factor for
prostate cancer growth and invasion. Prostate. 66:1311–1318. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li M, Knight DA, A Snyder L, Smyth MJ and
Stewart TJ: A role for CCL2 in both tumor progression and
immunosurveillance. Oncoimmunology. 2:e254742013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fang WB, Jokar I, Zou A, Lambert D,
Dendukuri P and Cheng N: CCL2/CCR2 chemokine signaling coordinates
survival and motility of breast cancer cells through Smad3 protein-
and p42/44 mitogen-activated protein kinase (MAPK)-dependent
mechanisms. J Biol Chem. 287:36593–36608. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma Y, Adjemian S, Galluzzi L, Zitvogel L
and Kroemer G: Chemokines and chemokine receptors required for
optimal responses to anticancer chemotherapy. Oncoimmunology.
3:e276632014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wolf MJ, Hoos A, Bauer J, Boettcher S,
Knust M, Weber A, Simonavicius N, Schneider C, Lang M, Stürzl M, et
al: Endothelial CCR2 signaling Induced by colon carcinoma cells
enables extravasation via the JAK2-Stat5 and p38MAPK Pathway.
Cancer Cell. 22:91–105. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gao J, Wang A, Zhang M, Li H, Wang K, Han
Y, Wang Z, Shi C and Wang W: RNAi targeting of CCR2 gene expression
induces apoptosis and inhibits the proliferation, migration and
invasion of PC-3M cells. Oncol Res. 21:73–82. 2013. View Article : Google Scholar
|
|
31
|
Lu Y, Chen Q, Corey E, Xie W, Fan J,
Mizokami A and Zhang J: Activation of MCP-1/CCR2 axis promotes
prostate cancer growth in bone. Clin Exp Metastasis. 26:161–169.
2009. View Article : Google Scholar
|
|
32
|
Furukawa S, Soeda S, Kiko Y, Suzuki O,
Hashimoto Y, Watanabe T, Nishiyama H, Tasaki K, Hojo H, Abe M and
Fujimori K: MCP-1 promotes invasion and adhesion of human ovarian
cancer cells. Anticancer Res. 33:4785–4790. 2013.PubMed/NCBI
|
|
33
|
Voulgari A and Pintzas A:
Epithelial-mesenchymal transition in cancer metastasis: Mechanisms,
markers and strategies to overcome drug resistance in the clinic.
Biochim Biophys Acta. 1796:75–90. 2009.PubMed/NCBI
|
|
34
|
Zhang H, Liu L, Wang Y, Zhao G, Xie R, Liu
C, Xiao X, Wu K, Nie Y, Zhang H and Fan D: KLF8 involves in
TGF-beta-induced EMT and promotes invasion and migration in gastric
cancer cells. J Cancer Res Clin Oncol. 139:1033–1042. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Peng Z, Wang CX, Fang EH, Wang GB and Tong
Q: Role of epithelial-mesenchymal transition in gastric cancer
initiation and progression. World J Gastroenterol. 20:5403–5410.
2014. View Article : Google Scholar : PubMed/NCBI
|