Introduction
Ewing's sarcoma belongs to the Ewing's sarcoma
family of tumors (ESFT). It is the second most frequent bone and
soft tissue sarcoma, affecting mostly adolescents and young adults.
Ewing's sarcoma is characterized by unique chromosomal
translocations involving fusion of the 5′segment of the Ewing's
sarcoma breakpoint region 1 (EWS) gene to the 3′segment of an E26
transformation-specific (ETS) family gene, with the most common
fusion taking place with friend leukemia integration 1
transcription factor (FLI1) (1).
Although the 5-year survival rate of Ewing's sarcoma has been
improved through multimodal therapy, patients with metastasis at
diagnosis still have poor clinical outcomes (2–4). It
has been hypothesized that the EWS-FLI1 fusion protein is involved
in a key oncogenic event in ESFT by inducing and repressing
specific target genes (5–7), thereby promoting tumor growth.
Although previous studies have identified the factors that modulate
or are modulated by EWS-FLI1 (8–10),
the mechanisms underlying the development of Ewing's sarcoma are
not yet fully understood.
Several studies have suggested that the EWS-FLI1
fusion gene is implicated in Ewing's sarcoma by modulating microRNA
(miRNA/miR) expression (11–13).
miRNAs are a class of small noncoding RNAs that regulate gene
expression by targeting mRNA (14,15).
Although the biological functions of miRNAs remain largely unknown,
there is some evidence that miRNAs can act as oncogenes or tumor
suppressor genes to regulate cell proliferation, differentiation,
apoptosis and metastasis by altering expression of their target
genes (16,17). Since miRNAs can be regulated by
EWS-FLI1, they may represent potential downstream targets in
Ewing's sarcoma (11–13,18,19).
The miR-34 family is a collection of evolutionarily
conserved miRNAs, which has been associated with tumor suppression
and is downregulated in human tumors (20–22).
miR-34a, a member of miR-34 family, has been reported to predict
the survival of patients with Ewing's sarcoma, and directly affects
the chemosensitivity of tumor cells and malignancy (23). However, miR-34b has not been
specifically detected in Ewing's sarcoma. The aim of the present
study was to investigate the effects of miR-34b, as well as those
of its direct target gene, Notch1, in promoting Ewing's sarcoma
growth and metastasis (24,25).
Materials and methods
Patients and cell culture
Tumor and normal tissue biopsy samples were
collected respectively from 14 patients with Ewing's sarcoma from
2010 to 2016 at the Qilu Hospital of Shandong University (Jinan,
China) and the Shandong Provincial Hospital Affiliated to Shandong
University (Jinan, China), including 5 females and 9 males whose
age from 12 to 23 years old. Patients did not receive therapy at
the time of sample collection. The present study was approved by
the Institutional Review Boards of Qilu Hospital of Shandong
University and Shandong Provincial Hospital Affiliated to Shandong
University. Written informed consent was provided by all patients
for the experimental use of surgical specimens.
Cell lines were obtained from American Type Culture
Collection (ATCC; Manassas, VA, USA). The NIH 3T3 cell line and the
human Ewing's sarcoma cell line A673 was cultured in Dulbecco's
modified Eagle's medium (DMEM; HyClone; GE Healthcare, Logan, UT,
USA) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), and RD-ES cells were
cultured in RPMI-1640 medium (HyClone; GE Healthcare) supplemented
with 15% FBS (Gibco; Thermo Fisher Scientific, Inc.). The cell
lines were maintained at 37°C in a humidified chamber containing 5%
CO2.
Transfection
RD-ES and A-673 Cells (1×105/L) were
transfected with 20 nM small interfering RNA (siRNA) (Shanghai
GeneChem Co., Ltd., Shanghai, China) targeting EWS-FLI-1 using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol. The RNA
sequences used are as follows: EWS-FLI-1-specific siRNA, sense
5′-GUACCCUUCUGACAUCUCCUTT-3′, antisense
5′-AGGAGUGUCAGAAGGGUACTT-3′; and non-silencing control siRNA, sense
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisense
5′-ACGUGACACGUUCGGAGAATT-3′. Infected cells were incubated for 24 h
at 37°C and used for further experiments.
The precursor (miRBase accession no. MI0000742) or
complementary miR-34b sequence (miRBase accession no. MIMAT0000685)
was inserted into a lentiviral vector (Shanghai Genechem Co., Ltd.,
Shanghai, China; cat. nos. PMUL217000742 and PMDL159000685,
respectively) to up- or downregulate miR-34b expression as
previously described (26).
[Anti-miRNA oligonucleotides (AMOs): Ammunition to target miRNAs
implicated in human disease]. The cells were infected with the
lentiviral vectors (1×108 TU/ml) according to
manufacturer's protocol.
Reverse transcription-polymerase chain
reaction (RT-PCR) and RT-quantitative PCR
Total RNA was extracted from cell lines and tumor
and normal tissue biopsy samples using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol, and the RNA was subsequently
reverse-transcribed into complementary DNA using
SuperScript™ First-Strand Synthesis System (Invitrogen;
Thermo Fisher Scientific, Inc.; cat. no. 11904–018; Table I). For reverse transcription
reactions, 10 ng total RNA was used in each reaction (5 µl) and
mixed with the RT primer (3 µl). The RT reaction was carried out at
16°C for 30 min; 42°C for 30 min; 85°C for 5 min; and then
maintained at 4°C. The specific primers used for EWS-FLI1, EWS-ERG
and miR amplification are summarized in Table II. The amplified PCR products were
separated by 1.5% agarose gel electrophoresis and visualized in gel
with ethidium bromide under ultraviolet light. RT-qPCR was
performed using an ABI Prism7300 Sequence Detection system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) using the GoTaq qPCR
Master Mix A6001 kit (Promega Corporation, Madison, WI, USA). The
following thermocycling conditions were used: Denaturation at 95°C
for 15 min, followed by 40 cycles of amplification at 95°C for 15
sec and 58°C for 30 sec for extension, followed by maintenance at
4°C. GAPDH and U6 was used as an endogenous control and reference
gene for relative quantifications of the 2−ΔΔCq analysis
method (27). Experiments were
performed in triplicate.
 | Table I.Stem-loop reverse transcription
primers for miR-34b and U6. |
Table I.
Stem-loop reverse transcription
primers for miR-34b and U6.
| Name | Reverse
transcription primer (5′-3′) |
|---|
| hsa-miR-34b |
GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGAC
ATGGCAGT |
| U6 |
AAAAATATGGAACGCTT |
| Table II.Specific primers for reverse
transcription-quantitative polymerase chain reaction. |
Table II.
Specific primers for reverse
transcription-quantitative polymerase chain reaction.
| Name | Forward primer
(5′-3′) | Reverse primer
(5′-3′) |
|---|
| hsa-miR-34b |
TGCGGTCAATCACTAACTCC |
CGTGCAGGGTCCGAGGT |
| U6 |
CTCGCTTCGGCAGCACA |
AACGCTTCACGAATTTGCGT |
| GAPDH |
GCACCGTCAAGGCTGAGAAC |
TGGTGAAGACGCCAGTGGA |
| EWS-FLI1 |
CCCAAGCTTATGGCGTCCACGGATTAC |
CCGCTCGAGCTAGTAGTAGCTGCCTAAGTGTG |
| EWS-ERG |
CGACTAGTTATGATCAGAGCAGT |
AGCAGCTCCAGGAGGAATTGCCA |
| Notch-1 |
TCAGCGGGATCCACTGTGAG |
ACACAGGCAGGTGAACGAGTTG |
| Notch-2 |
CAACCGCAATGGAGGCTATG |
GCGAAGGCACAATCATCAATGTT |
| Notch-3 |
TGGCGACCTCACTTACGACT |
CACTGGCAGTTATAGGTGTTGAC |
| Notch-4 |
CCTGGCTCCTTCAACTGCC |
GCAAGTAGGTCCAGACAGGT |
| Hes-1 |
GAGCACAGAAAGTCATCAAAGC |
ATTTCCAGAATGTCCGCCTTC |
| Hey-1 |
TTCAAGGCAGCTCGGTAACT |
GGGCATTTTACTTCCCCAAT |
Western blot analysis
Western blot analysis was performed as previously
described (13). Briefly, Ewing's
sarcoma cells and 3T3 cells were washed twice with ice-cold PBS and
lysed in radioimmunoprecipitation assay lysis buffer (20 mmol/l of
Tris-HCl, 150 mmol/l of NaCl, 1% NP-40, 5 mmol/l of EDTA, and 1
mmol/l of Na3VO4, pH 7.5) containing protease
inhibitor cocktail (Sigma-Aldrich; Merck KGaA) and then incubated
for 25 min at 4°C and centrifuged for 20 min at 13,000 × g at 4°C.
Thereafter, the supernatant was recovered and quantified using the
Pierce bicinchoninic acid protein quantification assay (Pierce;
Thermo Fisher Scientific, Inc.). An aliquot (50–100 mg of protein
per lane) of the total protein was loaded onto 10% SDS-PAGE and
then blotted to polyvinylidene fluoride membranes (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Individual immunoblots were
performed with primary antibodies against Notch1 (1:1,000; cat. no.
ab8925; Abcam, Cambridge, MA, USA), Hes family BHLH transcription
factor 1 (1:1,000; Hes1; cat. no. ab71559; Abcam), Hes-related
family BHLH transcription factor with YRPW motif 1 (1:1,000; Hey1;
cat. no. ab22614; Abcam) and β-actin (1:2,000; cat no. sc-130065;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA). Briefly,
membranes were blocked in Tris-buffered saline with 20% Tween
(TBST) buffer containing 5% nonfat dry milk for 2 h at room
temperature and incubated overnight with primary antibody.
Subsequently, membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies (Santa Cruz
Biotechnology, Inc.) for 1 h. Enhanced chemiluminescent reagent (GE
Healthcare, Chicago, IL, USA) was used for protein visualization.
Experiments were performed in triplicate.
Cell proliferation and adhesion
assays
Cell proliferation was determined by the MTT assay.
Following infection by EWS-FLI-1 siRNA and miR-34b precursor or
inhibitor, Cells were trypsinized and 5×103 cells/well
were seeded into 96-well plates in triplicate. Cellular
proliferation was determined once a day over the course of 5 days.
Briefly, 20 µl 5 mg/ml MTT was added to each well and cells were
incubated at 37°C in a humidified atmosphere containing 5%
CO2. Cells were lysed after 4–6 h using cell lysis
reagent (20% sodium dodecyl sulfate, 50% dimethyl sulfoxide, pH
4.7), and absorbance was measured at 570 nm with an EL-311SX
microplate reader (BioTek Instruments, Inc., Winooski, VT,
USA).
The adhesion assay was performed using MTT-stained
cells. At 24 h following infection by siRNA targeting EWS/FLI1 and
miR-34b precursor or inhibitor at 37°C, the Ewing sarcoma cells
(1×105) were resuspended in 200 ml medium without serum
and seeded into a Matrigel-precoated (100 mg/ml) 96-well plate.
Cells were washed after 30 and 60 min to remove non-adherent cells.
After the final wash, absorbance was measured at 490 nm to quantify
adherence. Experiments were repeated three times.
Cell migration and invasion
assays
Migration assays for A673 or RD-ES cells were
performed by seeding 3×105 cells in 200 µl DMEM or RPMI
serum-free medium in the upper chambers of 24-well Transwell
inserts with polyethylene terephthalate membranes (8.0-µm pore
size; Costar, Corning Inc., Corning, NY, USA). The lower chambers
were filled with 0.8 ml DMEM or RPMI supplemented with 15% FBS.
After 24 h incubation at 37°C, the non-migrating cells were
removed; and the membranes were fixed and stained using the
Differential Quik Stain kit (Sysmex Corporation, Kobe, Japan).
Cells that migrated through the membranes were quantified using
NIKON LABOPHOT optical light microscopic visualization
(magnification ×200) and by randomly selecting 10 regions for cell
counting. Experiments were performed in triplicate.
The procedure for the invasion assay was similar to
the migration assay. Briefly, the membranes in the Transwell
inserts were coated evenly with 25 µl Matrigel (100 mg/ml) per well
(BD Biosciences, Franklin Lakes, NJ, USA; cat. no. 356234) before
cells were seeded into the upper chamber. After incubation for 36
and 48 h for A673 and RD-ES cells, respectively, invaded cells on
the lower surface of the membrane were analyzed as previously
described in the migration assay.
Luciferase reporter assay
The 3′-untranslated region (UTR) of Notch1 was
amplified from human genomic DNA and cloned into the
pmiR-RB-REPORT™ reporter gene plasmid vector (Guangzhou
RiboBio Co., Ltd., Guangzhou, China) with restriction enzymes
XhoI and NotI. Similarly, a fragment representing
mutant Notch1 3′-UTR, harbouring the change of the seven
miRNA34b-binding sites (ACU GCC U→UGA CGG A), was cloned into the
pmiR-RB-REPORT vector as a control. For reporter assays,
1×105/l cells were plated in a 24-well culture plate at
a density of 8,000 cells/well and co-transfected with the reporter
plasmid and 100 nM miR-34b inhibitor or miR inhibitor negative
control (NC) (NC sequence: 5′-UUGUACUACACAAAAGUACUG-3′) which were
synthesized by Shanghai GenePharma Co., Ltd., with Lipofectamine
2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol Luciferase activity was measured 48 h
post-transfection using the Dual-Luciferase® Reporter
Assay system (Promega Corporation, Madison, WI, USA), according to
the manufacturer's protocol. Firefly luciferase activity was
normalized against Renilla luciferase activity for each
transfected well to normalize transfection efficiency.
Statistical analysis
The results of RT-qPCR, proliferation, adhesion,
migration and invasion assays are presented as the means ± standard
error. Each assay was performed in triplicate Data were analyzed by
one-way analysis of variance. All statistical analyses were
performed using the SPSS 13.0 statistical software package (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Expression levels of EWS-FLI1 and
miR-34b in biopsy samples
A large proportion (85–90%) of Ewing's sarcoma cases
are characterized by the EWS-FLI1 fusion gene (28). Therefore, the expression levels of
the fusion gene were detected in Ewing's sarcoma and normal tissue
(NT) biopsy samples using RT-PCR. The results demonstrated that NT
did not express the EWS-FLI1 fusion gene (Fig. 1A) and 3/14 tumor samples did
express EWS-ERG fusion gene (Fig.
1B). The results of 10 samples and 1 NT are presented in the
Fig. 1A because the lanes of gel
electrophoresis were limited in our laboratory.
The expression levels of miR-34b in tumor samples
and NT were measured using stem-loop RT-qPCR. The results
demonstrated that tumor samples expressed increased levels of
miR-34b compared with NT. Notably, miR-34b was expressed at higher
levels in EWS-FLI1-positive samples compared within
EWS-ERG-positive samples (Fig.
1C). These results indicated that the EWS-FLI1 gene may have
greater controlover miR-34 be xpression than the EWS-ERG gene.
EWS-FLI1 fusion gene may affect
miR-34b expression in Ewing's sarcoma cell lines
RD-ES and A673 cells are known to possess the
EWS-FLI1 fusion gene (29). Ewing
sarcoma cells secrete EWS/Fli-1 fusion mRNA via microvesicles.
NIH3T3 cells were detected as negative control. Therefore, EWS-FLI1
expression levels in these cell lines were detected using RT-PCR
(Fig. 2A). The expression of the
fusion gene was successfully downregulated by siRNA in RD-ES cells
and A673 cells (Fig. 2B). In
addition, the relative miR-34b expression levels were detected in
cell lines with or without the interrupted fusion gene. As
presented in the figure, miR-34b expression in RD-ES and A673 cells
was downregulated following siRNA transfection (Fig. 2C and D). These results indicated
that the EWS-FLI1 fusion gene may be associated with miR-34b
expression in Ewing's sarcoma cell lines, which is consistent with
the observations made in the tumor biopsy samples.
 | Figure 2.(A) Detection of EWS-FLI1 fusion gene
expression in RD-ES and A673 cell lines using reverse
transcription-polymerase chain reaction. (B) Expression levels of
the fusion gene were downregulated in RD-ES and A-673 cells
following siRNA transfection. (C) miR-34b expression in A673 cells
was downregulated following siRNA transfection. (D) miR-34b
expression in RD-ES cells was downregulated following siRNA
transfection. (E) miR-34b expression was upregulated by the
precursor of miR-34b in RD-ES and A-673 cells. (F) miR-34b
expression was downregulated by the inhibitor of miR-34b in RD-ES
and A-673 cells. BC, cell lines that were treated with lentiviral
vector alone; NC(+), normal cell lines that were not transfected
with siRNA; NS, cells transfected with non-targeting siRNA; siRNA,
siRNA-transfected cells; NC(−), normal cell lines that were
transfected with specific siRNA targeting the EWS-FLI1 fusion gene;
Micro-up, NC(−) cell lines treated with precursor miR-34b
sequences; Micro-down, NC(+) cell lines treated with complementary
sequences of miR-34b. **P<0.01, ##P<0.01. EWS,
Ewing's sarcoma breakpoint region 1; FLI1, friend leukemia
integration 1 transcription factor; miR, microRNA; siRNA, small
interfering RNA. |
Up- and downregulation of miR-34b
expression
miR-34b expression can be controlled by the EWS-FLI1
fusion gene; in addition, in the present study its expression was
altered using lentiviral vectors. The lentiviral vector with
complementary sequences of miR-34b was infected into normal cells
to downregulate miR-34b expression, whereas the lentiviral vector
with precursor sequences of miR-34b was infected into
siRNA-transfected cells to upregulate miR-34b expression. As shown
in Fig. 2E and F, miR-34b
expression was successfully up- and downregulated by precursor and
complementary miR-34b sequences, respectively.
miR-34b promotes proliferation of
Ewing's sarcoma cells in vitro
To investigate the effects of miR-34b on
proliferation of Ewing's sarcoma cell lines, an MTT assay was
performed. The results demonstrated that when miR-34b expression
was upregulated, the siRNA-transfected RD-ES and A673 cells in
which EWS-FLI1 was downregulated exhibited a significant increase
in proliferative capacity compared with the cells in the control
groups (Fig. 3A and B).
Conversely, the proliferative ability of normal RD-ES and A673
cells was significantly inhibited when miR-34b expression was
downregulated (Fig. 3C and D).
These results indicated that miR-34b may promote cell proliferation
in Ewing's sarcoma.
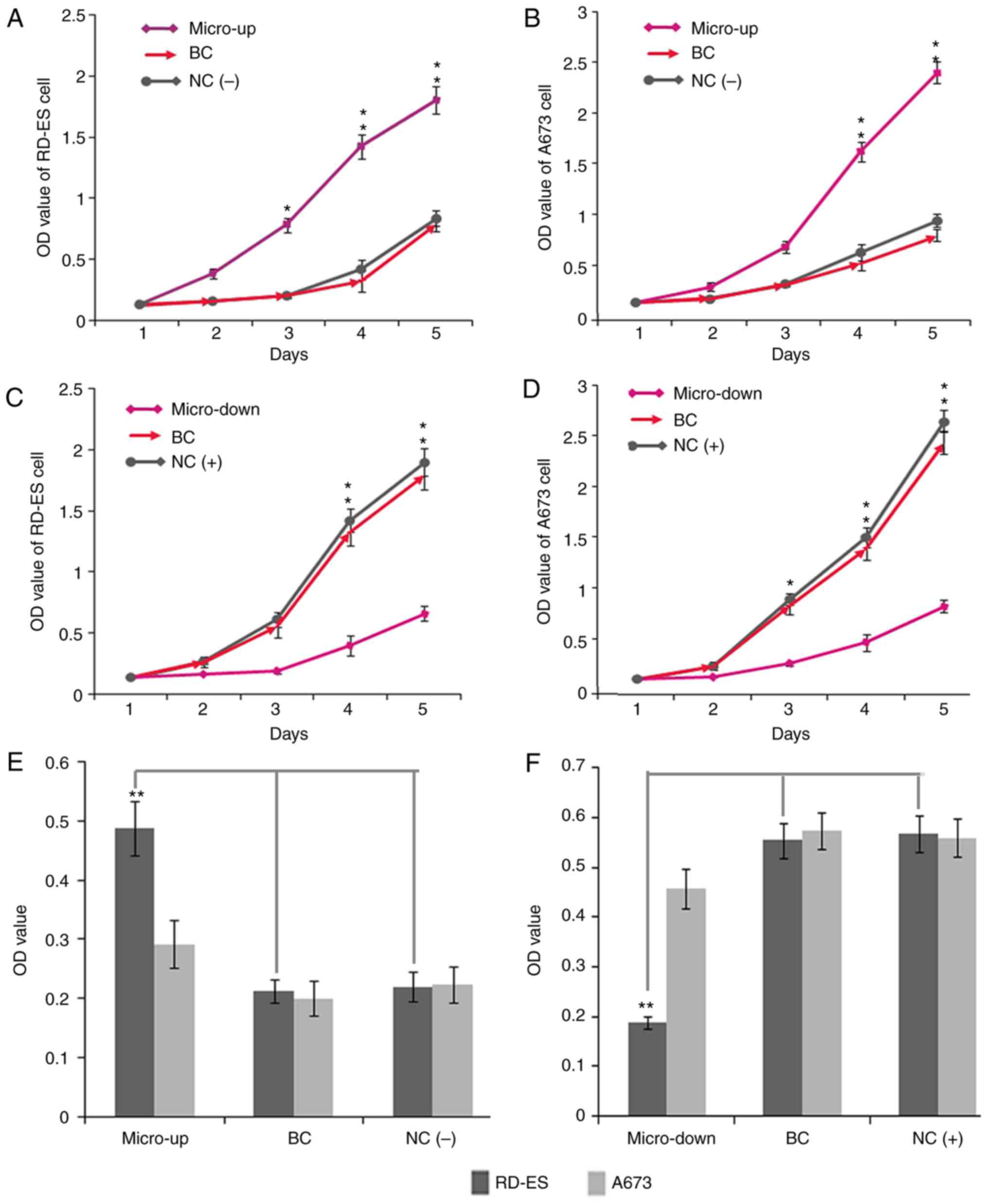 | Figure 3.Proliferation and adhesion of RD-ES
and A673 cells were detected using an MTT assay. (A) When miR-34b
expression was upregulated, the siRNA-transfected RD-ES cells
exhibited significantly increased proliferative capacity compared
with cells in the control groups. (B) When miR-34b expression was
upregulated, the siRNA-transfected A673 cells exhibited
significantly increased proliferative capacity. (C) Proliferative
ability of normal RD-ES cells was significantly inhibited when
miR-34b expression was downregulated. (D) Proliferative of normal
A673 cells was significantly inhibited when miR-34b expression was
downregulated. (E) Overexpression of miR-34b significantly improved
the adhesion of RD-ES cells, but had little effect on A673 cells.
(F) Knockdown of miR-34b reduced the adhesive ability of RD-ES
cells, but had little effect on A673 cells. BC, cell lines that
were treated with lentiviral vector alone; NC(+), normal cell lines
that were not transfected with siRNA; siRNA, siRNA-transfected
cells; NC(−), normal cell lines that were transfected with specific
siRNA; Micro-up, cell lines treated with precursor miR-34b
sequences; Micro-down, cell lines treated with complementary
sequences of miR-34b. *P<0.05 and **P<0.01. miR, microRNA;
OD, optical density; siRNA, small interfering RNA. |
miR-34b promotes RD-ES, but not A673,
cell adhesion in vitro
The adhesion of cancer cells to the extracellular
matrix has a significant role in metastasis after disaggregation
from the primary tumor. Therefore, an inhibition of adhesion may
contribute to a reduction in metastatic potential. The present
study demonstrated that upregulation of miR-34b expression
significantly increased the adhesion of RD-ES cells, whereas up- or
downregulation of miR-34b had little effect on A673 cell adhesion
(Fig. 3E and F).
miR-34b improves the migration and
invasion of Ewing's sarcoma cells in vitro
Migration and invasion of tumor cells are
indispensable for metastasis in vivo; therefore, inhibiting
the migratory and invasive capacity of Ewing's sarcoma cells may
decrease metastatic potential. In the present study, the effects of
miR-34b on the migratory and invasive properties of Ewing's sarcoma
cell lines were investigated using Transwell assays. As shown in
Fig. 4, the ability of RD-ES cells
to migrate and invade was significantly inhibited when miR-34b
expression was downregulated. Similar results were also obtained
usingA673 cells (data not shown).
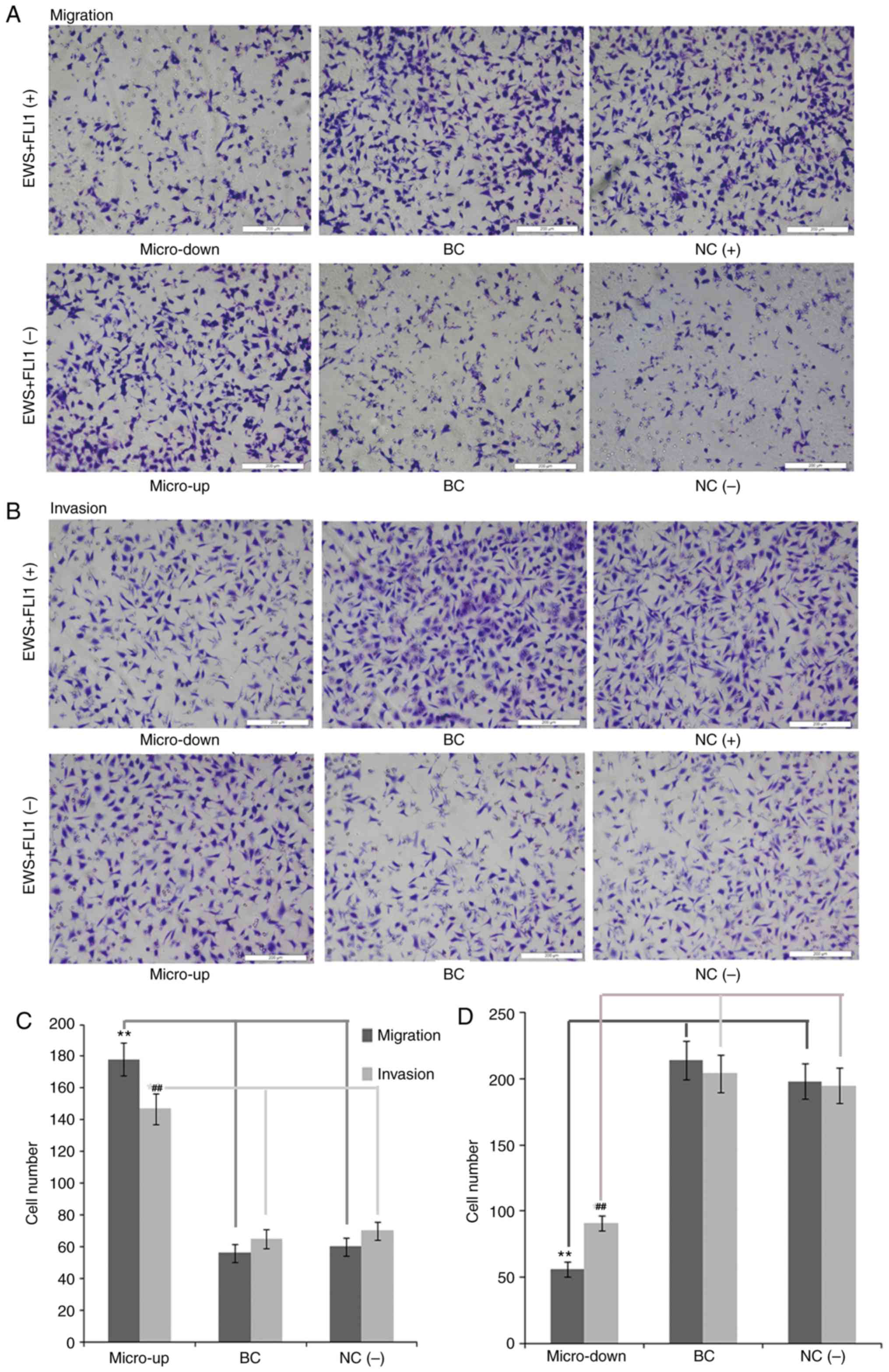 | Figure 4.Effects of miR-34b on the migration
and invasion of RD-ES cells in vitro. Magnification, ×200
(A) Migratory and (B) invasive ability of normal RD-ES cells was
inhibited when miR-34b expression was downregulated (top panel).
However, miR-34b overexpression improved the migratory and invasive
ability of siRNA-transfected-RD-ES cells (bottom panel).
Quantification of (C) EWS-FLI1 (−) and (D) EWS-FLI1 (+) cells. The
number of cells that passed through the membrane was counted. BC,
cell lines that were treated with lentiviral vector alone; NC(+),
normal cell lines that were not transfected with siRNA; siRNA,
siRNA-transfected cells; NC(−), normal cell lines that were
transfected with specific siRNA; Micro-up, cell lines treated with
precursor miR-34b sequences; Micro-down, cell lines treated with
complementary sequences of miR-34b; EWS-FLI1 (+), cells that were
not transfected with EWS-FLI1 fusion gene siRNA; EWS-FLI1 (−),
cells that were transfected with EWS-FLI1 fusion gene siRNA against
EWS-FLI1 gene. **P<0.01, ##P<0.01. EWS, Ewing's
sarcoma breakpoint region 1; FLI1, friend leukemia integration 1
transcription factor; miR, microRNA; siRNA, small interfering
RNA. |
Effects of miR-34b on Notch1, Hes1 and
Hey1 in Ewing's sarcoma cells
To determine the molecular mechanisms involved in
miR-34b-induced cell proliferation and migration, the Notch
signaling pathway was investigated as it is a target of miR-34b and
exhibits the opposite role in suppressing tumor development. When
miR-34b expression was inhibited, the RT-qPCR results demonstrated
that Notch1 expression was increased compared with the other three
Notch receptors (data not shown). Therefore, the expression levels
of Notch1, and its target genes, Hes1 and Hey1, in RD-ES and A673
cells were further assessed by RT-qPCR analysis. Compared with in
normal cells, the expression levels of Notch1, Hes1and Hey1 were
increased when the EWS-FLI1 fusion gene was silenced by siRNA in
RD-ES and A673 cells (Fig. 5A).
Conversely, the expression levels of Notch1, Hes1 and Hey1 were
decreased after the cells in which EWS/FLI1 expression was
downregulated with the siRNA were transfected with miR-34b
precursor sequences (Fig. 5B).
Furthermore, there was an increase in the expression levels of
Notch1, Hes1 and Hey1 after miR-34b downregulation (Fig. 5C).
 | Figure 5.mRNA expression levels of Notch1,
Hes-1 and Hey-1 demonstrated the negative association with that of
EWS-FLI1 and miR-34b in RD-ES and A673 cells. (A) mRNA expression
levels of Notch-1, Hes-1 and Hey-1 were increased when the
expression of the EWS-FLI1 fusion gene was downregulated by siRNA
in RD-ES and A673 cells. (B) Conversely, mRNA expression levels
were inhibited after the siRNA-transfected cells were treated with
miR-34b precursor sequences. (C) Notch-1, Hes-1 and Hey-1 mRNA
expression was increased after miR-34b was downregulated in normal
cells. BC, cell lines that were treated with lentiviral vector
alone; NC(+), normal cell lines that were not transfected with
siRNA; NS, cells transfected with non-targeting siRNA; siRNA,
siRNA-transfected cells; NC(−), normal cell lines that were
transfected with specific siRNA; MU, cell lines treated with
precursor miR-34b sequences; MD, cell lines treated with
complementary sequences of miR-34b. **P<0.01. EWS, Ewing's
sarcoma breakpoint region 1; FLI1, friend leukemia integration 1
transcription factor; Hes1, Hes family BHLH transcription factor 1;
Hey1, Hes-related family BHLH transcription factor with YRPW motif
1; miR, microRNA; siRNA, small interfering RNA. |
The protein expression levels of Notch1, Hes1 and
Hey1 were examined by western blot analysis. In agreement with the
RT-qPCR data, it was also demonstrated that miR-34b was able to
downregulate Notch1, Hes1 and Hey1 protein expression (Fig. 6). These results suggested that the
EWS-FLI1 fusion gene mayinactivate the Notch1 signaling pathway in
Ewing's sarcoma via miR-34b.
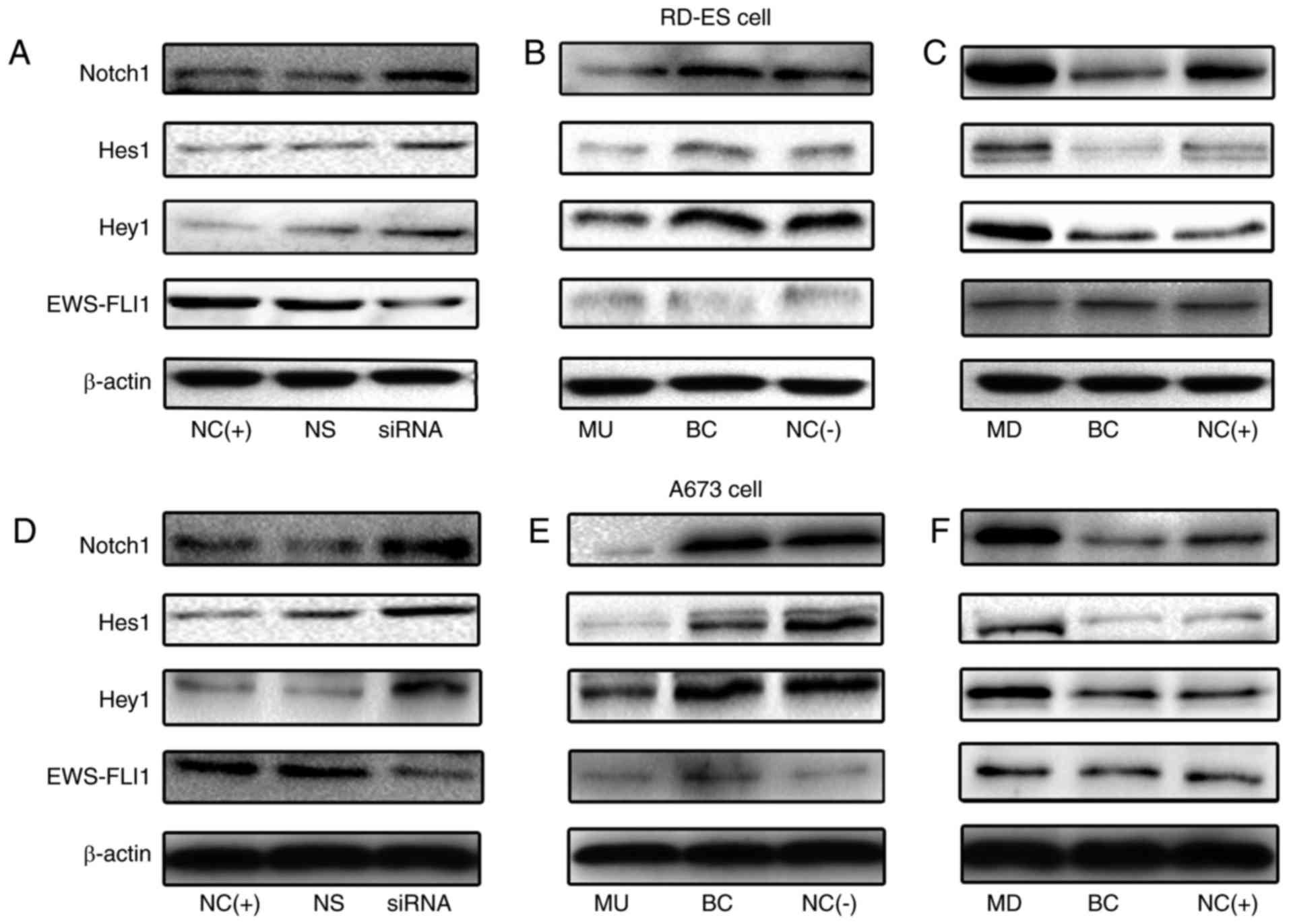 | Figure 6.Protein expression levels of Notch1,
Hes-1 and Hey-1 demonstrated the negative association with EWS-FLI1
and miR-34b in RD-ES and A673 cells. (A) The protein expression
levels of Notch-1, Hes-1 and Hey-1 were increased following
EWS-FLI1 repression in RD-ES cells. (B) The expression of Notch-1,
Hes-1 and Hey-1 were decreased after miR-34b was upregulated in
RD-ES cells in which EWS-FLI1 is repressed. (C) The expression of
Notch-1, Hes-1 and Hey-1 were increased after miR-34b was
downregulated in RD-ES cells. (D) The protein expression levels of
Notch-1, Hes-1 and Hey-1 were increased following EWS-FLI1
repression in A673 cells. (E) The expression of Notch-1, Hes-1 and
Hey-1 were decreased following miR-34b was upregulated in A673
cells in which EWS-FLI1 is repressed. (F) The expression of
Notch-1, Hes-1 and Hey-1 were increased after miR-34b was
downregulated in A673 cells. BC, cell lines that were treated with
lentiviral vector alone; NC(+), normal cell lines that were not
transfected with siRNA; NS, cells transfected with non-targeting
siRNA; siRNA, siRNA-transfected cells; NC(−), normal cell lines
that were transfected with specific siRNA; MU, cell lines treated
with precursor miR-34b sequences; MD, cell lines treated with
complementary sequences of miR-34b. EWS, Ewing's sarcoma breakpoint
region 1; FLI1, friend leukemia integration 1 transcription factor;
Hes1, Hes family BHLH transcription factor 1; Hey1, Hes-related
family BHLH transcription factor with YRPW motif 1; miR, microRNA;
siRNA, small interfering RNA. |
miR-34b directly modulates expression
of the Notch1 gene in Ewing's sarcoma cells in vitro
A dual-luciferase reporter assay was conducted to
confirm the effects of miR-34b on Notch1. Fragments containing the
miR-34b binding sequence or mutated sequence in the 3′-UTR of the
Notch1 mRNA were cloned into the pmiR-RB-REPORT luciferase reporter
vector to generate the pmiR-RB-REPORT-NOTCH1-3′UTR and
pmiR-RB-REPORT-NOTCH1-3′UTR-MUT plasmids, respectively. These
reporter constructs were co-transfected with miR-34b inhibitor or
miR-NC into RD-ES and A673 cells, and luciferase activity was
measured. It was revealed that miR-34b inhibition led to
significantly increased luciferase activity of
pmiR-RB-REPORT-NOTCH1-3′UTR (P<0.05; Fig. 7), but had no effect on
pmiR-RB-REPORT-NOTCH1-3′UTR-mut. Taken together, these results
suggested that miR-34b may directly target the 3′-UTR of
Notch1.
Discussion
Accumulating evidence has suggested that miRNA shave
a key role in tumorigenesis through the regulation of their target
genes. Previous studies involving in vitro experiments and
analysis of clinical samples have demonstrated a direct link
between miRNA function and the EWS-FLI1 fusion gene (11–13,23).
In particular one member of the miR-34 family, miR-34a, has been
reported to predict the survival of patients with Ewing's sarcoma
(23). Therefore, the present
study aimed to investigate miR-34b expression in Ewing's sarcoma, a
disease predominantly characterized by the EWS-FLI1 fusion gene
(1). Firstly, gene expression of
the EWS-FLI1 gene in the NT samples was detected by RT-PCR followed
by gel electrophoresis and the results indicated that the EWS-FLI1
fusion gene was not expressed in all NT samples. Subsequently,
RT-PCR was performed to detect the expression levels of miR-34b
using stem-loop primers. Notably, the expression levels of miR-34b
were higher in tumor samples compared within NT samples. In
addition, the expression levels of miR-34b were higher in
EWS-FLI1-positive samples compared with in EWS-FLI1-negative
samples. It has been demonstrated at the molecular level that ~10%
of Ewing's sarcoma cases harbor the EWS-ERG fusion transcript
(28,30). FLI1 and ERG belong to the ETS
transcription factor family, which all share a highly conserved
DNA-binding domain, and can act as aberrant transcription factors
when their genes fuse with the EWS gene (1,31–34).
The difference inmiR-34b expression inEWS-FLI1-and EWS-ERG-positive
samples may be associated with the differential regulatory
abilities of the FLI1 and ERG genes (35). miR-34b expression was also detected
in Ewing's sarcoma cell lines and miR-34b expression was inhibited
when the cells were transfected with EWS-FLI1-specific siRNA. These
results indicated that miR-34b may be upregulated in Ewing's
sarcoma biopsy samples, particularly those harboring the EWS-FLI1
fusion gene, and Ewing's sarcoma cell lines. The results were
consistent with other studies demonstrating the involvement of
miRNAs in Ewing's sarcoma as well as other solid tumors (11,36).
Studies have demonstrated that miRNAs can act as
oncogenes or tumor suppressor genes, and that widespread
alterations in miRNA expression patterns are highly associated with
various human cancers (37,38).
miR-34b is a member of the evolutionarily conserved miR-34family,
and can exert tumor suppressive effects by downregulating its
target genes, including Notch, B-cell lymphoma 2andhigh mobility
group AT-hook 2 (22). miR-34b
also has an important role in p53-induced cell cycle arrest, cell
senescence, apoptosis and other biological functions (39). Numerous studies have demonstrated
that miR-34b inhibits cell proliferation, migration and invasion
(20,21,40);
however, miR-34b also has an oncogenic role in esophageal squamous
cell carcinoma (41). To the best
of our knowledge, the functions of miR-34b in Ewing's sarcoma have
not yet been elucidated.
Uncontrolled cell proliferation leads to tumor
growth, and aggressive tumor cell metastasis promotes spreading of
tumor cells to distal sites. Therefore, the present study
investigated the role of miR-34b in proliferation, adhesion,
migration and invasion of tumor cells. Firstly, the precursor
sequence of miR-34b was infected into siRNA-transfected Ewing's
sarcoma cells. The proliferative, migratory and invasive abilities
of the cells were significantly enhanced following upregulation of
miR-34b expression. The adhesive ability was also enhanced in RD-ES
cells, but not in A673 cells. According to the ATCC, these cell
lines possess different culture characteristics that could account
for the differences observed. RD-ES cells grow as a loosely
attached monolayer in small clusters, whereas A673 cells are fully
adherent (42). Subsequently, the
complementary sequence was used to downregulate miR-34b expression
and, as expected, the adhesive abilities were inhibited. These
results indicated that miR-34b may serve an oncogenic role in
Ewing's sarcoma.
Notch is an evolutionarily conserved signaling
pathway that affects cell fate, proliferation, migration and
invasion (43). Notch can be
oncogenic or tumor suppressive in human cancer (44–47);
therefore, the role of Notch needs to be further clarified in the
context of different types of cancer. Previous studies have
demonstrated that suppression of EWS-FLI1 reactivates Notch
signaling in ESFT cells, resulting in cell cycle arrest (48), and using microarray analysis, Notch
signaling has been revealed to be crucial for the metastatic
phenotype (49). SinceNotch1 is
expressed in ESFT cell lines (48,50)
and is a target gene of miR-34b (25,51,52),
the present study investigated the expression levels of Notch1in
A673 and RD-ES cell lines. The results indicated that suppression
of EWS-FLI1 couldincreaseNotch1expression at both the mRNA and
protein levels. Conversely, when miR-34b expression was
upregulated, the mRNA and protein levels of Notch1 were decreased.
It has previously been reported that Hey1 is the main downstream
effector of Notch signaling and that Hes1 is uncoupled from the
Notch pathway in ESFT cells (53).
In the present study, the observation was made that the mRNA and
protein expression levels of Hes1 and Hey1 were downregulated by
miR-34b, indicating that the EWS-FLI1 fusion gene may suppress the
Notch1 signaling pathway, at least in part via miR-34b.
miRNAs can function as either tumor suppressor genes
or oncogenes depending on their target genes (54,55).
Tumor suppressive miRNAs that target tumor-promoting genes are
repressed in cancer, whereas oncogenic miRNAs that target tumor
suppressor genes are upregulated in cancer. For tumors where Notch1
serves an oncogenic role (44,56),
miR-34 has been demonstrated to be tumor suppressive (25,51,57).
Therefore, as Notch1 acts as a tumor suppressor in Ewing's sarcoma,
miR-34b may be exerting its oncogenic effects through this gene.
Using a dual-luciferase reporter assay, it was revealed that
miR-34b may directly modulate the expression of Notch1 by binding
to the 3′-UTR region (sequence 181–187) of Notch1.
Despite overexpression of miR-34b in Ewing's sarcoma
samples and cell lines, the underlying mechanisms by which EWS-FLI1
affects miR-34b remain unclear. miR-34 has been reported to be
directly induced by the c-MYC proto-oncogene, which is a known
EWS-FLI1 target gene (58), thus
suggesting that EWS-FLI1 may indirectly modulate miR-34b via c-Myc
induction. In conclusion, EWS-FLI1 may modulate miR-34b expression
through direct or indirect mechanisms, and miR-34b appears to serve
an oncogenic role in Ewing's sarcoma by downregulating Notch1.
Acknowledgements
The authors would like to thank the Department of
Science & Technology of Shandong province for their continued
support.
Funding
The present study was supported by the Nature
Science Foundation of Shandong Province for Young Scholars (China;
grant no. 2015ZRE27529).
Availability of data and materials
The datasets used or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QL and SZ conceived and designed the study. QL and
SZ performed the experiments. DL and ML provided some of the
samples and experiment methods. QL wrote the paper. SZ reviewed and
edited the manuscript. All authors read and approved the
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Boards of Qilu Hospital of Shandong University (Jinan,
China) and Shandong Provincial Hospital Affiliated to Shandong
University (Jinan, China). Written informed consent was obtained
from all patients.
Consent for publication
The study was done following agreement from the
local ethics committee and with the patients' informed consent.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Riggi N and Stamenkovic I: The Biology of
Ewing sarcoma. Cancer Lett. 254:1–10. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bacci G, Forni C, Longhi A, Ferrari S,
Donati D, De Paolis M, Barbieri E, Pignotti E, Rosito P and Versari
M: Long-term outcome for patients with non-metastatic Ewing's
sarcoma treated with adjuvant and neoadjuvant chemotherapies. 402
patients treated at Rizzoli between 1972 and 1992. Eur J Cancer.
40:73–83. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Grier HE, Krailo MD, Tarbell NJ, Link MP,
Fryer CJ, Pritchard DJ, Gebhardt MC, Dickman PS, Perlman EJ, Meyers
PA, et al: Addition of ifosfamide and etoposide to standard
chemotherapy for Ewing's sarcoma and primitive neuroectodermal
tumor of bone. N Engl J Med. 348:694–701. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Esiashvili N, Goodman M and Marcus RB Jr:
Changes in incidence and survival of Ewing sarcoma patients over
the past 3 decades: Surveillance epidemiology and end results data.
J Pediatr Hematol Oncol. 30:425–430. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Riggi N, Suva ML, Suva D, Cironi L,
Provero P, Tercier S, Joseph JM, Stehle JC, Baumer K, Kindler V and
Stamenkovic I: EWS-FLI-1 expression triggers a Ewing's sarcoma
initiation program in primary human mesenchymal stem cells. Cancer
Res. 68:2176–2185. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Riggi N, Cironi L, Provero P, Suva ML,
Kaloulis K, Garcia-Echeverria C, Hoffmann F, Trumpp A and
Stamenkovic I: Development of Ewing's sarcoma from primary bone
marrow-derived mesenchymal progenitor cells. Cancer Res.
65:11459–1168. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jedlicka P: Ewing Sarcoma, an enigmatic
malignancy of likely progenitor cell origin, driven by
transcription factor oncogenic fusions. Int J Clin Exp Pathol.
3:338–347. 2010.PubMed/NCBI
|
|
8
|
Aryee DN, Niedan S, Kauer M, Schwentner R,
Bennani-Baiti IM, Ban J, Muehlbacher K, Kreppel M, Walker RL,
Meltzer P, et al: Hypoxia modulates EWS-FLI1 transcriptional
signature and enhances the malignant properties of Ewing's sarcoma
cells in vitro. Cancer Res. 70:4015–4023. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bachmaier R, Aryee DN, Jug G, Kauer M,
Kreppel M, Lee KA and Kovar H: O-GlcNAcylation is involved in the
transcriptional activity of EWS-FLI1 in Ewing's sarcoma. Oncogene.
28:1280–1284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hancock JD and Lessnick SL: A
transcriptional profiling meta-analysis reveals a core EWS-FLI gene
expression signature. Cell Cycle. 7:250–256. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
De Vito C, Riggi N, Suva ML, Janiszewska
M, Horlbeck J, Baumer K, Provero P and Stamenkovic I: Let-7a is a
direct EWS-FLI-1 target implicated in Ewing's sarcoma development.
PLoS One. 6:e235922011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ban J, Jug G, Mestdagh P, Schwentner R,
Kauer M, Aryee DN, Schaefer KL, Nakatani F, Scotlandi K, Reiter M,
et al: Hsa-mir-145 is the top EWS-FLI1-repressed microRNA involved
in a positive feedback loop in Ewing's sarcoma. Oncogene.
30:2173–2180. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Riggi N, Suva ML, De Vito C, Provero P,
Stehle JC, Baumer K, Cironi L, Janiszewska M, Petricevic T, Suvà D,
et al: EWS-FLI-1 modulates miRNA145 and SOX2 expression to initiate
mesenchymal stem cell reprogramming toward Ewing sarcoma cancer
stem cells. Genes Dev. 24:916–932. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Papagiannakopoulos T and Kosik KS:
MicroRNAs: Regulators of oncogenesis and stemness. BMC Med.
6:152008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sandberg R, Neilson JR, Sarma A, Sharp PA
and Burge CB: Proliferating cells express mRNAs with shortened 3′
untranslated regions and fewer microRNA target sites. Science.
320:1643–1647. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McKinsey EL, Parrish JK, Irwin AE,
Niemeyer BF, Kern HB, Birks DK and Jedlicka P: A novel oncogenic
mechanism in Ewing sarcoma involving IGF pathway targeting by
EWS/Fli1-regulated microRNAs. Oncogene. 30:4910–4920. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Robin TP, Smith A, McKinsey E, Reaves L,
Jedlicka P and Ford HL: EWS/FLI1 regulates EYA3 in Ewing sarcoma
via modulation of miRNA-708, resulting in increased cell survival
and chemoresistance. Mol Cancer Res. 10:1098–1108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dong F and Lou D: MicroRNA-34b/c
suppresses uveal melanoma cell proliferation and migration through
multiple targets. Mol Vis. 18:537–546. 2012.PubMed/NCBI
|
|
21
|
Majid S, Dar AA, Saini S, Shahryari V,
Arora S, Zaman MS, Chang I, Yamamura S, Tanaka Y, Chiyomaru T, et
al: miRNA-34b inhibits prostate cancer through demethylation,
active chromatin modifications, and AKT pathways. Clin Cancer Res.
19:73–84. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hermeking H: The miR-34 family in cancer
and apoptosis. Cell Death Differ. 17:193–199. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nakatani F, Ferracin M, Manara MC, Ventura
S, Del Monaco V, Ferrari S, Alberghini M, Grilli A, Knuutila S,
Schaefer KL, et al: miR-34a predicts survival of Ewing's sarcoma
patients and directly influences cell chemo-sensitivity and
malignancy. J Pathol. 226:796–805. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zweidler-McKay PA: Notch signaling in
pediatric malignancies. Current Oncol Rep. 10:459–468. 2008.
View Article : Google Scholar
|
|
25
|
Ji Q, Hao X, Zhang M, Tang W, Yang M, Li
L, Xiang D, Desano JT, Bommer GT, Fan D, et al: MicroRNA miR-34
inhibits human pancreatic cancer tumor-initiating cells. PLoS One.
4:e68162009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Weiler J, Hunziker J and Hall J:
Anti-miRNA oligonucleotides (AMOs): Ammunition to target miRNAs
implicated in human disease? Gene Ther. 13:496–502. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Delattre O, Zucman J, Melot T, Garau XS,
Zucker JM, Lenoir GM, Ambros PF, Sheer D, Turc-Carel C, Triche TJ,
et al: The Ewing family of tumors-a subgroup of small-round-cell
tumors defined by specific chimeric transcripts. N Engl J Med.
331:294–299. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tsugita M, Yamada N, Noguchi S, Yamada K,
Moritake H, Shimizu K, Akao Y and Ohno T: Ewing sarcoma cells
secrete EWS/Fli-1 fusion mRNA via microvesicles. PLoS One.
8:e774162013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zucman J, Melot T, Desmaze C, Ghysdael J,
Plougastel B, Peter M, Zucker JM, Triche TJ, Sheer D, Turc-Carel C,
et al: Combinatorial generation of variable fusion proteins in the
Ewing family of tumours. EMBO J. 12:4481–4487. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Turc-Carel C, Aurias A, Mugneret F, Lizard
S, Sidaner I, Volk C, Thiery JP, Olschwang S, Philip I, Berger MP,
et al: Chromosomes in Ewing's sarcoma. I. An evaluation of 85 cases
of remarkable consistency of t(11;22)(q24;q12). Cancer Genet
Cytogenet. 32:229–238. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Delattre O, Zucman J, Plougastel B,
Desmaze C, Melot T, Peter M, Kovar H, Joubert I, de Jong P, Rouleau
G, et al: Gene fusion with an ETS DNA-binding domain caused by
chromosome translocation in human tumours. Nature. 359:162–165.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Douglass EC, Valentine M, Green AA, Hayes
FA and Thompson EI: t(11;22) and other chromosomal rearrangements
in Ewing's sarcoma. J Natl Cancer Inst. 77:1211–1215.
1986.PubMed/NCBI
|
|
34
|
May WA, Gishizky ML, Lessnick SL, Lunsford
LB, Lewis BC, Delattre O, Zucman J, Thomas G and Denny CT: Ewing
sarcoma 11;22 translocation produces a chimeric transcription
factor that requires the DNA-binding domain encoded by FLI1 for
transformation. Proc Natl Acad Sci USA. 90:pp. 5752–5756. 1993;
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Im YH, Kim HT, Lee C, Poulin D, Welford S,
Sorensen PH, et al: EWS-FLI1, EWS-ERG, and EWS-ETV1 oncoproteins of
Ewing tumor family all suppress transcription of transforming
growth factor beta type II receptor gene. Cancer Res. 60:1536–1540.
2000.PubMed/NCBI
|
|
36
|
Mendell JT: miRiad roles for the miR-17-92
cluster in development and disease. Cell. 133:217–22. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Esquela-Kerscher A and Slack FJ:
Oncomirs-microRNAs with a role in cancer. Nat Rev Cancer.
6:259–269. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hermeking H: p53 enters the microRNA
world. Cancer Cell. 12:414–418. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yamazaki H, Chijiwa T, Inoue Y, Abe Y,
Suemizu H, Kawai K, Wakui M, Furukawa D, Mukai M, Kuwao S, et al:
Overexpression of the miR-34 family suppresses invasive growth of
malignant melanoma with the wild-type p53 gene. Exp Ther Med.
3:793–796. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Harata K, Ishiguro H, Kuwabara Y, Kimura
M, Mitsui A, Ogawa R, Katada T, Tanaka T, Shiozaki M and Fujii Y:
MicroRNA-34b has an oncogenic role in esophageal squamous cell
carcinoma. Oncol Lett. 1:685–689. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Giard DJ, Aaronson SA, Todaro GJ, Arnstein
P, Kersey JH, Dosik H and Parks WP: In vitro cultivation of human
tumors: Establishment of cell lines derived from a series of solid
tumors. J Natl Cancer Inst. 51:1417–1423. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Artavanis-Tsakonas S, Rand MD and Lake RJ:
Notch signaling: Cell fate control and signal integration in
development. Science. 284:770–776. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lobry C, Oh P and Aifantis I: Oncogenic
and tumor suppressor functions of Notch in cancer: It's NOTCH what
you think. J Exp Med. 208:1931–1935. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Avila JL and Kissil JL: Notch signaling in
pancreatic cancer: Oncogene or tumor suppressor? Trends Mol Med.
19:320–327. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yap LF, Lee D, Khairuddin A, Pairan MF,
Puspita B, Siar CH and Paterson IC: The opposing roles of NOTCH
signalling in head and neck cancer: A mini review. Oral Dis.
21:850–857. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rampias T, Vgenopoulou P, Avgeris M,
Polyzos A, Stravodimos K, Valavanis C, Scorilas A and Klinakis A: A
new tumor suppressor role for the Notch pathway in bladder cancer.
Nat Med. 20:1199–1205. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ban J, Bennani-Baiti IM, Kauer M, Schaefer
KL, Poremba C, Jug G, Schwentner R, Smrzka O, Muehlbacher K, Aryee
DN and Kovar H: EWS-FLI1 suppresses NOTCH-activated p53 in Ewing's
sarcoma. Cancer Res. 68:7100–7109. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Schaefer KL, Eisenacher M, Braun Y,
Brachwitz K, Wai DH, Dirksen U, Lanvers-Kaminsky C, Juergens H,
Herrero D, Stegmaier S, et al: Microarray analysis of Ewing's
sarcoma family of tumours reveals characteristic gene expression
signatures associated with metastasis and resistance to
chemotherapy. Eur J Cancer. 44:699–709. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Baliko F, Bright T, Poon R, Cohen B, Egan
SE and Alman BA: Inhibition of notch signaling induces neural
differentiation in Ewing sarcoma. Am J Pathol. 170:1686–1694. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ji Q, Hao X, Meng Y, Zhang M, Desano J,
Fan D and Xu L: Restoration of tumor suppressor miR-34 inhibits
human p53-mutant gastric cancer tumorspheres. BMC Cancer.
8:2662008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tang Y, Tang Y and Cheng YS: miR-34a
inhibits pancreatic cancer progression through Snail1-mediated
epithelial-mesenchymal transition and the Notch signaling pathway.
Sci Rep. 7:382322017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bennani-Baiti IM, Aryee DN, Ban J, Machado
I, Kauer M, Muhlbacher K, Amann G, Llombart-Bosch A and Kovar H:
Notch signalling is off and is uncoupled from HES1 expression in
Ewing's sarcoma. J Pathol. 225:353–363. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sharma S, Kelly TK and Jones PA:
Epigenetics in cancer. Carcinogenesis. 31:27–36. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lee YS and Dutta A: MicroRNAs in cancer.
Annu Rev Pathol. 4:199–227. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Groth C and Fortini ME: Therapeutic
approaches to modulating Notch signaling: Current challenges and
future prospects. Semin Cell Dev Biol. 23:465–472. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Vrba L, Munoz-Rodriguez JL, Stampfer MR
and Futscher BW: miRNA gene promoters are frequent targets of
aberrant DNA methylation in human breast cancer. PLoS One.
8:e543982013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
O'Donnell KA, Wentzel EA, Zeller KI, Dang
CV and Mendell JT: c-Myc-regulated microRNAs modulate E2F1
expression. Nature. 435:839–843. 2005. View Article : Google Scholar : PubMed/NCBI
|
















