|
1
|
Hershberger RE and Siegfried JD: Update
2011: Clinical and genetic issues in familial dilated
cardiomyopathy. J Am Coll Cardiol. 57:1641–1649. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hershberger RE, Hedges DJ and Morales A:
Dilated cardiomyopathy: The complexity of a diverse genetic
architecture. Nat Rev Cardiol. 10:531–547. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zahr HC and Jaalouk DE: Exploring the
Crosstalk Between LMNA and Splicing Machinery Gene Mutations in
Dilated Cardiomyopathy. Front Genet. 9:2312018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Skrzynia C, Berg JS, Willis MS and Jensen
BC: Genetics and heart failure: A concise guide for the clinician.
Curr Cardiol Rev. 11:10–17. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang J, Xu WW and Hu SJ: Heart failure:
advanced development in genetics and epigenetics. Biomed Res Int.
2015:3527342015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arbustini E, Pilotto A, Repetto A, Grasso
M, Negri A, Diegoli M, Campana C, Scelsi L, Baldini E, Gavazzi A
and Tavazzi L: Autosomal dominant dilated cardiomyopathy with
atrioventricular block: A lamin A/C defect-related disease. J Am
Coll Cardiol. 39:1–990. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
van Tintelen JP, Hofstra RM, Katerberg H,
Rossenbacker T, Wiesfeld AC, du Marchie Sarvaas GJ, Wilde AA, van
Langen IM, Nannenberg EA, van der Kooi AJ, et al: High yield of
LMNA mutations in patients with dilated cardiomyopathy and/or
conduction disease referred to cardiogenetics outpatient clinics.
Am Heart J. 154:1130–1139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
van Spaendonck-Zwarts KY, van Rijsingen
IA, van den Berg MP, Lekanne Deprez RH, Post JG, van Mil AM,
Asselbergs FW, Christiaans I, van Langen IM, Wilde AA, et al:
Genetic analysis in 418 index patients with idiopathic dilated
cardiomyopathy: Overview of 10 years' experience. Eur J Heart Fail.
15:628–636. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hutchison CJ and Worman HJ: A-type lamins:
guardians of the soma? Nat Cell Biol. 6:1062–1067. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Scaffidi P and Misteli T: Lamin
A-dependent nuclear defects in human aging. Science. 312:1059–1063.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Al-Haboubi T, Shumaker DK, Köser J,
Wehnert M and Fahrenkrog B: Distinct association of the nuclear
pore protein Nup153 with A- and B-type lamins. Nucleus. 2:500–509.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Worman HJ and Bonne G: ‘Laminopathies’: A
wide spectrum of human diseases. Exp Cell. 313:2121–2133. 2007.
View Article : Google Scholar
|
|
13
|
Freeman AR and Levine SA: Clinical
significance of systolic murmurs: Study of 1000 consecutive
‘noncardiac’ cases. Ann Intern Med. 6:1371–1379. 1933. View Article : Google Scholar
|
|
14
|
Mestroni L, Maisch B, McKenna WJ, Schwartz
K, Charron P, Rocco C, Tesson F, Richter A, Wilke A and Komajda M:
Guidelines for the study of familial dilated cardiomyopathies.
Collaborative research group of the European human and capital.
mobility project on familial dilated cardiomyopathy. Eur Heart J.
20:93–102. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
AHA Medical/Scientific Statement, . 1994
revisions to classification of functional capacity and objective
assessment of patients with diseases of the heart. Circulation.
90:644–645. 1994.PubMed/NCBI
|
|
16
|
Li Z, Huang J, Zhao J, Chen C, Wang H,
Ding H and Wang DW and Wang DW: Rapid molecular genetic diagnosis
of hypertrophic cardiomyopathy by semiconductor sequencing. J
Transl Med. 12:1732014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
1000 GenomesProject Consortium, ; Abecasis
GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang
HM, Marth GT and McVean GA: An integrated map of genetic variation
from 1,092 human genomes. Nature. 491:56–65. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Smith RJH and Hildebrand M: DFNA2
Nonsyndromic Hearing LossGeneReviews® [Internet]. Pagon
RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD,
Fong CT, Mefford HC, Smith RJH and Stephens K: University of
Washington; Seattle, WA: pp. 1993–2016. 2008
|
|
19
|
Su CC, Yang JJ, Shieh JC, Su MC and Li SY:
Identification of novel mutations in the KCNQ4 gene of patients
with nonsyndromic deafness from Taiwan. Audiol Neurootol. 12:20–26.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Azuma N, Hirakiyama A, Inoue T, Asaka A
and Yamada M: Mutations of a human homologue of the Drosophila eyes
absent gene (EYA1) detected in patients with congenital cataracts
and ocular anterior segment anomalies. Hum Mol Genet. 9:363–366.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sullivan T, Escalante-Alcalde D, Bhatt H,
Anver M, Bhat N, Nagashima K, Stewart CL and Burke B: Loss of
A-type lamin expression compromises nuclear envelope integrity
leading to muscular dystrophy. J Cell Biol. 147:913–920. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jahn D, Schramm S, Schnölzer M, Heilmann
CJ, de Koster CG, Schütz W, Benavente R and Alsheimer M: A
truncated lamin A in the Lmna −/− mouse line: Implications for the
understanding of laminopathies. Nucleus. 3:463–474. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kubben N, Voncken JW, Konings G, van
Weeghel M, van den Hoogenhof MM, Gijbels M, van Erk A,
Schoonderwoerd K, van den Bosch B, Dahlmans V, et al: Post-natal
myogenic and adipogenic developmental: Defects and metabolic
impairment upon loss of A-type lamins. Nucleus. 2:195–207. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kang SM, Yoon MH and Park BJ:
Laminopathies; Mutations on single gene and various human genetic
diseases. BMB Rep. 51:327–337. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McNally EM, Golbus JR and Puckelwartz MJ:
Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin
Invest. 123:19–26. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Worman HJ, Ostlund C and Wang Y: Diseases
of the nuclear envelope. Cold Spring Harb Perspect Biol.
2:a0007602010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bilińska ZT, Sylvius N, Grzybowski J,
Fidziańska A, Michalak E, Walczak E, Walski M, Bieganowska K,
Szymaniak E, Kuśmierczyk-Droszcz B, et al: Dilated cardiomyopathy
caused by LMNA mutations. Clinical and morphological studies.
Kardiol Pol. 64:812–821. 2006.PubMed/NCBI
|
|
28
|
Sun LP, Wang L, Wang H, Zhang YH and Pu
JL: Connexin 43 remodeling induced by LMNA gene mutation Glu82Lys
in familial dilated cardiomyopathy with atrial ventricular block.
Chin Med J (Engl). 123:1058–1062. 2010.PubMed/NCBI
|
|
29
|
van Berlo JH, de Voogt WG, van der Kooi
AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T,
Heidbüchel H, de Visser M, et al: Meta-analysis of clinical
characteristics of 299 carriers of LMNA gene mutations: do lamin
A/C mutations portend a high risk of sudden death? J Mol Med
(Berl). 83:79–83. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Meune C, Van Berlo JH, Anselme F, Bonne G,
Pinto YM and Duboc D: Primary prevention of sudden death in
patients with lamin A/C gene mutations. N Engl J Med. 354:209–210.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu W, Shan J, Bonne G, Worman HJ and
Muchir A: Pharmacological inhibition of c-Jun N-terminal kinase
signaling prevents cardiomyopathy caused by mutation in LMNA gene.
Biochim Biophys Acta. 1802:632–638. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu W, Muchir A, Shan J, Bonne G and Worman
HJ: Mitogen-activated protein kinase inhibitors improve heart
function and prevent fibrosis in cardiomyopathy caused by mutation
in lamin A/C gene. Circulation. 123:53–61. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mattout A, Pike BL, Towbin BD, Bank EM,
Gonzalez-Sandoval A, Stadler MB, Meister P, Gruenbaum Y and Gasser
SM: An EDMD mutation in C. elegans lamin blocks muscle-specific
gene relocation and compromises muscle integrity. Curr Biol.
21:1603–1614. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Novelli G and D'Apice MR: The strange case
of the ‘lumper’ lamin A/C gene and human premature ageing. Trends
Mol Med. 9:370–375. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang H, Zheng WY, Wang JZ, Wang XJ, Zhen
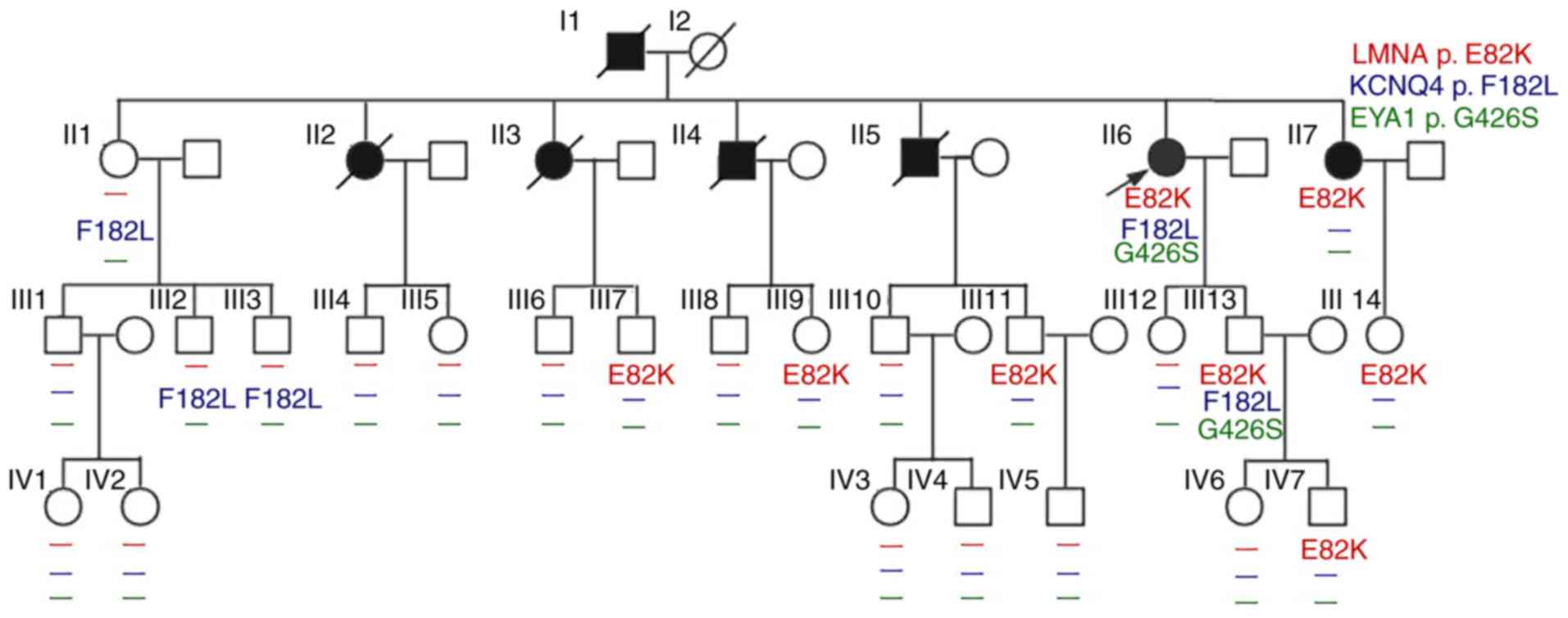
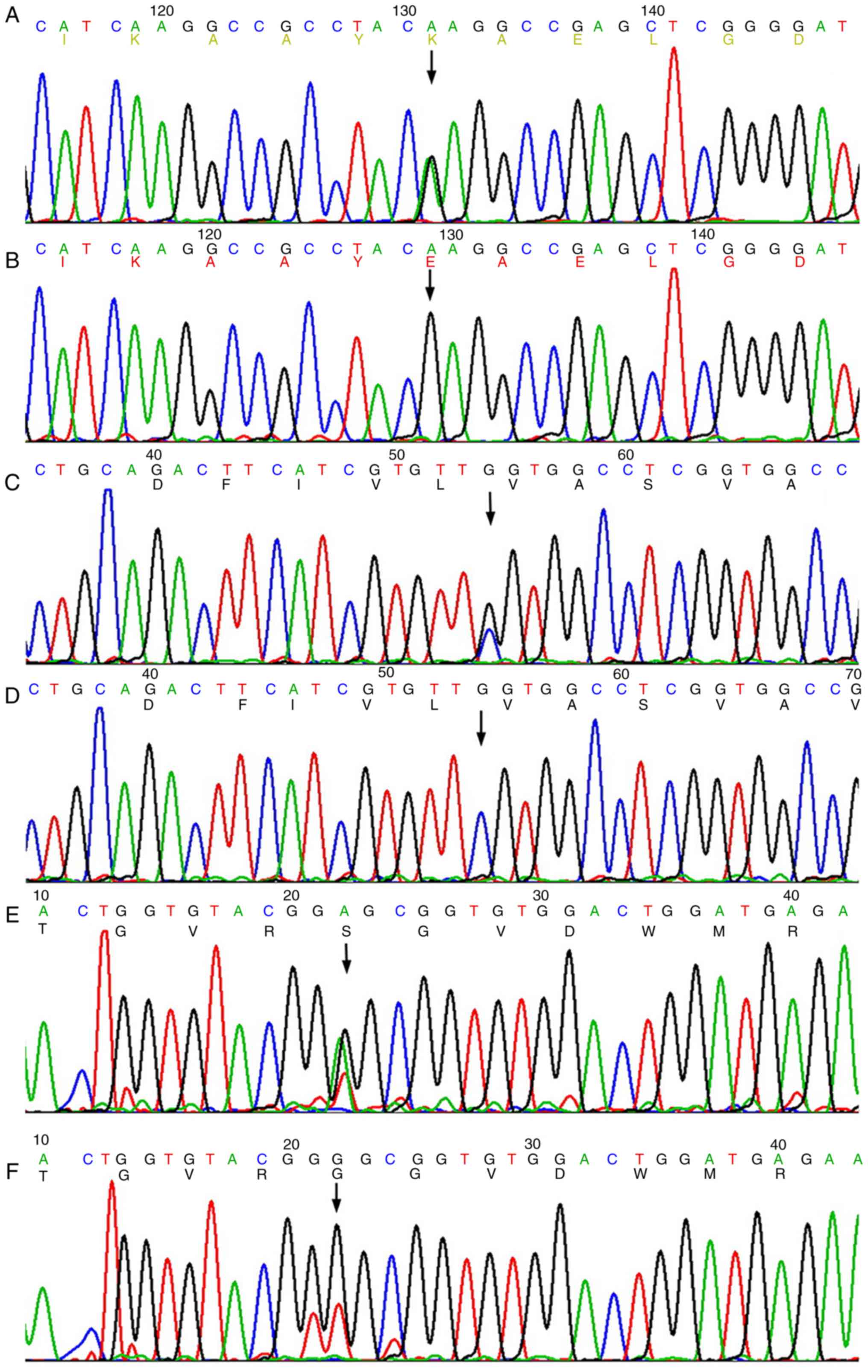
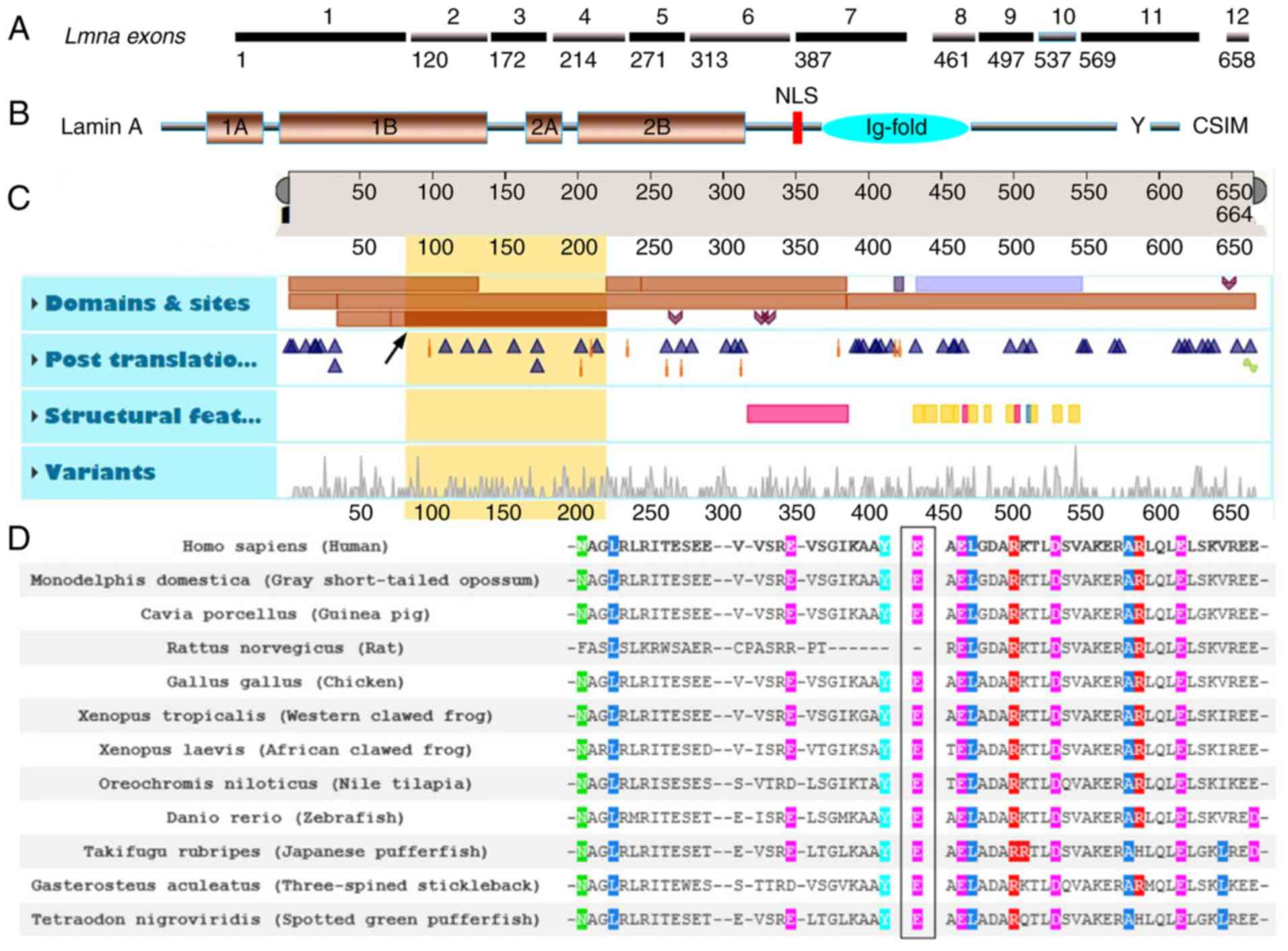
YS, Song L, Zou YB and Hui RT: A novel LMNA gene mutation E82K
associated with familial dilated cardiomyopathy. Zhonghua Xin Xue
Guan Bing Za Zhi. 33:875–879. 2005.(In Chinese). PubMed/NCBI
|
|
36
|
Wu X, Wang QK, Gui L, Liu M, Zhang X, Jin
R, Li W, Yan L, Du R, Wang Q, et al: Identification of a new lamin
A/C mutation in a Chinese family affected with atrioventricular
block as the prominent phenotype. J Huazhong Univ Sci Technolog Med
Sci. 30:103–107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang H, Song XD, Wang SX, Fu CY, Sun K and
Hui RT: Effects of a novel familial dilated cardiomyopathy
associated LMNA gene mutation E82K on cell cycle of HEK293 cells.
Zhonghua Xin Xue Guan Bing Za Zhi. 35:21–23. 2007.(In Chinese).
PubMed/NCBI
|
|
38
|
Lu D, Lian H, Zhang X, Shao H, Huang L,
Qin C and Zhang L: LMNA E82K mutation activates FAS and
mitochondrial pathways of apoptosis in heart tissue specific
transgenic mice. PLoS One. 5:e151672010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Manrai AK, Ioannidis JP and Kohane IS:
Clinical genomics: from pathogenicity claims to quantitative risk
estimates. JAMA. 315:1233–1234. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Duzkale H, Shen J, McLaughlin H, Alfares
A, Kelly MA, Pugh TJ, Funke BH, Rehm HL and Lebo MS: A systematic
approach to assessing the clinical significance of genetic
variants. Clin Genet. 84:453–463. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Severs NJ, Bruce AF, Dupont E and Rothery
S: Remodelling of gap junctions and connexin expression in diseased
myocardium. Cardiovasc Res. 80:9–19. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Petrich BG, Eloff BC, Lerner DL, Kovacs A,
Saffitz JE, Rosenbaum DS and Wang Y: Targeted activation of c-Jun
N-terminal kinase in vivo induces restrictive cardiomyopathy and
conduction defects. J Biol Chem. 279:15330–15338. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cho JH, Cho SD, Hu H, Kim SH, Lee SK, Lee
YS and Kang KS: The roles of ERK1/2 and p38 MAP kinases in the
preventive mechanisms of mushroom Phellinus linteus against the
inhibition of gap junctional intercellular communication by
hydrogen peroxide. Carcinogenesis. 23:1163–1169. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Muchir A, Shan J, Bonne G, Lehnart SE and
Worman HJ: Inhibition of extracellular signal-regulated kinase
signaling to prevent cardiomyopathy caused by mutation in the gene
encoding A-type lamins. Hum Mol Genet. 18:241–247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang H, Wang J, Zheng W, Wang X, Wang S,
Song L, Zou Y, Yao Y and Hui R: Mutation Glu82Lys in lamin A/C gene
is associated with cardiomyopathy and conduction defect. Biochem
Biophys Res Commun. 344:17–24. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang H, Zheng WY, Wang JZ, Song L, Zou YB,
Zhen YS, Wang XJ and Hui RT: The effect of cell mitosis of LMNA
gene mutation E82K associated with familial dilated cardiomyopathy.
Mol Cardiol China. 5:777–779. 2005.
|
|
47
|
Sylvius N, Bilinska ZT, Veinot JP,
Fidzianska A, Bolongo PM, Poon S, McKeown P, Davies RA, Chan KL,
Tang AS, et al: In vivo and in vitro examination of the functional
significances of novel lamin gene mutations in heart failure
patients. J Med Genet. 42:639–647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Malhotra R and Mason PK: Lamin A/C
deficiency as a cause of familial dilated cardiomyopathy. Curr Opin
Cardiol. 24:203–208. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Małek LA, Labib S, Mazurkiewicz L, Saj M,
Płoski R, Tesson F and Bilińska ZT: A new c C > G, p.R541G lamin
A/C mutation in a family with DCM and regional wall motion
abnormalities (akinesis/dyskinesis): Genotype-phenotype
correlation. J Hum Genet. 56:83–86. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Şahan E, Şahan S, Karamanlıoğlu M, Gul M
and Tufekcioğlu O: The MOGE(S) classification: A TNM-like
classification for cardiomyopathies. Herz. 41:503–506. 2016.
View Article : Google Scholar : PubMed/NCBI
|