Introduction
Esophageal cancer is one of the most common lethal
malignancies worldwide (1).
Esophageal cancer is the sixth most common cause of
cancer-associated mortality, causing >400,000 mortalities per
year (2). Esophageal cancer
presents as two principal types: Esophageal squamous cell carcinoma
(ESCC) and esophageal adenocarcinoma. Although the incidence rate
of Barrett's adenocarcinoma is increasing in Western countries, the
incidence of ESCC is increasing at a fast rate in East Asian
populations (3). Due to the lack
of specific symptoms and effective early diagnostic methods, the
5-year survival rate of patients with ESCC remains low, ranging
between 10 and 25% (4). Current
biomarkers, such as serum squamous cell carcinoma antigen,
carbohydrate antigen 19-9, carcinoembryonic antigen and
cytokeratin-19 fragments, are commonly used in the diagnosis of
patients with ESCC. However, these tumor markers are not used in
the early detection of ESCC, due to insufficient diagnostic
sensitivity and specificity (5,6).
Therefore, understanding the molecular mechanisms underlying ESCC
tumorigenesis would facilitate the identification and the
development of novel biomarkers with high sensitivity and
specificity that may be able to improve the early detection and
prognosis of ESCC.
The development and progression of cancer involve
various types of genomic alterations, including DNA mutations,
epigenetic modifications, changes in gene expression and the
complex interplay of these processes (7). Epigenetic modifications, such as DNA
methylation, histone deacetylation, chromatin remodeling and
non-coding RNA (ncRNA) regulation, are critical for the development
and metastasis of various types of cancer, including ESCC (8,9).
ncRNAs have attracted increasing interest over the past decade.
Long ncRNAs (lncRNAs) are a large class of ncRNAs of >200
nucleotides in length, and lncRNA genes are interspersed within the
genome (10–12). lncRNAs have been shown to serve
critical roles in cancer initiation and progression, mediating
oncogenic or tumor suppressing effects at the transcriptional and
post-transcriptional levels (13,14).
Aberrantly expressed lncRNAs have been reported to serve as
potential biomarkers for cancer diagnosis and prognosis (15). For example, the increased
expression of HOX transcript antisense RNA in metastatic breast
cancer (16), CDKN2B antisense RNA
1-induced epigenetic silencing of cyclin dependent kinase inhibitor
2B in leukemia (17) and the
increased expression of metastasis associated lung adenocarcinoma
transcript 1 in metastatic non-small cell lung cancer are
lncRNA-mediated processes associated with the development or
progression of cancer (18,19).
Dysregulated lncRNAs are frequently observed in ESCC (20), but the functions of most lncRNAs in
ESCC remain unclear.
Systems biology approaches can facilitate the
understanding of the pathogenesis of ESCC and the identification of
potential novel biomarkers. Many transcriptome analyses and
datasets of ESCC samples have been generated, and several lncRNAs
have been identified as ESCC-associated lncRNAs (21–25).
Nevertheless, compared with coding genes and microRNAs, the
specific lncRNAs involved in the onset and development of ESCC
remain unknown.
The main aim of the present study was to identify
effective biomarkers or therapeutic targets associated with ESCC.
An ESCC gene expression profile dataset was downloaded from the
Gene Expression Omnibus (26)
(GEO; accession no. GSE53625) and was used as a training dataset.
Additionally, expression profiles were downloaded from The Cancer
Genome Atlas (27) (TCGA) and were
used as a validation dataset. Tumor samples were then divided into
early- and advanced-stage ESCC, according to the
Tumor-Node-Metastasis (TNM) staging system (28), and differentially expressed lncRNAs
(DElncRs) between early- and advanced-stage tumor samples were
identified. A random forest algorithm was used to select optimal
lncRNA biomarkers, which were then analyzed via the support vector
machine (SVM) algorithm in R. The identified lncRNA biomarkers were
also examined in the validation dataset by performing bidirectional
hierarchical clustering and classification using an SVM classifier.
Univariate and multivariate Cox regression analyses were then
performed to determine the ability of the identified lncRNAs to
predict the patient survival rates.
Materials and methods
RNA expression data
RNA expression profiles and patient information from
the GSE53625 dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM1296956)
were downloaded from the GEO (http://www.ncbi.nlm.nih.gov/geo/) database, which is
based on the Affymetrix (Thermo Fisher Scientific, Inc.) GPL18109
platform. In the original study, tumor tissues were collected from
179 patients with ESCC (29). In
the present study, the GSE53625 dataset was used as a training
dataset. RNA-Seq expression profiling data generated using the
Illumina HiSeq 2000 (Illumina, Inc.) and ESCC patient information
(http://www.cbioportal.org/study?id=hnsc_tcga#clinical)
were downloaded from TCGA (https://gdc-portal.nci.nih.gov/). The TCGA dataset was
used as a validation dataset. The GSE53625 dataset contained
normalized gene expression data. The TCGA RNA-Seq expression data
were quantified using an Expectation-Maximization algorithm
(30) of normalized read counts
(log2 transformed). The demographic and clinical data of
patients in both the training and validation datasets are presented
in Table I.
 | Table I.Demographic and clinical information
of patients in the training dataset GSE53625 and in the validation
dataset downloaded from TCGA. |
Table I.
Demographic and clinical information
of patients in the training dataset GSE53625 and in the validation
dataset downloaded from TCGA.
| Patient
characteristics | GSE53625 dataset,
n=179 | TCGA dataset,
n=86 |
|---|
| Age, years (mean ±
SD) | 59.34±9.03 | 58.29±10.63 |
| Gender |
|
|
|
Male | 146 | 75 |
|
Female | 33 | 11 |
| Alcohol use |
|
|
|
Yes | 106 | 62 |
| No | 73 | 23 |
|
Unavailable | 0 | 1 |
| Tobacco use |
|
|
|
Yes | 114 | 57 |
| No | 65 | 27 |
|
Unavailable | 0 | 2 |
| Pathological grade
N |
|
|
| N0 | 83 | 49 |
| N1 | 62 | 26 |
| N2 | 22 | 6 |
| N3 | 12 | 2 |
|
Unavailable | 0 | 3 |
| Pathological grade
T |
|
|
| T1 | 12 | 7 |
| T2 | 27 | 25 |
| T3 | 110 | 48 |
| T4 | 30 | 4 |
|
Unavailable | 0 | 2 |
| Tumor stage |
|
|
| I | 10 | 7 |
| II | 77 | 47 |
|
III | 92 | 27 |
| IV | 0 | 3 |
|
Unavailable | 0 | 2 |
| Adjuvant
therapy |
|
|
|
Yes | 104 | 0 |
| No | 45 | 0 |
|
Unavailable | 30 | 86 |
| Survival
status |
|
|
|
Deceased | 106 | 30 |
|
Alive | 73 | 54 |
|
Unavailable | 0 | 2 |
| Overall survival,
months (mean ± SD) | 36.25±22.86 | 13.67±11.82 |
DElncR identification
According to the pathological disease stage of
patients in the training dataset, tumor tissues were divided into
early-stage (stage I and II) and advanced-stage (stage III and IV)
ESCC. The limma software (version 3.34.9) package of R/Bioconductor
(version 3.6) (31) was used to
analyze DElncRs between early- and advanced-stage ESCC. False
discovery rate <5% was used as the cutoff value, based on a
permutation test, as previously described (32). The fold-change (FC) values of gene
expression between tumor tissues and normal esophageal tissues were
calculated, and |log2FC|>0.263 was used as the cutoff
value.
DElncR clustering analysis
Bidirectional hierarchical clustering based on the
expression profile of the DElncRs identified in the GSE53625
dataset was performed by calculating the centered Pearson
correlation coefficient (33). A
heatmap was then constructed using the R package pheatmap (version
1.0.12) (34). To determine
whether the Pearson correlation coefficient was appropriate for
hierarchical clustering, the chisq.test (χ2) function in
R and the Kaplan-Meier method in the R survival package (version
2.43–3; http://cran.r-project.org/web/packages/survival/index.html)
were used for further evaluation. Specifically, the associations
between the clusters classified by hierarchical clustering and the
stages of ESCC were analyzed using the chisq.test function in R.
Subsequently, the Kaplan-Meier method in the R survival package was
used to estimate the associations between clusters and patient
survival rates based on the patient information in the different
clusters.
Identification of the optimal
combination of lncRNAs using a random forest algorithm
Optimal lncRNA biomarkers for ESCC were selected
using a random forest algorithm, which was calculated using
bootstrap methods (35). The
random forest prediction model was generated using the R package
randomForest (version 4.6–14) (36). In the bootstrap method, out-of-bag
(OBB) error rates were used to evaluate the selection performance
of the random forest algorithm, and lower OBB error rates indicated
higher prediction accuracy.
Construction of a SVM classifier
The optimal combination of lncRNAs was further
analyzed using the SVM function in the e1071 package of R, and a
SVM classifier was constructed (37). The SVM classifier was used to
separate the data points from the two classification groups using a
decision surface. To evaluate the robustness of the SVM model, a
10-fold cross-validation was performed (38). The SVM classifier was then used to
distinguish between patients with early- and advanced-stage-like
ESCC. Moreover, Kaplan-Meier curves were plotted for patients with
early- and advanced-stage ESCC and compared using the log-rank
test.
Validation of optimal lncRNA
biomarkers
The identified optimal lncRNA biomarkers were
hierarchically clustered by calculating Pearson's correlation
coefficient. The SVM classifier, generated using the training
dataset, was used to distinguish between patients with early- and
advanced-stage ESCC in the validation dataset.
Univariate and multivariate Cox
regression analysis
Univariate and multivariate Cox regression analyses
were performed to identify independent prognostic factors
associated with patient survival rates. The tumor stages were
classified using the SVM classifier. Then, demographic and clinical
information, including age, gender, alcohol use, tobacco use, TNM
grade and adjuvant therapy, were analyzed using univariate and
multivariate analyses. In addition, the patients were separated
based on age, gender, alcohol use, tobacco use, TNM grade and
adjuvant therapy and the relationship between the tumor stage, as
classified by the SVM classifier, and patient prognosis was
analyzed.
Functional enrichment analysis of
genes associated with the identified lncRNA biomarkers
The correlation between optimal lncRNA biomarkers
and the expression of all genes in the training dataset was
evaluated by calculating Pearson correlation coefficients using the
cor.test function in R (39). Each
lncRNA was associated with ≥1 gene. The genes were arranged in
descending order based on the absolute value of the Pearson
coefficient, and an lncRNA-mRNA network was generated using the top
1% of lncRNA-mRNA gene pairs.
Next, the mRNA-mRNA interactions of the top 1% of
genes were identified using the Search Tool for the Retrieval of
Interacting Genes/Proteins (STRING) (40). Using a STRING score >0.8, the
genetic interactions predicted by the protein-protein interaction
network were further analyzed. Finally, Kyoto Encyclopedia of Genes
and Genomes (KEGG) (41) pathway
enrichment analysis was performed for all mRNAs that correlated
with the expression of the identified lncRNAs using the Database
for Annotation, Visualization and Integrated Discovery (version
6.8) bioinformatics resources (42), and P<0.05 was considered to
indicate a statistically significant difference.
Results
DElncR screening
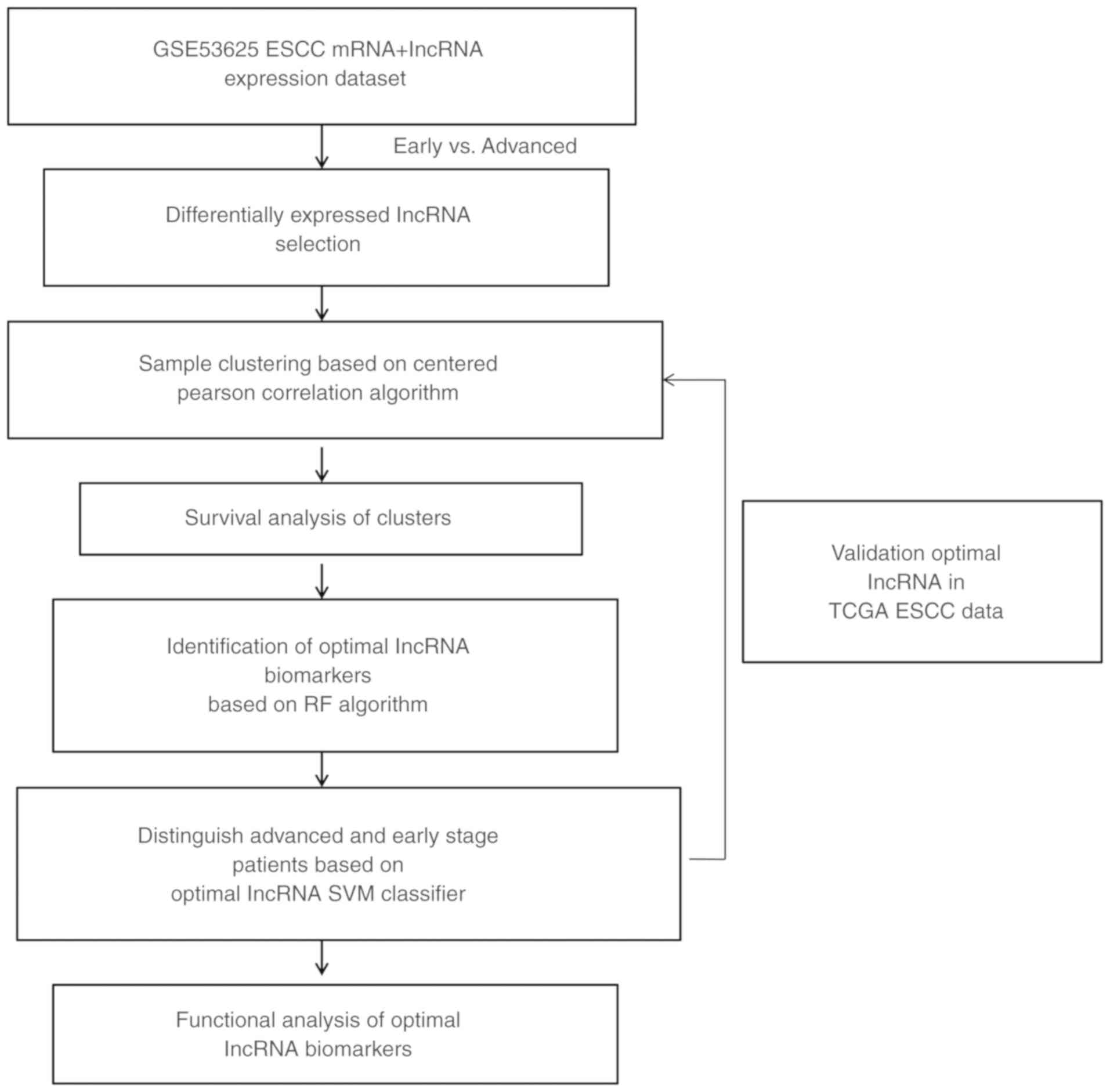
A flow chart of the present study is shown in
Fig. 1. According to the
pathological disease stage of patients in the training dataset, the
tumor samples collected from 179 patients were divided into early-
and advanced-stage ESCC, and the two groups consisted of 87 and 92
patients, respectively. Using the limma R package, a total of 259
DElncRs were identified, including 175 downregulated and 84
upregulated lncRNAs.
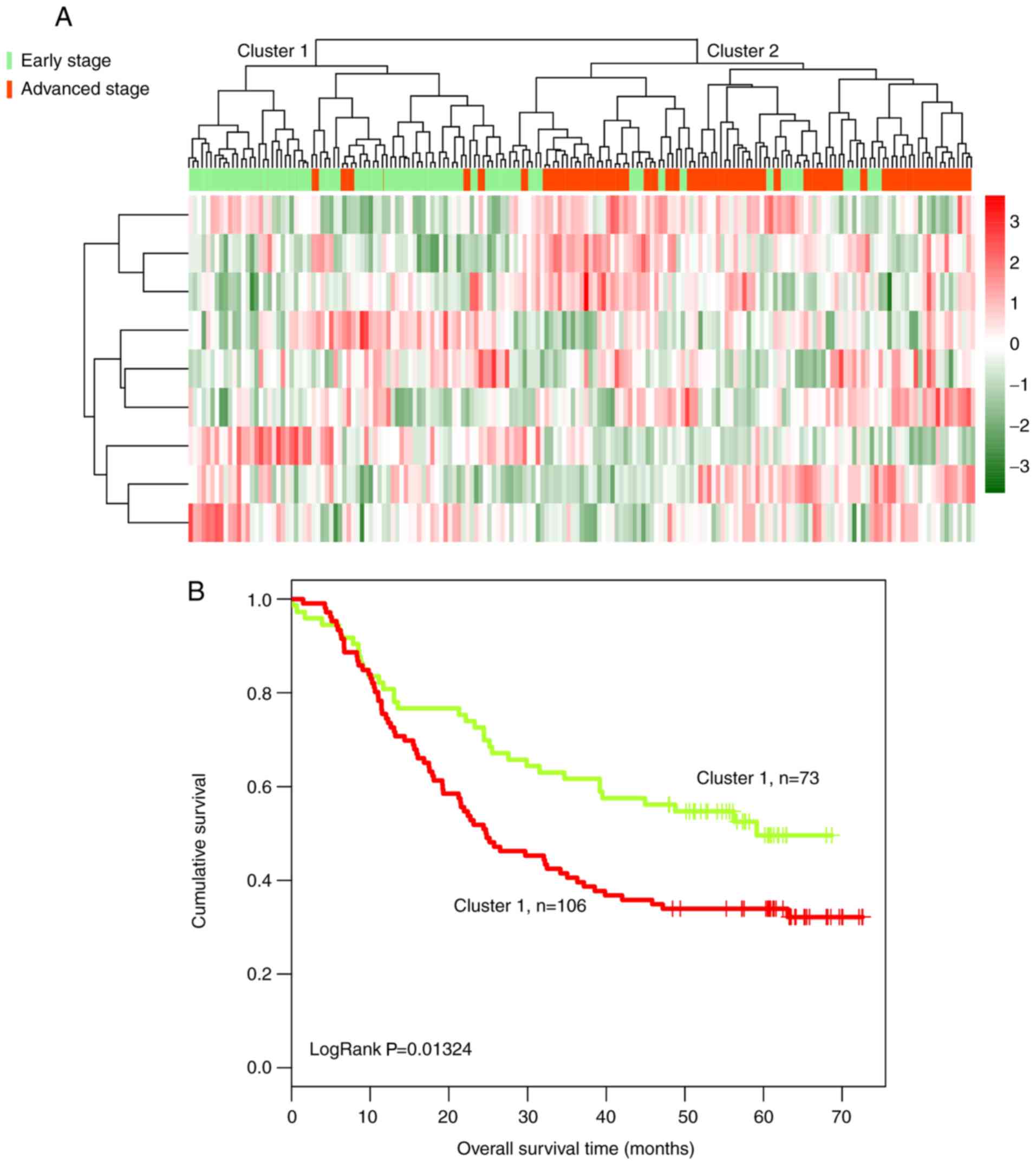
Subsequently, bidirectional hierarchical clustering
was performed based on the expression profiles of 259 DElncRs in
179 tumor tissue samples, by calculating the centered Pearson
correlation. Samples were separated into two clusters (Fig. 2A). In cluster 1, 65 of 67 tumor
samples presented at an early stage. In cluster 2, most of the
tumor samples (90/112) presented at an advanced stage, whereas 22
samples presented at an early stage. The accuracy of tumor stage
identification was 86.59% (155/179; χ2=97.39;
P=2.20×10−16).
Kaplan-Meier analysis suggested that the patients in
cluster 1 exhibited a significantly longer survival time compared
with the patients in cluster 2 (43.50±21.51 and 31.92±22.64 months,
respectively; P=2.639×10−4; Fig. 2B).
Identification of lncRNA biomarkers
for early-stage ESCC
A random forest algorithm was used to identify the
most important lncRNAs. The optimal lncRNA combination was obtained
using the smallest OBB error rate (OBB error=0.183; Fig. 3). A total of nine optimal lncRNA
biomarkers of ESCC were identified, including AC098973, AL133493,
RP11-51M24, RP11-317N8, RP11-834C11, RP11-69C17, LINC00471,
LINC01193 and RP1-124C. The expression levels of AC098973,
AL133493, RP11-51M24 and RP11-317N8 were significantly increased in
early-stage tumors compared with advanced-stage tumors, whereas the
expression levels of the remaining five lncRNAs were significantly
decreased in early-stage tumors (Table II).
| Table II.lncRNA biomarkers associated with the
progression of esophageal squamous cell carcinoma. |
Table II.
lncRNA biomarkers associated with the
progression of esophageal squamous cell carcinoma.
| Ensembl ID | Gene name | Genomic
coordinates | P-value | False discovery
rate | Log2
fold change |
|---|
|
ENSG00000225548 | AC098973 | Chr3:
27,802,762-27,891,301(+) | 0.0002 | 0.0078 | −0.5822 |
|
ENSG00000233922 | AL133493 | Chr21:
45,593,654-45,603,056(+) | 0.0008 | 0.0340 | −0.5420 |
|
ENSG00000249875 | RP11-51M24 | Chr4:
174,354,854-174,376,445(−) | <0.0001 | 0.0012 | −0.3375 |
|
ENSG00000257272 | RP11-317N8 | Chr14:
35,873,857-35,875,303(+) | 0.0001 | 0.0043 | −0.3017 |
|
ENSG00000249388 | RP11-834C11 | Chr12:
54,082,118-54,102,693(+) | 0.0002 | 0.0079 | 0.2818 |
|
ENSG00000227912 | RP11-69C17 | Chr10:
2,166,332-2,169,460(+) | 0.0003 | 0.0139 | 0.2981 |
|
ENSG00000181798 | LINC00471 | Chr2:
231,508,426-231,514,339(+) | <0.0001 | 0.0014 | 0.4400 |
|
ENSG00000258710 | LINC01193 | Chr15:
20,940,438-20,993,303(+) | 0.0002 | 0.0083 | 0.5258 |
|
ENSG00000232316 | RP1-124C6 | Chr6:
113,428,540-113,433,421(−) | <0.0001 | 0.0002 | 0.5867 |
All tumor samples were hierarchically clustered
based on the expression level of the nine identified lncRNAs, by
calculating Pearson correlation coefficients. Tumor samples were
divided into two clusters (Fig.
4A). In cluster 1, 68 of 73 tumor samples presented at an early
stage and only five tumor samples presented at an advanced stage.
By contrast, in cluster 2, 87 of 106 tumor samples presented at an
advanced stage, and 19 samples presented at an early stage. The
clusters exhibited an overall accuracy of 86.59% (155/179). The
patients in cluster 1 had a significantly longer overall survival
time compared with the patients in cluster 2 (40.25±21.61 and
33.50±23.39 months, respectively; Fig.
4B). The present results suggested that these nine lncRNAs
could be used to predict the survival outcome of patients with
ESCC.
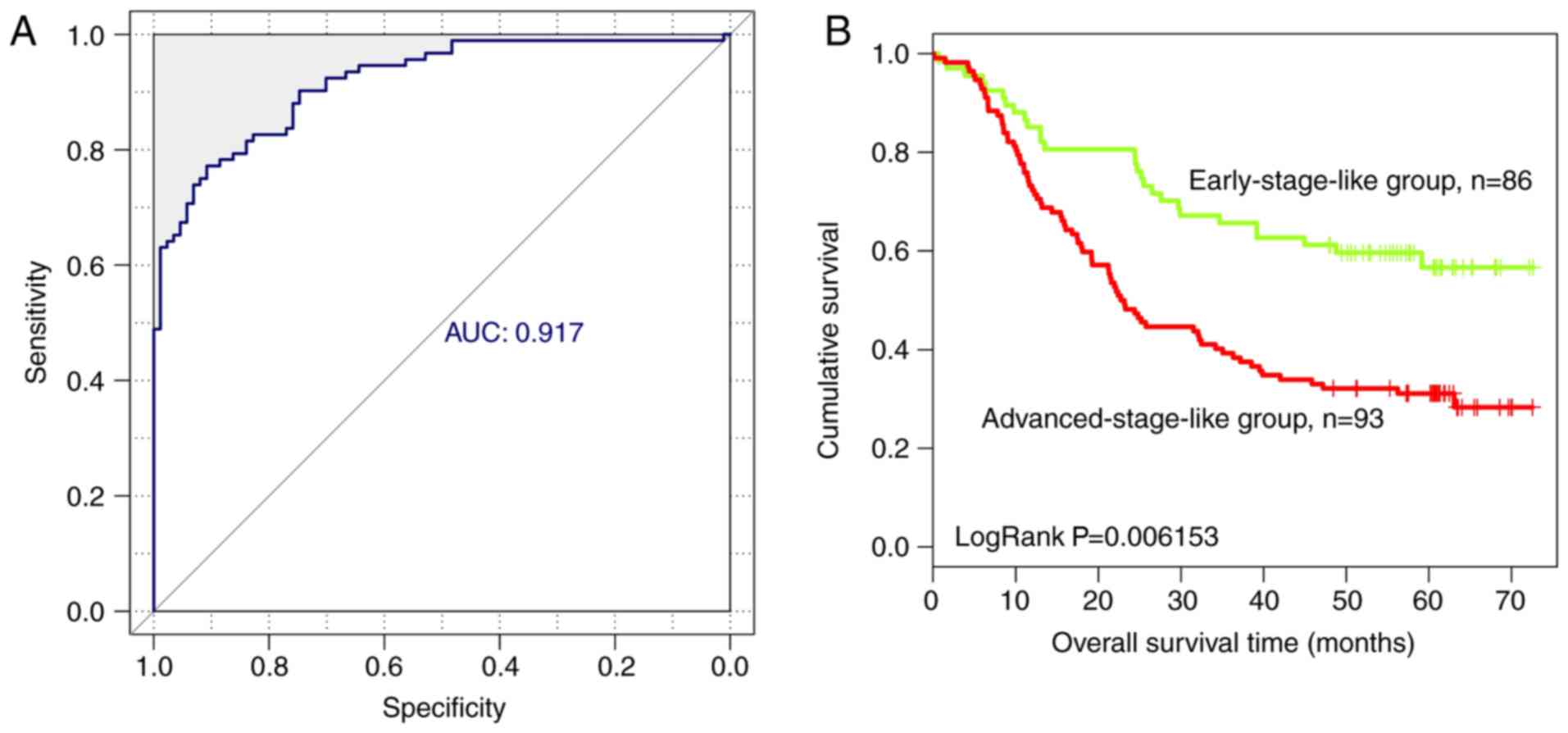
lncRNA classification by SVM
classifier
Based on the expression level of the nine optimal
lncRNAs identified in the present study, an SVM classifier was
established to identify tumors at different stages. The resulting
SVM classifier could distinguish the progression stage of ESCC in
160 of 179 samples, exhibiting an overall accuracy of 89.39%, a
sensitivity of 90.22%, a specificity of 88.51%, a positive
predictive value (PPV) of 89.25%, a negative predictive value (NPV)
of 89.53% and a receiver operating characteristic area under the
curve (AUC) of 0.917 (Fig.
5A).
In addition, overall survival in patients with
early-stage-like and advanced-stage-like tumors, as defined by the
SVM classifier, was calculated using the Kaplan-Meier method and
compared using the log-rank test. Patients with early-stage-like
tumors exhibited a significantly longer overall survival time
compared with patients with advanced-stage-like tumors (40.15±22.46
and 32.81±22.78 months, respectively; Fig. 5B). The present results suggested
that the tumor progression stage identified by the SVM classifier
on the basis of the expression levels of the nine identified
lncRNAs was associated with patient survival rate.
Validation of optimal lncRNA
biomarkers in ESCC
Additional RNA-Seq expression profiling datasets
associated with ESCC were downloaded from TCGA. Specifically,
transcriptomic data from 86 tumor samples and survival data from 71
patients with ESCC were downloaded. Therefore, 71 ESCC samples were
used to validate the ability of the nine identified lncRNAs to
predict the progression of ESCC.
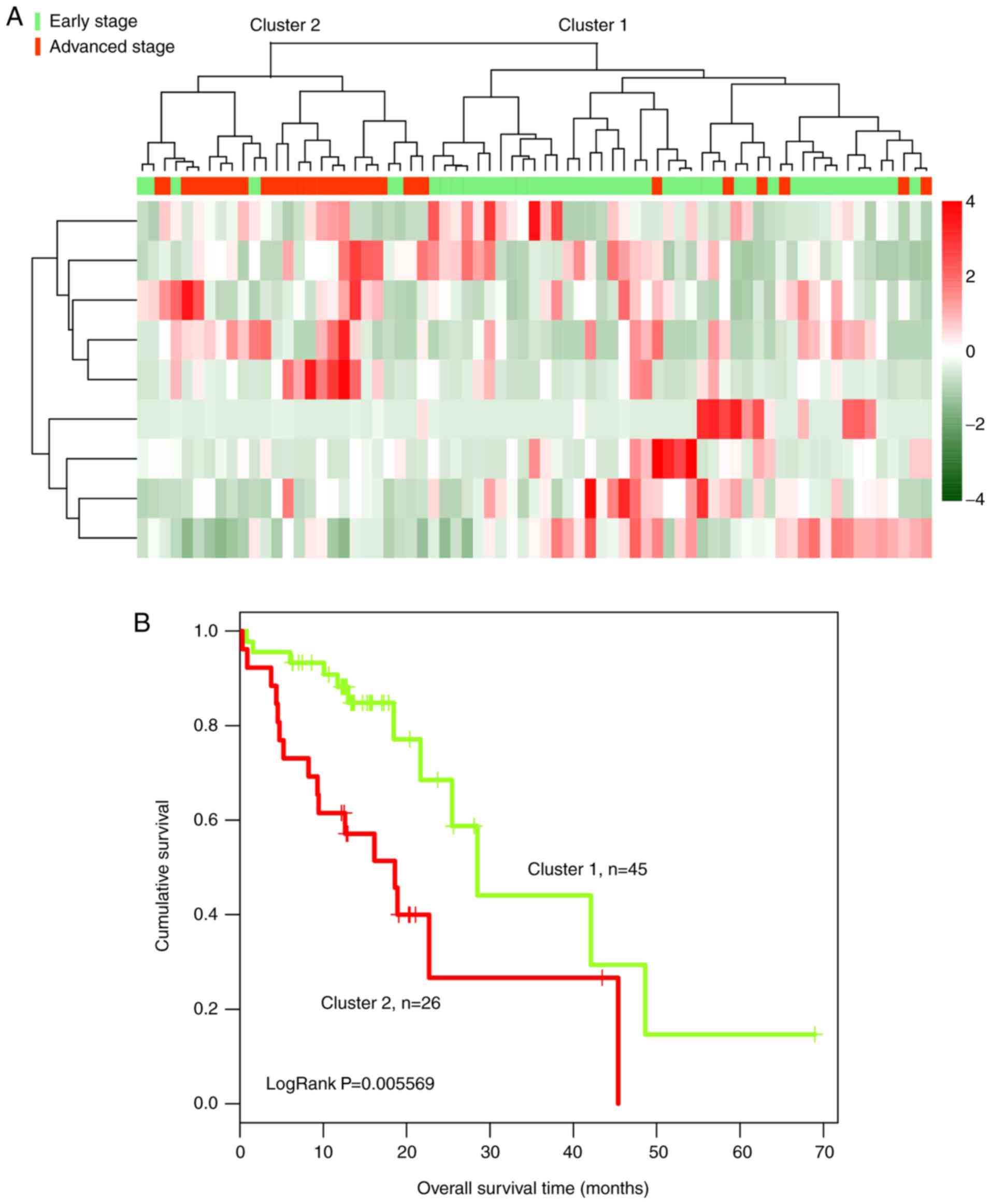
By calculating Pearson correlation coefficients, 71
tumor samples, including 45 at an early stage and 26 at an advanced
stage, were hierarchically clustered based on the expression levels
of the nine identified lncRNAs in the TCGA dataset (Fig. 6A). In the validation dataset, tumor
samples were divided into two clusters based on the expression
levels of the nine lncRNAs. Cluster 1 consisted of 45 samples,
including 39 at an early stage and six at an advanced stage.
Cluster 2 consisted of 26 samples, including six at an early stage
and 20 at an advanced stage. The overall accuracy of the identified
clusters was 83.10% (59/71). The patients in cluster 1 exhibited a
significantly longer overall survival time than patients in cluster
2 (16.70±11.96 and 14.32±11.01 months, respectively; Fig. 6B).
In addition, the SVM classifier based on the
expression levels of the nine identified lncRNAs was used to
discriminate different tumor progression stages in the validation
dataset. The SVM classifier could accurately distinguish the
progression stage of ESCC in 64/71 samples, exhibiting an overall
accuracy of 90.14%. The sensitivity, specificity, PPV and NPV of
the SVM classifier were 73.08, 100, 100 and 86.54%, respectively,
with an AUC of 0.915 (Fig. 7A).
Furthermore, after performing Kaplan-Meier survival analysis
followed by log-rank test, patients with early-stage-like ESCC, as
classified by the SVM classifier, exhibited a significantly longer
overall survival time compared with patients with
advanced-stage-like ESCC (16.51±11.97 and 13.96±10.59 months,
respectively; P=0.038; Fig.
7B).
Based on the results from the bidirectional
hierarchical clustering and the SVM classification, the optimal
combination of lncRNAs was able to reliably predict the survival
time of patients with ESCC.
Identification of independent
prognostic factors associated with patient survival rates
Univariate and multivariate Cox regression analyses
were performed to identify independent prognostic factors
associated with patient survival rates (Table III). The SVM classification,
tobacco use and adjuvant therapy were significantly correlated with
overall patient survival time.
| Table III.Univariate and multivariate Cox
regression analysis for SVM prediction model and clinical
features. |
Table III.
Univariate and multivariate Cox
regression analysis for SVM prediction model and clinical
features.
|
| Univariate
analysis | Multivariate
analysis |
|---|
|
|
|
|
|---|
| Variables | HR | P-value | HR | P-value |
|---|
| SVM prediction
(Early/advanced stage like) | 1.723 | 0.006a | 1.637 | 0.005a |
| Age (≤60 or
>60) | 1.681 | 0.007a | 1.416 | 0.140 |
| Gender | 0.782 | 0.305 | 1.366 | 0.390 |
| Alcohol use | 0.864 | 0.455 | 0.967 | 0.916 |
| Tobacco use | 0.750 | 0.014 | 0.456 | 0.010a |
| Pathological grade
(N0+N1) or (N2+N3) | 1.645 | 0.028a | 1.079 | 0.794 |
| Pathological grade
(T1+T2) or (T3+T4) | 1.091 | 0.711 | 0.834 | 0.528 |
| Adjuvant
therapy | 2.264 | 0.003a | 2.492 | 0.002a |
Additionally, the hazard ratios of the SVM
classifier and stratified clinical factors were calculated,
including age, gender, alcohol use, tobacco use, tumor grade and
adjuvant therapy (Table IV).
Patients with early-stage-like tumors exhibited a longer survival
time than patients with advanced-stage-like tumors. According to
the present results, the tumor progression stage predicted by the
constructed SVM classifier was significantly correlated with the
patient survival rate in the following subgroups: Male patients,
patients <60 years old, alcohol consumers, smokers, patients
with tumors at TNM stages of N0+N1 or T3+T4 and patients who did
not receive adjuvant therapy.
| Table IV.Univariate regression analysis for
each clinicopathological characteristic in the training
dataset. |
Table IV.
Univariate regression analysis for
each clinicopathological characteristic in the training
dataset.
|
| Univariate
analysis |
|
|---|
|
|
|
|
|---|
| Variables | HR | 95% CI | P-value |
|---|
| Age |
| ≤60,
n=99 | 1.966 | 1.113–3.473 | 0.0176a |
| >60,
n=80 | 1.433 | 0.829–2.477 | 0.1954 |
| Gender |
| Male,
n=146 | 1.705 | 1.099–2.645 | 0.0160a |
| Female,
n=33 | 1.707 | 0.691–4.219 | 0.2413 |
| Alcohol use |
| Yes,
n=106 | 1.767 | 1.038–3.007 | 0.0335a |
| No,
n=73 | 1.771 | 0.9799–3.199 | 0.0552 |
| Tobacco use |
| Yes,
n=114 | 1.714 | 1.025–2.865 | 0.0375a |
| No,
n=65 | 1.805 | 0.973–3.35 | 0.0576 |
| Pathological
grade |
| N0+N1,
n=34 | 1.640 | 1.058–2.543 | 0.0256a |
| N2+N3,
n=145 | 1.497 | 0.447–5.015 | 0.5099 |
| Pathological
grade |
| T1+T2,
n=39 | 0.896 | 0.348–2.309 | 0.8198 |
| T3+T4,
n=140 | 1.998 | 1.24–3.219 | 0.0038a |
| Adjuvant
therapy |
| Yes,
n=104 | 1.197 | 0.742–1.932 | 0.4609 |
| No,
n=45 | 4.890 | 1.687–14.22 | 0.0013a |
Functional analysis of genes
associated with the nine optimal lncRNAs
A total of 1,656 genes were identified to be
significantly correlated with the nine identified lncRNAs. Notably,
728 genes were positively correlated, and 928 genes were negatively
correlated. A co-expression network of lncRNAs and genes was then
established. After screening for gene-gene interaction pairs in the
STRING database, lncRNA-mRNA co-expression networks were
constructed (data not shown).
The genes significantly correlated with the nine
identified lncRNA were involved in several KEGG pathways, such as
‘cell cycle’ and ‘DNA replication’, indicating that these lncRNAs
may be involved in the progression of ESCC by regulating these
cellular processes (Fig. 8).
Specifically, the present analysis identified the enriched KEGG
pathways that were negatively and positively associated with
lncRNAs (Fig. 8A and B,
respectively).
Discussion
ESCC is a neoplastic diseases with one of the
highest mortality rates worldwide, which exhibits a particularly
high incidence in certain regions of China (43). However, the etiology of ESCC
remains poorly understood. The present study analyzed public
databases in order to identify novel effective biomarkers or
therapeutic targets involved in the pathogenesis of ESCC.
In the present study, 259 DElncRs between early- and
advanced-stage ESCC were identified. These 259 lncRNAs were used to
predict the tumor stage in the training dataset with high accuracy.
Using a random forest algorithm, a total of nine lncRNA biomarkers
associated with ESCC were identified, including AC098973, AL133493,
RP11-51M24, RP11-317N8, RP11-834C11, RP11-69C17, LINC00471,
LINC01193 and RP1-124C. In addition, the present results suggested
that the combination of these nine lncRNAs was able to predict
tumor stage and patient survival rate in the training dataset.
These nine lncRNA biomarkers associated with ESCC were subsequently
validated. In the validation dataset, the nine lncRNAs were used to
predict the tumor stage and patient survival rate with high
reliability and accuracy. Furthermore, these nine lncRNA biomarkers
were identified to be involved in regulating ‘cell cycle’ and ‘DNA
replication’, which were previously identified to be associated
with the progression of ESCC (44,45).
Collectively, the present study identified nine candidate lncRNAs
associated with the progression and prognosis of ESCC.
Additionally, data enrichment analysis identified the possible
molecular mechanism underlying their function.
The association between the dysregulation of certain
lncRNAs and the prognosis of patients with cancer has been reported
for several malignancies, such as hepatocellular carcinoma
(46), breast cancer (16) and colorectal cancer (47). In addition, many previous
transcriptome analyses of ESCC samples have been performed
(48–50). Several groups have reported the
aberrant expression of various lncRNAs in ESCC and multiple
ESCC-associated lncRNAs have been identified, some of which may be
used as biomarkers for the diagnosis or prognosis of ESCC (21–25).
Li et al (29) compared the
expression levels of lncRNAs in ESCC tissues with paired adjacent
normal tissues and identified a three-lncRNA signature, consisting
of ENST00000435885.1, XLOC_013014 and ENST00000547963.1, which was
identified to be associated with the prognosis of patients with
ESCC (GEO accession no. GSE53625). By analyzing the datasets
generated by Li et al (29), a nine-lncRNA signature was
identified in the present study. The nine identified lncRNAs were
able to predict the tumor stage and survival time of patients with
ESCC. In addition, the nine-lncRNA signature identified in the
training dataset showed reliable prognostic ability in the
validation dataset downloaded from ATCG. Therefore, the identified
lncRNA signature may be used to determine the prognosis of patients
with ESCC.
To the best of our knowledge, the lncRNAs identified
in the present study, including AC098973, AL133493, RP11-51M24,
RP11-317N8, RP11-834C11, RP11-69C17, LINC00471, LINC01193 and
RP1-124C have not been functionally annotated. However, in the
present study, the possible functions of these lncRNAs were
predicted using mRNA expression data from the same group of
patients. The genes correlated with the signature lncRNAs were
identified to be involved in several KEGG pathways, such as ‘cell
cycle’ and ‘DNA replication’, suggesting that these lncRNAs may be
involved in the progression of ESCC by regulating these cellular
processes.
Notably, the present study presents certain
limitations. Although the nine-lncRNA signature identified in the
present study was generated and tested in a large cohort of
patients with ESCC, datasets from other institutions and other
countries are required to verify its clinical application. The
training and validation datasets used in the present study
exhibited differences in the survival rates, possibly due to the
different tumor stages. In particular, the training dataset
contained no ESCC at stage IV. Therefore, the validity of the nine
lncRNAs identified in the present study should be confirmed in
additional prospective studies. Further studies are needed to
validate the prognostic ability of these nine lncRNAs in an
independent cohort of patients with ESCC. In the present study, a
nine-lncRNA signature associated with tumor stage was identified.
Notably, these nine lncRNAs were able to predict the survival time
of patients with ESCC. However, the prognostic ability of the
nine-lncRNA signature identified in the present study should be
validated in further prospective studies in order to use it in
clinical settings.
Acknowledgements
Not applicable.
Funding
The present work was supported by The Jiangsu
Natural Science Foundation (grant no. BK20161598), The Jiangsu
Province Health and Family Planning Commission (project no.
H2017035), The Science and Technology of Nanjing Science Committee
(project no. 201605006) and The Jiangsu Provincial Science and
Technology Department (project no. BE2017759).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
JY and XW performed data analyses and wrote the
manuscript. KH, MZ, XZ, YZ and SC contributed significantly to data
analyses and manuscript revision. QZ and XX conceived and designed
the study. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
van Hagen P, Hulshof M, van Lanschot J,
Steyerberg EW, van Berge Henegouwen MI, Wijnhoven BP, Richel DJ,
Nieuwenhuijzen GA, Hospers GA, Bonenkamp JJ, et al: Preoperative
chemoradiotherapy for esophageal or junctional cancer. N Engl J
Med. 366:2074–2084. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Matsushima K, Isomoto H, Yamaguchi N,
Inoue N, Machida H, Nakayama T, Hayashi T, Kunizaki M, Hidaka S,
Nagayasu T, et al: MiRNA-205 modulates cellular invasion and
migration via regulating zinc finger E-box binding homeobox 2
expression in esophageal squamous cell carcinoma cells. J Transl
Med. 9:302011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ohashi S, Miyamoto S, Kikuchi O, Goto T,
Amanuma Y and Muto M: Recent advances from basic and clinical
studies of esophageal squamous cell carcinoma. Gastroenterology.
149:1700–1715. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hirajima S, Komatsu S, Ichikawa D,
Takeshita H, Konishi H, Shiozaki A, Morimura R, Tsujiura M, Nagata
H, Kawaguchi T, et al: Clinical impact of circulating miR-18a in
plasma of patients with oesophageal squamous cell carcinoma. Br J
Cancer. 108:1822–1829. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kosugi S, Nishimaki T, Kanda T, Nakagawa
S, Ohashi M and Hatakeyama K: Clinical significance of serum
carcinoembryonic antigen, carbohydrate antigen 19-9, and squamous
cell carcinoma antigen levels in esophageal cancer patients. World
J Surg. 28:680–685. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Evans JR, Feng FY and Chinnaiyan AM: The
bright side of dark matter: lncRNAs in cancer. J Clin Invest.
126:2775–2782. 2016. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schmitt AM and Chang HY: Long noncoding
RNAs in cancer pathways. Cancer Cell. 29:452–463. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Clark MB, Johnston RL, Inostroza-Ponta M,
Fox AH, Fortini E, Moscato P, Dinger ME and Mattick JS: Genome-wide
analysis of long noncoding RNA stability. Genome Res. 22:885–898.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rinn JL and Chang HY: Genome regulation by
long noncoding RNAs. Annu Rev Biochem. 81:145–166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
ENCODE Project Consortium, ; Birney E,
Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH,
Weng Z, Snyder M, Dermitzakis ET, et al: Identification and
analysis of functional elements in 1% of the human genome by the
ENCODE pilot project. Nature. 447:799–816. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nagano T and Fraser P: No-nonsense
functions for long noncoding RNAs. Cell. 145:178–181. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yoon JH, Abdelmohsen K, Srikantan S, Yang
X, Martindale JL, De S, Huarte M, Zhan M, Becker KG and Gorospe M:
LincRNA-p21 suppresses target mRNA translation. Mol Cell.
47:648–655. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guttman M and Rinn JL: Modular regulatory
principles of large non-coding RNAs. Nature. 482:339–346. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gupta RA, Shah N, Wang KC, Kim J, Horlings
HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL, et al: Long
non-coding RNA HOTAIR reprograms chromatin state to promote cancer
metastasis. Nature. 464:1071–1076. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu W, Gius D, Onyango P, Muldoon-Jacobs K,
Karp J, Feinberg AP and Cui H: Epigenetic silencing of tumour
suppressor gene p15 by its antisense RNA. Nature. 451:202–206.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ren S, Wang F, Shen J, Sun Y, Xu W, Lu J,
Wei M, Xu C, Wu C, Zhang Z, et al: Long non-coding RNA metastasis
associated in lung adenocarcinoma transcript 1 derived miniRNA as a
novel plasma-based biomarker for diagnosing prostate cancer. Eur J
Cancer. 49:2949–2959. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ji P, Diederichs S, Wang W, Böing S,
Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, et
al: MALAT-1, a novel noncoding RNA, and thymosin beta4 predict
metastasis and survival in early-stage non-small cell lung cancer.
Oncogene. 22:8031–8041. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li CQ, Huang GW, Wu ZY, Xu YJ, Li XC, Xue
YJ, Zhu Y, Zhao JM, Li M, Zhang J, et al: Integrative analyses of
transcriptome sequencing identify novel functional lncRNAs in
esophageal squamous cell carcinoma. Oncogenesis. 6:e2972017.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cao W, Wu W, Shi F, Chen X, Wu L, Yang K,
Tian F, Zhu M, Chen G, Wang W, et al: Integrated analysis of long
noncoding RNA and coding RNA expression in esophageal squamous cell
carcinoma. Int J Genomics. 2013:4805342013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pan Z, Mao W, Bao Y, Zhang M, Su X and Xu
X: The long noncoding RNA CASC9 regulates migration and invasion in
esophageal cancer. Cancer Med. 5:2442–2447. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yao J, Huang JX, Lin M, Wu ZD, Yu H, Wang
PC, Ye J, Chen P, Wu J and Zhao GJ: Microarray expression profile
analysis of aberrant long non-coding RNAs in esophageal squamous
cell carcinoma. Int J Oncol. 48:2543–2557. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang W, Wei C, Li P, Wang L, Li W, Chen K,
Zhang J, Zhang W and Jiang G: Integrative analysis of mRNA and
lncRNA profiles identified pathogenetic lncRNAs in esophageal
squamous cell carcinoma. Gene. 661:169–175. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mathé EA, Nguyen GH, Bowman ED, Zhao Y,
Budhu A, Schetter AJ, Braun R, Reimers M, Kumamoto K, Hughes D, et
al: MicroRNA expression in squamous cell carcinoma and
adenocarcinoma of the esophagus: Associations with survival. Clin
Cancer Res. 15:6192–6200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Clough E and Barrett T: The gene
expression omnibus database. Methods Mol Biol. 1418:93–110. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tomczak K, Czerwinska P and Wiznerowicz M:
The cancer genome atlas (TCGA): An immeasurable source of
knowledge. Contemp Oncol (Pozn). 19:A68–A77. 2015.PubMed/NCBI
|
|
28
|
Huang Y, Guo W, Shi S and He J: Evaluation
of the 7(th) edition of the UICC-AJCC tumor, node, metastasis
classification for esophageal cancer in a Chinese cohort. J Thorac
Dis. 8:1672–1680. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li J, Chen Z, Tian L, Zhou C, He MY, Gao
Y, Wang S, Zhou F, Shi S, Feng X, et al: LncRNA profile study
reveals a three-lncRNA signature associated with the survival of
patients with oesophageal squamous cell carcinoma. Gut.
63:1700–1710. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Moon TK: The expectation maximization
algorithm. IEEE Signal Process Mag. 13:47–60. 1996. View Article : Google Scholar
|
|
31
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: Limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jiao S and Zhang S: On correcting the
overestimation of the permutation-based false discovery rate
estimator. Bioinformatics. 24:1655–1661. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Eisen MB, Spellman PT, Brown PO and
Botstein D: Cluster analysis and display of genome-wide expression
patterns. Proc Natl Acad Sci USA. 95:14863–14868. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang L, Cao C, Ma Q, Zeng Q, Wang H, Cheng
Z, Zhu G, Qi J, Ma H, Nian H and Wang Y: RNA-seq analyses of
multiple meristems of soybean: Novel and alternative transcripts,
evolutionary and functional implications. BMC Plant Biol.
14:1692014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zapf A, Brunner E and Konietschke F: A
wild bootstrap approach for the selection of biomarkers in early
diagnostic trials. BMC Med Res Methodol. 15:432015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cutler A and Stevens JR: Random forests
for microarrays. Methods Enzymol. 411:422–432. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Q and Liu X: Screening of feature
genes in distinguishing different types of breast cancer using
support vector machine. OncoTargets Ther. 8:2311–2317. 2015.
|
|
38
|
Fushiki T: Estimation of prediction error
by using K-fold cross-validation. Statistics Computing. 21:137–146.
2011. View Article : Google Scholar
|
|
39
|
Langfelder P and Horvath S: Fast R
functions for robust correlations and hierarchical clustering. J
Stat Softw. 46(pii): i112012.PubMed/NCBI
|
|
40
|
Szklarczyk D, Franceschini A, Wyder S,
Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos
A, Tsafou KP, et al: STRING v10: Protein-protein interaction
networks, integrated over the tree of life. Nucleic Acids Res 43
(Database Issue). D447–D452. 2015. View Article : Google Scholar
|
|
41
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Enzinger PC and Mayer RJ: Esophageal
cancer. N Engl J Med. 349:2241–2252. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dai L, Li JL, Liang XQ, Li L, Feng Y, Liu
HZ, Wei WE, Ning SF and Zhang LT: Flowers of Camellia nitidissima
cause growth inhibition, cell-cycle dysregulation and apoptosis in
a human esophageal squamous cell carcinoma cell line. Mol Med Rep.
14:1117–1122. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Dadkhah E, Naseh H, Farshchian M, Memar B,
Sankian M, Bagheri R, Forghanifard MM, Montazer M, Kazemi Noughabi
M, Hashemi M and Abbaszadegan MR: A cancer-array approach
elucidates the immune escape mechanism and defects in the DNA
repair system in esophageal squamous cell carcinoma. Arch Iran Med.
16:463–470. 2013.PubMed/NCBI
|
|
46
|
Yang F, Zhang L, Huo XS, Yuan JH, Xu D,
Yuan SX, Zhu N, Zhou WP, Yang GS, Wang YZ, et al: Long noncoding
RNA high expression in hepatocellular carcinoma facilitates tumor
growth through enhancer of zeste homolog 2 in humans. Hepatology.
54:1679–1689. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kogo R, Shimamura T, Mimori K, Kawahara K,
Imoto S, Sudo T, Tanaka F, Shibata K, Suzuki A, Komune S, et al:
Long non-coding RNA HOTAIR regulates Polycomb-dependent chromatin
modification and is associated with poor prognosis in colorectal
cancers. Cancer Res. 71:6320–6326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ito T, Shimada Y, Kan T, David S, Cheng Y,
Mori Y, Agarwal R, Paun B, Jin Z, Olaru A, et al: Pituitary
tumor-transforming 1 increases cell motility and promotes lymph
node metastasis in esophageal squamous cell carcinoma. Cancer Res.
68:3214–3224. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ma S, Bao JYJ, Kwan PS, Chan YP, Tong CM,
Fu L, Zhang N, Tong AHY, Qin YR, Tsao SW, et al: Identification of
PTK6, via RNA sequencing analysis, as a suppressor of esophageal
squamous cell carcinoma. Gastroenterology. 143:675–686.e12. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sawada G, Niida A, Uchi R, Hirata H,
Shimamura T, Suzuki Y, Shiraishi Y, Chiba K, Imoto S, Takahashi Y,
et al: Genomic landscape of esophageal squamous cell carcinoma in a
Japanese population. Gastroenterology. 150:1171–1182. 2016.
View Article : Google Scholar : PubMed/NCBI
|