Introduction
Erastin is a classic ferroptosis inducer that
suppresses the glutamate/cystine antiporter (system
XC−), subsequently inhibits cellular cystine
uptake and depletes glutathione (GSH) (1). GSH is a primary cellular antioxidant
that serves critical functions in maintaining the redox balance and
defending against oxidative stress, including reactive oxygen
species (ROS) (1). Erastin has
been widely found to induce ferroptosis in several types of cancer
cells. Yang et al (2) found
that in fibrosarcoma, a fatal dose of erastin induces ferroptosis
by depleting GSH, confirmed to be an important target of erastin.
DeHart et al (3) found that
in the hepatocarcinoma cell lines HepG2 and Huh7, erastin binds to
voltage-dependent anion channels and thus increases ΔΨ, leading to
mitochondrial ROS generation followed by ferroptosis induction. Pan
et al (4) reported that in
non-small cell lung cancer cells, erastin can induce ferroptosis,
which can be reversed by ferrostatin-1, a ferroptosis inhibitor,
but not by Z-VAD-FMK, an apoptosis inhibitor, indicating that
erastin-induced cell death is a non-apoptotic cell death.
ROS are a heterogeneous group of highly reactive
ions and molecules that are derived from molecular oxygen (5). ROS are believed to act as an
antioxidant system that is critical for maintaining redox
homeostasis (6) and to be toxic
and closely associated with various pathological mechanisms
(7). The dual roles of ROS can be
briefly described as follows: A relatively low or controlled
increase in ROS regulates cell proliferation, apoptosis,
angiogenesis and other physiological processes (5), and an abnormal generation or
accumulation of ROS can induce oxidative stress and damage
programmed physiological processes by damaging cellular lipids,
proteins and DNA (8). Thus, the
balance of ROS is critical for regulating physiological processes
or predicting oxidative stress. Considering the ROS accumulation
caused by erastin, the present study hypothesized that erastin
potentially regulated physiological processes by affecting the
balance of ROS levels.
Gastric cancer has been the second leading cause of
cancer-related mortality worldwide in recent decades (9). Numerous therapeutic strategies have
been developed, but most patients are asymptomatic in the early
stages and more than half of the patients suffer from distant
metastasis; therefore, gastric cancer has a poor prognosis and the
5-year survival rate is <10% (10,11).
Thus, substantial effort has been made to determine the mechanisms
of gastric carcinogenesis. By considering the relatively high
levels of ROS in cancer cells compared with normal cells, cancer
cells are believed to be more sensitive to oxidative stress and
this sensitivity has become an important therapeutic target for
cancer treatment, including treatment of gastric cancer (12). Chen et al (12) revealed that in gastric cancer, the
stimulation of ROS by oxidative stress activates the pro-apoptotic
pathway and this was hypothesized to be a novel therapeutic
strategy for the treatment of gastric cancer. Thus, it is worth
determining whether erastin-induced ROS contribute to the
physiological processes of gastric cancer cells.
The present study examined the effects of a
relatively low dose of erastin on the malignant behaviors of HGC-27
gastric cancer cells, including proliferation, migration, invasion,
colony formation and soft agar tumor formation. It shows that
erastin-induced ROS disrupted mitochondrial function. Together,
these results suggest that erastin-induced ROS may contribute to
antitumor effects and provide an effective alternative for gastric
cancer therapy.
Materials and methods
Cell culture and treatment
Human gastric cancer cells HGC-27 were obtained from
the Institute of Biochemistry and Cell Biology, Chinese Academy of
Sciences. Cells were maintained in RPMI-1640, supplemented with 10%
fetal bovine serum (FBS) at 37°C and 5% CO2 humidified
incubator. All reagents were purchased from Thermo Fisher
Scientific, Inc.
For cell treatment, 0–50 µM of erastin was
supplemented to the original medium and cell viability was analyzed
after 24 h. For apoptosis analysis, the cells were treated with
6.23 µM of erastin for 7 days followed by further analysis. To
abolish ROS, 10 µM NAC was supplemented in original medium; to
inhibit ferroptosis, 10 µM of ferrostatin-1 was employed; to
inhibit apoptosis, 10 µM of zVAD was employed.
CCK-8 assay
Cell viability was measured using CCK-8 assay. The
cells were plated at a density of 5×103 cells per well
in 96-well microtiter culture plates, incubated overnight at 37°C
and treated with specified concentrations of erastin for 24 h.
Then, 10 µl of CCK-8 was added into each well for another 4 h
incubation and scanned by a multi-spectrophotometer (Thermo Fisher
Scientific, Inc.) at 470 nm.
5-Ethynyl-20-deoxyuridine (EdU)
staining
Cell proliferation was detected by the incorporation
of EdU with an EdU Cell Proliferation Assay kit (Guangzhou RiboBio
Co., Ltd.). Briefly, cells were incubated with 50 mM EdU for 4 h
and then fixed with 4% paraformaldehyde for 30 min at 25°C. Then,
EdU staining was performed according to the manufacturer's
protocol. Cell nuclei were stained with Hoechst-33342
(Sigma-Aldrich; Merck KGaA) at a concentration of 1 mg/ml for 20
min. The proportion of cells incorporating EdU was determined using
an X71 (U-RFL-T) fluorescence microscope (Olympus Corporation;
magnification, ×40).
Propidium iodide (PI) staining
followed by flow cytometry
The cells were trypsinized and suspended in ice-cold
PBS. After centrifugation (200 × g) for 5 min at room temperature,
the cells were fixed with ice-cold 70% ethanol. After 4 h, the
cells were pelleted by centrifugation (200 × g) for 5 min at room
temperature and washed with ice-cold PBS and suspended with 5 µg/ml
PI (Sigma-Aldrich; Merck KGaA) and 10 µg/ml RNase A (Sigma-Aldrich;
Merck KGaA) for 10 min in darkness. Then, the stained cells were
pelleted by centrifugation (200 × g) for 5 min at room temperature
and washed with three washes using ice-cold PBS and then analyzed
using three laser Navios flow cytometers (Beckman Coulter, Inc.)
according to the manufacturer's instructions.
Cell migration and invasion assay
Cells were seeded in a 6-well plate at a density of
2.5×105 cells/well. A scratch was made with a sterile
10-µl pipette tip. After 0 and 24 h, images were captured.
To measure invasion ability, 5×103 cells
were suspended and plated into upper chamber (8-µm pore size;
Corning Inc.) coated with Matrigel for 24 h at 37°C (Sigma-Aldrich;
Merck KGaA). Then, the chamber was fixed with 4% paraformaldehyde
at 25°C for 20 min and stained with 0.1% crystal violet for 20 min
at 25°C (Sigma-Aldrich).
Colony formation
The cells were trypsinized and suspended in PBS.
Subsequently, 1,000 cells were seeded in 6-well plates and cultured
for 2–3 weeks. When the colonies were visible to the naked eye, the
plate was washed twice with PBS. The colonies were fixed with 4%
paraformaldehyde for 10 min and stained by 0.25% crystal violet for
20 min at 25°C.
Tumor formation in soft agar
Cells were resuspended with 0.3% soft agar in
RPMI-1640 containing 10% FBS, then layered onto 0.6% solidified
agar in RPMI-1640 containing 10% FBS in 6-well plates
(5×103 cells/well). These plates were incubated at 37°C
for 2–3 weeks. Colonies containing ≥50 cells were counted using
Image J software (version-2.0; National Institutes of Health).
ROS detection
The specific fluorescent probe H2DCFDA
(Sigma-Aldrich) was used to detect the intracellular ROS level.
H2DCFDA (5 µl) was added for 15 min at 25°C. The cells
in 6-well plates were washed three times with PBS and imaged using
an X71 (U-RFL-T) fluorescence microscope (Olympus Corporation,
magnification, ×40).
Mitochondrial transcriptional
activity
RNA was isolated from cells following the
manufacturer's instructions using RNeasy kit (Qiagen GmbH). Reverse
transcription was performed using a high capacity RNA-to-cDNA kit
(Thermo Fisher Scientific, Inc.). Quantitative PCR was performed
using SYBR Green PCR Master Mix (Thermo Fisher Scientific, Inc.)
and the primers were as follows: COXI: Forward,
5′-ATGCGGCCATAGGTTCTGC-3′ and reverse,
5′-TCCTCAAGATGTCTCAGTTCCAT-3′; ND1: Forward,
5′-TCGTCATAATCTGTCCCTACACA-3′ and reverse,
5′-CGGCTTCGGCTCTTAGCAAA-3′; and β-actin: Forward,
5′-GTGACGTTGACATCCGTAAAGA-3′ and reverse,
5′-GCCGGACTCATCGTACTCC-3′. The thermocycling conditions were as
follows: 95°C for 5 min, 40 cycles of 95°C for 10 sec, 60°C for 50
sec. The amplification and analysis were performed using an ABI
Prism 7500 Real-Time PCR System (Applied Biosystems; Thermo Fisher
Scientific, Inc.). Samples were normalized to a housekeeping gene
using the 2−ΔΔCq method (13).
ATP measurement
Cells (1×106) were suspended and washed
with ice-cold PBS. Cells pelleted by centrifugation (200 × g) for 5
min at room temperature were suspended in ATP measuring buffer and
ATP level was measured using the ATP Bioluminescence Assay kit
(Roche Diagnostics) following the manufacturer's instructions.
JC-1 staining
JC-1 probe (Sigma-Aldrich; Merck KGaA) was used to
investigate the mitochondrial membrane potential following erastin
treatment. HGC-27 cells were incubated with JC-1 (20 µg/ml) at 37°C
for 20 min and then washed twice with PBS. Treated cells were
observed by an X71 (U-RFL-T) fluorescence microscope (Olympus
Corporation, magnification, ×40).
Carboxyfluorescein diacetate,
succinimidyl ester (CFSE)/PI double staining
Cells (5×105) were seeded in a 6-well
plate at a density of 2.5×105 cells/well, suspended and
washed with ice-cold PBS and stained with 10 µM CFSE at 37°C for 2
h. Then cells were seeded in 12-well plates and treated with
erastin. After 24 h, the medium was removed and the cells were
washed with ice-cold PBS twice and incubated with 5 µg/ml PI for 10
min at 25°C in dark. Then the supernatant was removed and cells
were imaged using an X71 (U-RFL-T) fluorescence microscope (Olympus
Corporation, magnification, ×40).
Annexin V/PI double staining
Apoptosis was measured using Annexin V-FITC/PI
apoptosis detection kit (BD Biosciences) according to the
manufacturer's instructions. The cells were washed with ice-cold
PBS and pelleted by centrifugation (200 × g) for 5 min at room
temperature. Following removal of the supernatant, the pellet was
resuspended in 1X binding buffer and stained with 5 µl fluorescein
isothiocyanate-labeled Annexin V at 4°C for 15 min in the dark.
Then 10 µl of PI was added at 4°C for 5 min in the dark. The cells
were analyzed by flow cytometry using 3 laser Navios flow
cytometers (Beckman Coulter, Inc.).
Western blot analysis
The cells suspended in RIPA buffer (Sigma-Aldrich;
Merck KGaA) and lysed using SoniConvert® Sonicator
(DocSense) and protein concentration was determined via BCA assay
(Sigma-Aldrich; Merck KGaA) according to the manufacturer's
instruction. In each lane, 20 µg total protein was applied to a 10%
SDS-PAGE gel. Fractionated proteins were transferred onto a
nitrocellulose membrane, and blocked using 5% BSA containing PBS at
room temperature for 1 h. The membrane was incubated with the
primary antibody specific to cleaved PARP, cleaved caspase-3
(1:1,000; cat. no. ab136812; Abcam), at 4°C overnight, then
incubated with the secondary antibody goat anti-mouse IgG H&L
antibody (HRP-labeled; 1:10,000; ab97040; Abcam) at 37°C for 1 h.
Mouse anti-β-actin (1:5,000; cat. no. ab8226; Abcam) was used as an
internal reference. Then blots were visualized with
chemiluminescence kit (Thermo Scientific, Inc.) and quantitatively
measured using Image J software (version-2.0; National Institutes
of Health).
Statistical analysis
All experiments were repeated three times
independently. The Student's t-test was used to determine the
statistical significance between two groups. One-way analysis of
variance was performed to compare multiple groups followed by
Tukey's post-hoc test. Data in the present study were presented as
the mean ± standard error of the mean. P<0.05 was considered to
indicate a statistically significant difference.
Results
Relative low dose of erastin inhibits
cell proliferation in gastric cancer cells HGC-27
To investigate the cellular cytotoxicity of erastin
on gastric cancer cells HGC-27, HGC-27 cells were treated with DMOS
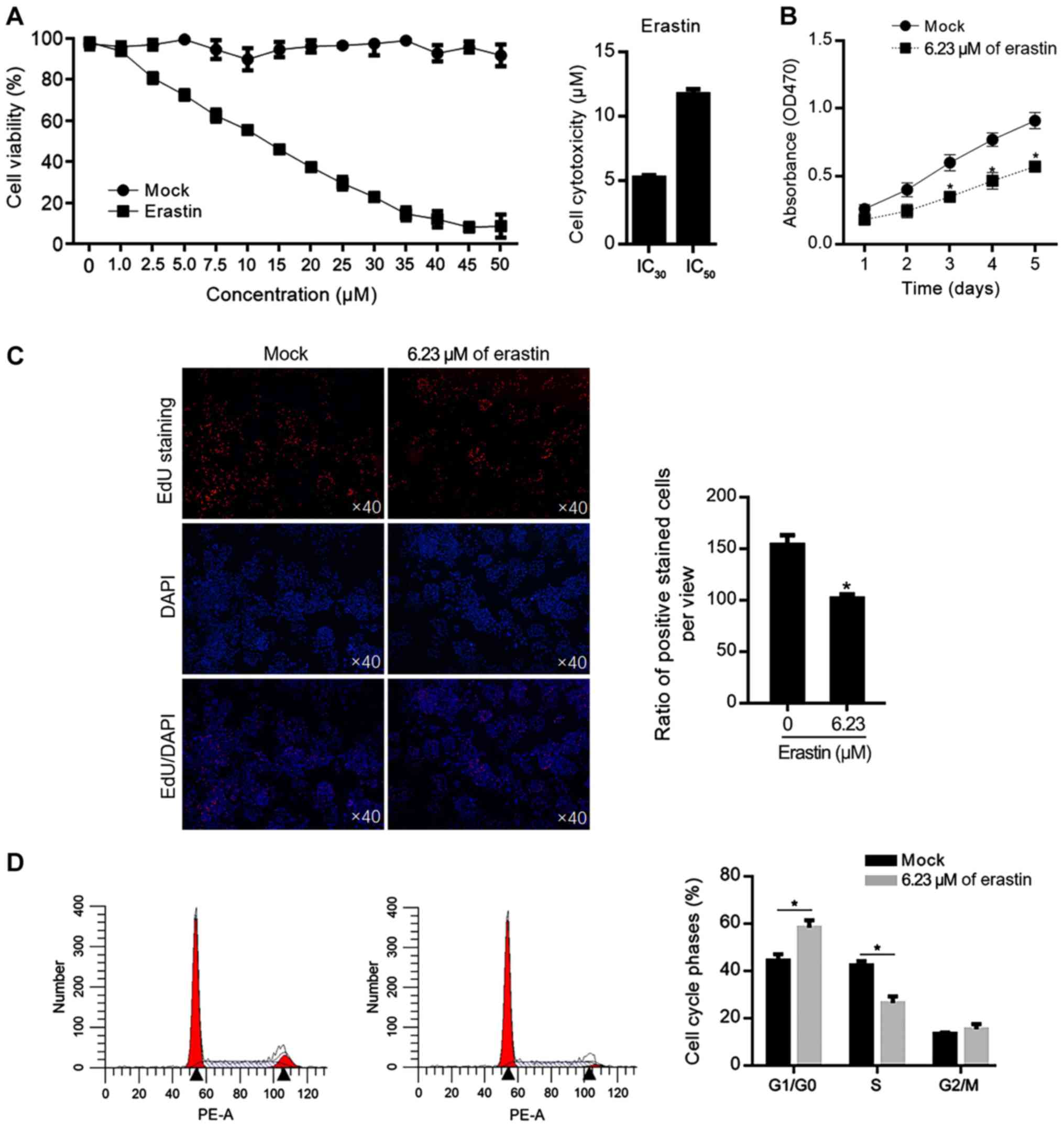
(Mock) or erastin at different concentration. As shown in Fig. 1A, the 30% inhibitory concentration
(IC30) of erastin was 6.23±0.09 µM and the 50%
inhibitory concentration (IC50) of erastin was
14.39±0.38 µM. To evaluate the effects of relative low
concentration of erastin, 6.23 µM of erastin was added in HGC-27
culture medium for 5 days and cell viability was measured each day.
The decreased cell viability from 1–5 days was confirmed (Fig. 1B). To further confirm the effect of
erastin on cell proliferation, EdU staining was performed after
erastin treatment for 24 h and fewer positively stained cells were
observed in erastin-treated group (Fig. 1C). Then the cell cycle phases were
also analyzed; 6.23 µM of erastin treatment increased the
proportion of G1/G0 phase and the decreased proportion of S phase
in HGC-27 cells, which demonstrated that a relatively low dose of
erastin inhibited proliferation via block cell cycle at G1/G0
(Fig. 1D).
Amount of 6.23 µM of erastin inhibits
malignant behaviors in HGC-27 cells
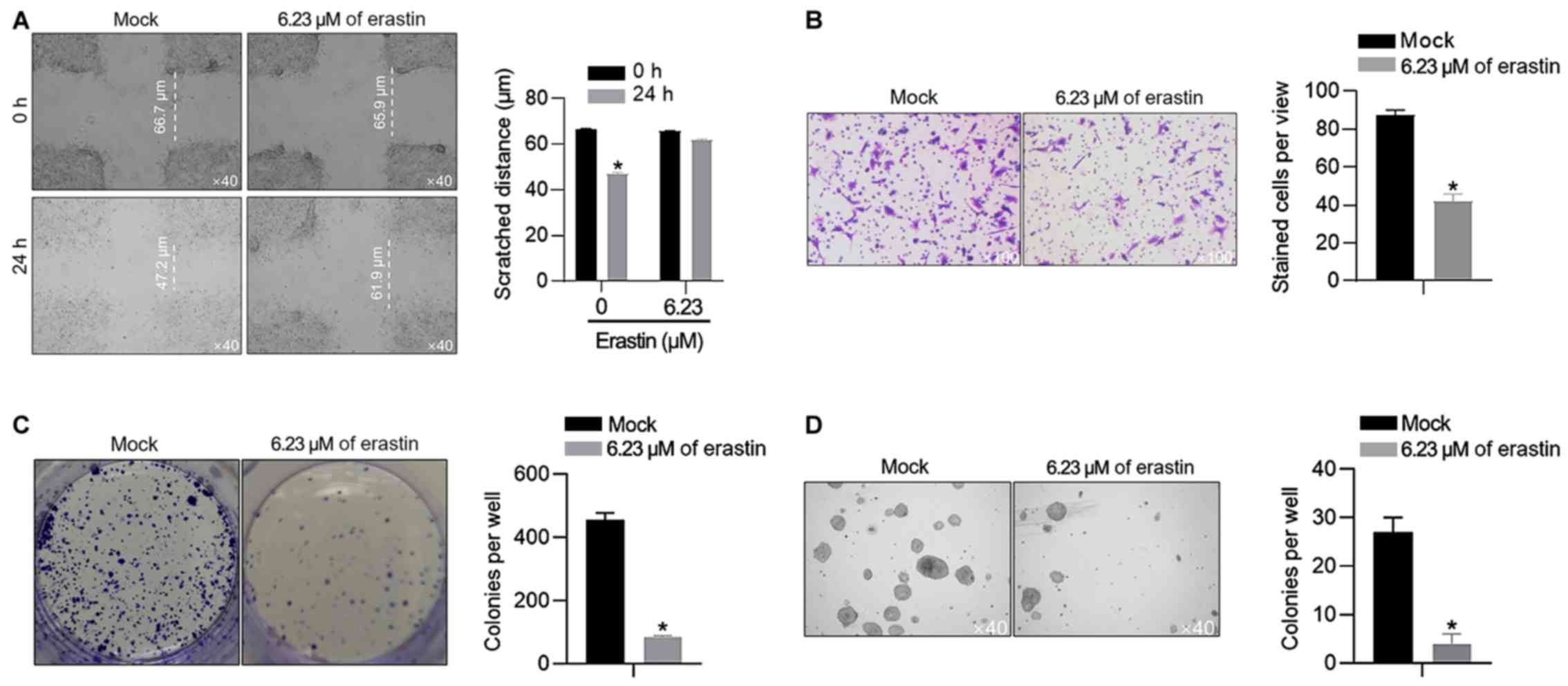
The present study also confirmed the effects of 6.23
µM of erastin on malignant behaviors in HGC-27 cells, including
migration, invasion, colony formation and tumor formation in soft
agar. In scratch assay, after 24-h healing, the gap in the
erastin-treated group was 66.1±2.4 µm, which was larger than that
in the mock group (50.3±1.5 µm; Fig.
2A). It was also observed that 6.23 µM of erastin treatment
inhibited invasion, colony formation and tumor formation (Fig. 2B-D), without knowing whether it was
associated with the proliferation inhibition caused by erastin
treatment.
Amount of 6.23 µM of erastin
depolarizes mitochondrial potential and leads to mitochondrial
dysfunction
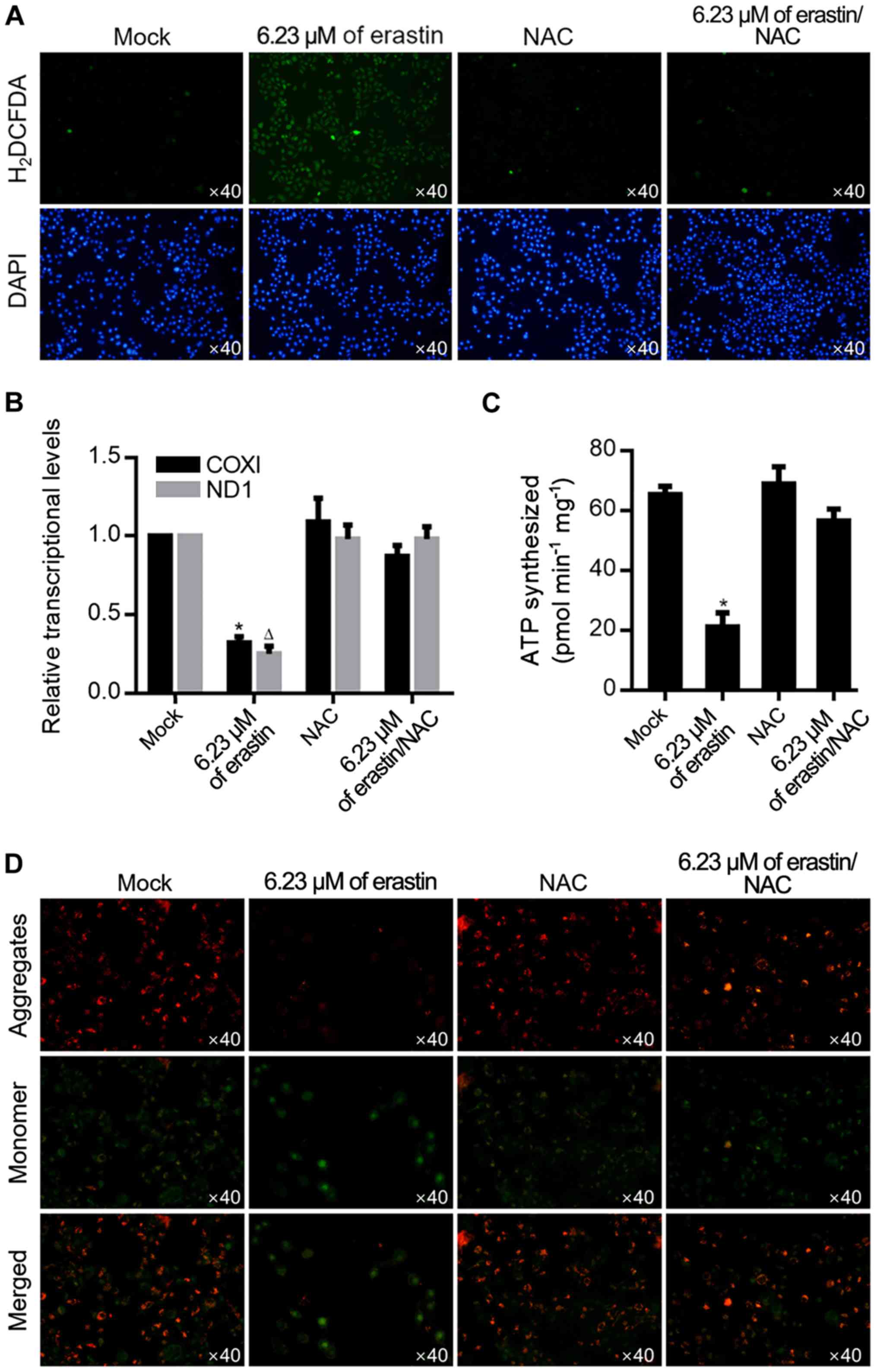
By considering the ROS accumulation effect of
erastin treatment, it was then investigated whether 6.23 µM of
erastin could also induce ROS accumulation. As shown in Fig. 3A, after ROS probe staining using
H2DCFDA, erastin treatment induced an evident increase
of positive signal, which was abolished by the addition of 10 µM
NAC, a ROS scavenger. ROS is a critical factor in the maintenance
of mitochondrial homeostasis, thus mitochondrial function was
measured by transcriptional activity and ATP production. As shown
in Fig. 3B, erastin treatment
significantly decreased transcripts COXI and ND1 in mitochondria
and decreased the amount of ATP synthesized (Fig. 3C), demonstrating that 6.23 µM of
erastin clearly caused mitochondrial dysfunction. Accumulated ROS
modulates mitochondrial function mainly by regulating mitochondrial
potential, so mitochondrial potential was detected following
erastin treatment by JC-1 staining. As shown in Fig. 3D, erastin treatment clearly
decreased JC-1 aggregates and increased JC-1 monomers and this was
reversed by 10 µM NAC treatment.
Long-term treatment of low dose of
erastin induces apoptosis, but not ferroptosis, and results in ROS
accumulation
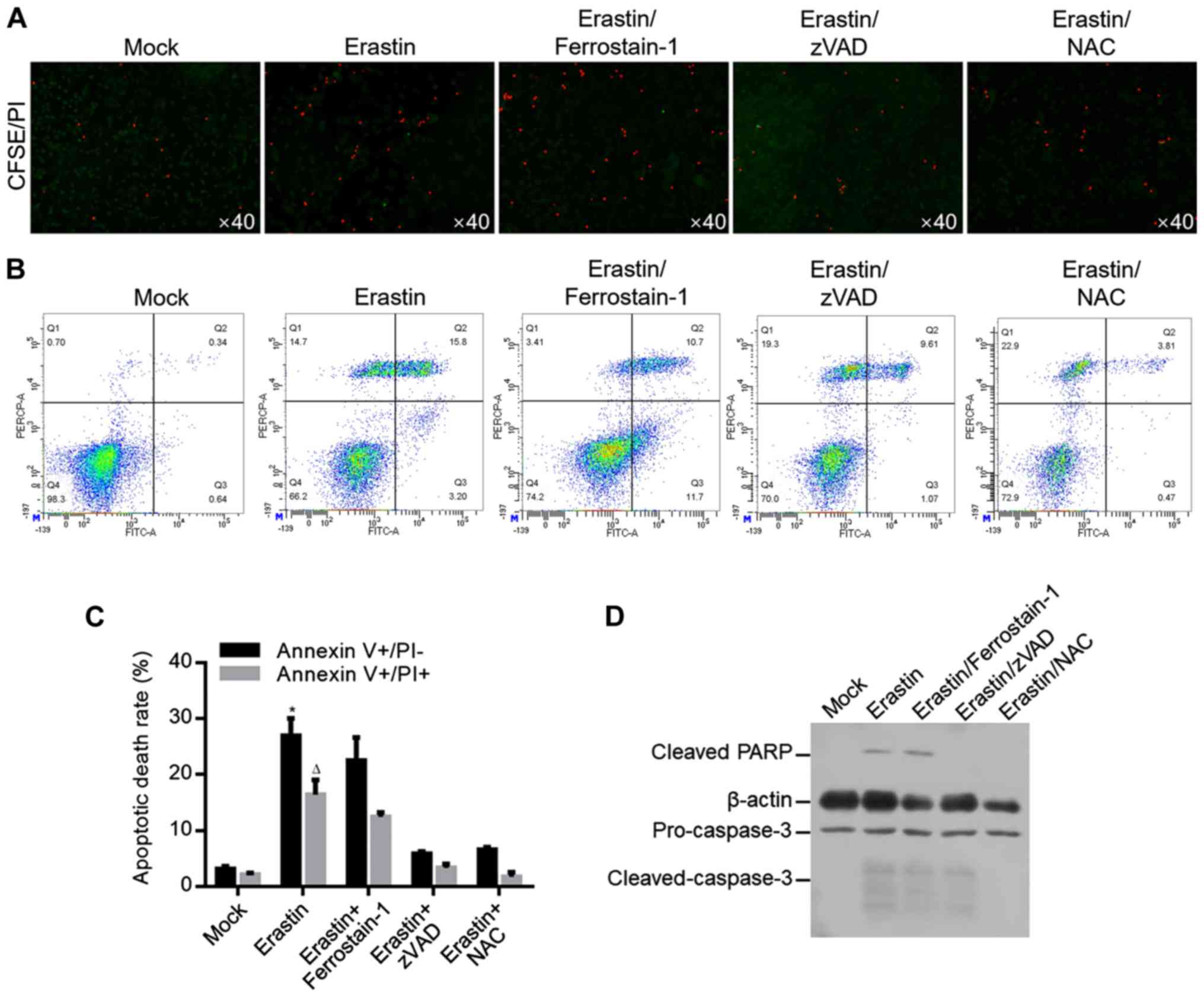
As shown above, 6.23 µM of erastin inhibited
proliferation from 1–5 days (Fig.
1) potentially by causing mitochondrial dysfunction. By
hypothesizing that mitochondrial dysfunction may cause oxidative
stress and subsequently lead to cell death, whether 6.23 µM of
erastin treatment causes cell death in long term was investigated.
Cells treated with 6.23 µM of erastin for 7 days were analyzed by
CFSE/PI double staining. As shown in Fig. 4A, erastin evidently increased
CFSE/PI double-stained cells, which was reversed by apoptosis
inhibitor, zVAD and ROS scavenger NAC, but not by ferroptosis
inhibitor ferrostatin-1, indicating that erastin-induced cell death
occurred mainly by inducing apoptosis, but not by inducing
ferroptosis. For specific analysis of apoptosis, annexin V/PI
double staining was performed and the results showed that erastin
treatment significantly increased proportion of Annexin V+/PI- and
Annexin V+/PI+, which was reversed by zVAD or NAC co-treatment, but
not by ferrostatin-1 co-treatment (Fig. 4B and C). To further confirm
apoptosis induced by erastin, apoptotic hallmarks including cleaved
PARP and cleaved caspase-3 were imaged by western blot and
consistent with previous results, erastin treatment evidently
increased cleaved PARP and cleaved caspase-3 (Fig. 4D).
Discussion
Erastin was first revealed as a small molecule
compound that specifically targets human tumor cells and induces
non-apoptotic cell death without affecting the isogenic normal cell
counterparts (14). The
non-apoptotic cell death induced by erastin was later identified as
ferroptosis, which is a novel form of cell death that is dependent
on the regulation of iron (15).
Researchers have also shown that erastin-induced ferroptosis occurs
mainly by inhibiting the cystine/glutamate antiporter (system
XC−), thus blocking the uptake of cystine and
resulting in the accumulation of ROS (15,16).
Considering the important dual effects of ROS in maintaining the
homeostasis of physiological processes and causing oxidant stress,
the accumulated ROS induced by erastin are thought to be closely
associated with cell behavior.
The present study observed the effects of a
relatively low dose of erastin on HGC-27 gastric cancer cells. To
avoid inducing acute cell death in a ferroptotic manner, the HGC-27
cells were treated with a 30% inhibitory concentration
(IC30) of erastin. Treatment with the 6.23 µM of erastin
resulted in the accumulation of ROS and inhibition of cell
proliferation by blocking the cell cycle at the G1/G0 phase. It
also clearly inhibited the malignant behaviors of HGC-27 cells,
including migration, invasion, colony formation and tumor formation
in soft agar. It has been reported that the inhibition of cystine
and decrease in GSH causes mitochondrial dysfunction by causing ROS
accumulation (17,18); thus, mitochondrial function was
measured. Erastin treatment notably decreased the mitochondrial
potential, transcriptional activity and ATP production, which were
all restored by cotreatment with 10 µM NAC, a ROS scavenger,
demonstrating that the erastin-induced ROS accumulation markedly
depolarized mitochondria and inhibited mitochondrial function.
According to the results, ROS accumulation induced by erastin
successfully induced mitochondrial dysfunction, although whether
mitochondrial mass was affected was unknown (17,18).
It is worth investigating the mitochondrial mass by measuring
mitochondrial genome following erastin treatment.
Erastin-induced ferroptosis has been widely studied;
however, the mechanism of a relatively low dose of erastin
treatment in the long term remains to be elucidated. To this end,
HGC-27 cells were treated with the 6.23 µM of erastin for 1, 3 and
7 days, and cell death was evaluated. As shown in the results,
although there was no evident induction of death at 1 and 3 days
(data not shown), erastin significantly induced cell death after 7
days of treatment, which was not reversed by ferrostatin-1, but was
reversed by zVAD, a mitochondria-related apoptosis inhibitor and
NAC, a ROS scavenger. Annexin V-PI double staining analysis by flow
cytometry and detection of cleaved PARP and cleaved caspase-3,
which are hallmarks of apoptosis, by western blotting further
confirmed that long-term treatment with erastin mainly induced
apoptosis. Although the results focused on apoptosis and failed to
determine whether long-term treatment with a low dose of erastin
induced ferroptosis, the addition of the ferrostatin-1 failed to
clearly affect cell death, indicating the slight effect of low-dose
erastin treatment on inducing ferroptosis.
In summary, the present study determined that
low-dose erastin treatment resulted in ROS accumulation and thus
induced mitochondrial dysfunction. It was also observed that
low-dose erastin treatment decreased the malignant behaviors of
HGC-27 cells in the short term and mainly induced apoptosis, but
not ferroptosis, in the long term. These results may provide an
additional understanding of the antitumor effects of erastin.
Acknowledgements
The authors wish to thank Dr Huimin Shi (Sichuan
University) for language editing.
Funding
The present study was supported by Scientific
Research Startup Fund for Scholars, Medicine School of Medical and
Life Sciences, Chengdu University of Traditional Chinese (grant no.
18110) and Scientific Fund of the Chengdu University of Traditional
Chinese Medicine (grant no. 077035012).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YS, RD and CZ contributed to conception, design and
acquisition of data. CZ contributed to acquisition, analysis and
interpretation of data. YS, RD and CZ contributed to cell culture.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differ. 23:2826–379. 2016. View Article : Google Scholar
|
|
2
|
Yang WS, SriRamaratnam R, Welsch ME,
Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji
AF, Clish CB, et al: Regulation of ferroptotic cancer cell death by
GPX4. Cell. 156:317–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
DeHart DN, Fang D, Heslop K, Li L,
Lemasters JJ and Maldonado EN: Opening of voltage dependent anion
channels promotes reactive oxygen species generation, mitochondrial
dysfunction and cell death in cancer cells. Biochem Pharmacol.
148:155–162. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pan X, Lin Z, Jiang D, Yu Y, Yang D, Zhou
H, Zhan D, Liu S, Peng G, Chen Z and Yu Z: Erastin decreases
radioresistance of NSCLC cells partially by inducing GPX4-mediated
ferroptosis. Oncol Lett. 17:3001–3008. 2019.PubMed/NCBI
|
|
5
|
Schieber M and Chandel NS: ROS function in
redox signaling and oxidative stress. Curr Biol. 24:R453–R462.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
D'Autreaux B and Toledano MB: ROS as
signalling molecules: Mechanisms that generate specificity in ROS
homeostasis. Nat Rev Mol Cell Biol. 8:813–824. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Valko M, Leibfritz D, Moncol J, Cronin MT,
Mazur M and Telser J: Free radicals and antioxidants in normal
physiological functions and human disease. Int J Biochem Cell Biol.
39:44–84. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Trachootham D, Alexandre J and Huang P:
Targeting cancer cells by ROS-mediated mechanisms: A radical
therapeutic approach? Nat Rev Drug Discov. 8:579–591. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ferlay J, Shin HR, Bray F, Forman D,
Mathers C and Parkin DM: Estimates of worldwide burden of cancer in
2008: GLOBOCAN 2008. Int J Cancer. 127:2893–2917. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wagner AD, Grothe W, Haerting J, Kleber G,
Grothey A and Fleig WE: Chemotherapy in advanced gastric cancer: A
systematic review and meta-analysis based on aggregate data. J Clin
Oncol. 24:2903–2909. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Power DG, Kelsen DP and Shah MA: Advanced
gastric cancer-slow but steady progress. Cancer Treat Rev.
36:384–392. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen W, Zou P, Zhao Z, Chen X, Fan X,
Vinothkumar R, Cui R, Wu F, Zhang Q, Liang G and Ji J: Synergistic
antitumor activity of rapamycin and EF24 via increasing ROS for the
treatment of gastric cancer. Redox Biol. 10:78–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dolma S, Lessnick SL, Hahn WC and
Stockwell BR: Identification of genotype-selective antitumor agents
using synthetic lethal chemical screening in engineered human tumor
cells. Cancer Cell. 3:285–296. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dixon SJ, Patel DN, Welsch M, Skouta R,
Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS
and Stockwell BR: Pharmacological inhibition of cystine-glutamate
exchange induces endoplasmic reticulum stress and ferroptosis.
Elife. 3:e25232014. View Article : Google Scholar
|
|
17
|
Diotte NM, Xiong Y, Gao J, Chua BH and Ho
YS: Attenuation of doxorubicin-induced cardiac injury by
mitochondrial glutaredoxin 2. Biochim Biophys Acta. 1793:427–438.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang J, Pan S and Berk BC: Glutaredoxin
mediates Akt and eNOS activation by flow in a glutathione
reductase-dependent manner. Arterioscler Thromb Vasc Biol.
27:1283–1288. 2007. View Article : Google Scholar : PubMed/NCBI
|