Introduction
According to the cancer statistics reported in 2021,
an estimated 1.93 million new colorectal carcinoma (CRC) cases
occurred in 2020, making it the third most common malignancy
globally (1). Although the
development of surgery, chemotherapy, radiotherapy, targeted
therapy and immunotherapy has improved the treatment effectiveness
for patients with CRC, the 5-year overall survival rate of patients
is still poor (2,3). Since the 1990s, 5-fluorouracil
(5-FU) or the 5-FU prodrug capecitabine has become the systemic
treatment for CRC (4).
Subsequently, the combination of 5-FU and leucovorin is the
first-line treatment option for metastatic CRC, which can prolong
the overall 2-year survival of the patients (4). However, more than half of the
patients develop resistance to this treatment (5). Understanding the mechanism may help
overcome drug resistance.
Development of CRC is driven by environmental
exposure and genetic variants. Based on genomics, transcriptomics
and proteomics studies, great progress has been made regarding the
molecular insights of CRC (6). An
inactivation mutation or downregulation of adenomatous polyposis
coli (APC) has been found to contribute to approximately half of
the cases of CRC (7,8). When APC is normally expressed in
cells, it interacts with β-catenin, and regulates GSK3β-mediated
phosphorylation of β-catenin, thus maintaining its protein
stability (9). By contrast, loss
of APC promotes the upregulation of β-catenin, which enters into
the cell nucleus and exerts a transcriptional activation function
(10). Based on these findings,
inhibitors of Wnt/β-catenin, such as LGK-974, are effective for the
treatment of CRC in vitro and in a mouse model (11,12). As mentioned in previous studies,
several WNT ligand antagonists and porcupine inhibitors have been
investigated in clinicals trials of WNT-associated human cancer
types (13–15). However, blockage of Wnt/β-catenin
has not been applied for CRC treatment in clinical settings, mainly
due to the side effects, including impairment of tissue homeostasis
and regeneration, as well as the bioavailability of flavonoids
during cellular metabolism, which need to be resolved (13). Therefore, it is urgent to
investigate the downstream target of the Wnt/β-catenin signaling
cascade, which can promote CRC growth and metastasis.
Pleckstrin 2 (PLEK2) is a 40-kDa protein which has
been primarily identified as a substrate for protein kinase C. It
is widely expressed in multiple organs (16). Recent studies have demonstrated
that overexpression of PLEK2 is associated with the development of
cancer (17–19). The expression levels of PLEK2 are
positively regulated by Janus kinase 2 (JAK2)/STAT5 and are
elevated in philadelphia-chromosome-negative myeloproliferative
neoplasms (MPNs) (20). Depletion
of PLEK2 reverted lethality for MPNs tumorigenesis in
JAK2V617F-knockin mice (21).
Upregulation of PLEK2 has also been observed in gallbladder cancer
(GBC) and serves a tumor-promoting role in GBC via regulation of
the EGFR/C-C motif chemokine ligand 2 signaling pathway (17). In addition, high expression levels
of PLEK2 are associated with the migration and invasion of
non-small cell lung cancer (NSCLC) induced by TGF-β1 (22). These results suggest that PLEK2
may serve a role in tumor growth and metastasis. However, to the
best of our knowledge, the significance of PLEK2 in CRC remains
unclear.
The present study aimed to investigate the
connection between Wnt/APC/β-catenin and PLEK2 and the role of
PLEK2 in CRC growth and chemotherapy. ‘Gain of function’ and ‘loss
of function’ strategies were employed to explore the biological
functions of PLEK2 in CRC cell lines. It was demonstrated that
activation of Wnt/APC/β-catenin signaling stimulated PLEK2
expression, which was upregulated in CRC tissues. Gain and loss of
function assay results revealed that PLEK2 was critical for CRC
cell proliferation and apoptosis. Finally, PLEK2 overexpression
conferred 5-FU resistance to CRC cells. Therefore, PLEK2 may be a
novel target for the treatment of CRC with hyper-activated
Wnt/APC/β-catenin.
Materials and methods
Analysis of PLEK2 expression in
patients with CRC based on The Cancer Genome Atlas (TCGA)
database
Using TCGA (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga),
PLEK2 expression in patients with CRC was analysed. To determine
the clinical relevance of PLEK2 in patients with CRC, the
expression levels of PLEK2 were analyzed in cancer and normal
samples using Gene Expression Profiling Interactive Analysis
(GEPIA2021; http://gepia.cancer-pku.cn/) (23). A total of 257 colon adenocarcinoma
samples and 349 normal tissues were included in the analysis.
Cell culture
Human normal colorectal cells (HIEC) and human CRC
cells (RKO and HCT-116) were purchased from American Type Culture
Collection. The cells were cultured in RPMI-1640 complete medium
(Gibco; Thermo Fisher Scientific, Inc.), supplemented with 10% FBS
(Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin solution (Corning, Inc.). The cell culture
was kept in a cell incubator at 37°C with 5% CO2. For
the 5-FU treatment, cells were treated with different
concentrations (0, 15.4, 38.5, 77, 308 and 616 µM) of 5-FU (cat.
no. HY-90006; MedChemExpress) for 48 h at 37°C. DMSO was used as a
vehicle to dissolve 5-FU.
APC, catenin β1 (β-catenin) and PLEK2
knockdown using small interfering RNA (siRNA/si)
siRNAs were synthesized by Huzhou Hippo
Biotechnology Co., Ltd. siRNAs were dissolved in RNase-free water.
Cells were seeded in 6-well plates at a density of 5×106 cells/well
and then transfected with 10 nM siRNAs (scrambled siCtrl,
5′-UUCUCCGAACGUGUCACGU-3′; siAPC, 5′-GGAAUCAACCCUCAAAAGU-3′;
siβ-catenin, 5′-CACTAACCAAGCTGAGTTT-3′; siPLEK2#1,
5′-ACCUCUUCAAAGUGAUUACUA-3′; and siPLEK2#2,
5′-CCAGCUUUCCUGCAUUACUAU-3′) using RNAiMAX (Invitrogen; Thermo
Fisher Scientific, Inc.). Transfection was performed for 48 h at
37°C. Total RNA and protein were harvested after 48 h. Cellular
function assays, including Cell Counting Kit-8 (CCK8), colony
formation, apoptosis and cell cycle assays, were performed at 48 h
after siRNA transfection.
PLEK2 overexpression
To overexpress PLEK2, the coding sequence of PLEK2
was cloned into pCDNA3.1 vectors (Beijing Tianyi Huiyuan
Biotechnology Co., Ltd.). HCT-116 and RKO cells were seeded into
6-well plates at a density of 5×105 cells per well.
After 12 h, the cells were transfected with 5 µg pCDNA3.1-Ctrl and
pCDNA3.1-PLEK2 vectors using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) for 48 h at 37°C.
Subsequently, PLEK2 overexpression was checked using immunoblotting
and the cells were subjected to cellular biology function
assessment.
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR)
To detect mRNA expression, total RNA was isolated
from CRC cells using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's protocol.
mRNA was reverse transcribed into cDNA using the reverse
transcriptional kit (Promega Corporation) according to the
manufacturer's protocol. Subsequently, mRNA expression was
quantified by detecting its cDNA abundance using SYBR Green master
mixture (Qiagen, Inc.) on a Biorad qPCR system (Bio-Rad
Laboratories, Inc.). The following steps were used for qPCR:
Initial denaturation at 95°C for 5 min; followed by 40 cycles of
95°C for 15 sec, 60°C for 30 sec and 70°C for 10 sec. The qPCR
primer sequences were as follows: PLEK2 forward,
5′-CCGAAGCATGGGAGCCATT-3′ and reverse,
5′-AGTGCTCAGGCTAATTTCTTCC-3′; β-catenin forward,
5′-AGCTTCCAGACACGCTATCAT-3′ and reverse,
5′-CGGTACAACGAGCTGTTTCTAC-3′; and GAPDH forward,
5′-TGACTTCAACAGCGACACCCA-3′ and reverse,
5′-CACCCTGTTGCTGTAGCCAAA-3. GAPDH was used as an internal control.
The 2−∆∆Cq method was used to calculate relative
expression (24).
Immunoblotting assay
Protein was obtained from CRC cells using RIPA
buffer (Beyotime Institute of Biotechnology) supplemented with
protease inhibitor cocktail. The concentration of total protein was
checked using a BCA assay kit (Beyotime Institute of
Biotechnology). Nuclear fractions of the total protein were
separated using NE-PER Nuclear and Cytoplasmic Extraction Reagent
(Thermo Fisher Scientific, Inc.). Subsequently, proteins (30 µg)
were subjected to immunoblotting experiments according to the
procedures described previously (25). Briefly, proteins were separated
via 12% SDS-PAGE and then transferred to PVDF membranes. After
transfection, the membranes were blocked with 5% skim milk for 2 h
at room temperature. Then, the membranes were incubated with the
primary antibodies at 4°C overnight. Subsequently, the membranes
were incubated with the secondary antibodies for 2 h at room
temperature. The following primary and secondary antibodies were
used: PLEK2 (1:1,000; cat. no. ab121131; Abcam), APC (1:1,000; cat.
no. 2504; Cell Signaling Technology, Inc.), β-catenin (1:1,000;
cat. no. 8480; Cell Signaling Technology, Inc.), GAPDH (1:1,000;
cat. no. 2118; Cell Signaling Technology, Inc.), Histone H3
(1:1,000; cat. no. 4499; Cell Signaling Technology, Inc.),
anti-rabbit IgG HRP-conjugated (1:2,000; cat. no. 7074; Cell
Signaling Technology, Inc.) and anti-mouse HRP-conjugated (1:2,000;
cat. no. 7076; Cell Signaling Technology, Inc.). The proteins were
detected using Pierce™ ECL Western Blotting substrate (Thermo
Fisher Scientific, Inc.) and analyzed using Image Lab 6.1 software
(Bio Rad Laboratories, Inc.).
CCK8 assay
The proliferation rate of CRC cells was determined
using a CCK8 assay. In brief, 2,000 CRC cells were seeded into
96-well plates supplemented with 100 µl RPMI-1640 complete medium.
After 1, 2, 3 and 4 days, 10 µl CCK8 (Beyotime Institute of
Biotechnology) reagent was added into each well and the cells were
incubated for 3 h in the cell incubator. Subsequently, the optical
density value at 450 nm was measured to assess cell
proliferation.
Colony formation
Colony formation was examined by seeding a density
of 1.5×103 CRC cells/well into 6-well plates. After
transfection with siRNAs for 48 h at 37°C, cell colonies were
cultured for 10 days. Then the culture medium was removed, and the
cells were washed with PBS, fixed with 100% methanol at room
temperature for 15 min and stained with 0.2% crystal violet
solution at room temperature for 30 min. Images were captured using
a camera (Nikon Corporation) to count the cell colonies (>50
cells) manually.
Cell cycle analysis
To detect the cell cycle distribution, the cells
were first fixed with iced 70% ethanol overnight at 4°C and then
stained with PI using the Cell Cycle and Apoptosis Analysis kit
(Shanghai Yeasen Biotechnology Co., Ltd.) according to the
manufacturer's instructions. Subsequently, cell cycle distribution
was analyzed using a flow cytometry system (cytoFlex; Beckman
Coulter, Inc.). Data were analyzed using Cytexpert software
(version 2.4.0.28; Beckman Coulter, Inc.).
Cell apoptosis detection
To examine cell apoptosis, CRC cells were carefully
collected using EDTA-free trypsin (Thermo Fisher Scientific, Inc.).
Subsequently, cells were washed with PBS and stained with PI and
Annexin V using the Annexin V-FITC/PI Apoptosis Detection kit
(Shanghai Yeasen Biotechnology Co., Ltd.) according to the
manufacturer's instructions. Cell apoptosis was analyzed using a
flow cytometry system (cytoFlex; Beckman Coulter, Inc.). Data were
analyzed using Cytexpert software (version 2.4.0.28; Beckman
Coulter, Inc.).
Luciferase reporter assay
Genomic DNA was extracted from HCT116 cells using
genomic DNA extraction kit (cat. no. ab156900; Abcam). For binding
assessment of transcription factor β-catenin to the PLEK2 gene
promoter, the promoter sequence of PLEK2 was amplified by PCR assay
using a Taq kit (New England Biolabs). The primers of PLEK2 were as
follows: Forward, 5′-TGCCAAAGAAAATGCCACTTCTTCTAAGCCTCAG-3′ and
reverse, 5′-AGCTCCGACGCGGCAGGG-3′. The thermocycling conditions
were as follows: Initial denaturation at 95°C for 30 sec;
denaturation at 95°C for 15 sec, annealing at 60°C for 30 sec and
extension at 68°C for 2 min for 39 cycles; a final extension at
68°C for 5 min; and then hold at 4°C. The PCR product was cloned
into the luciferase reporter vector, psi-CHECK vector (Promega
Corporation). Subsequently, the luciferase reporter vectors and
siRNAs of β-catenin were transfected into RKO and HCT116 cells
using Lipo2000 (Invitrogen; Thermo Fisher Scientific, Inc.). At 48
h post transfection, the relative luciferase activity was examined
using a dual luciferase reporter assay system (Promega Corporation)
according to the manufacturer's instructions. The firefly
luciferase activity was normalized to Renilla luciferase
activity.
Statistical analysis
Statistical data are presented as the mean ± SEM and
were analyzed using GraphPad prism software (v8.0; GraphPad
Software, Inc.). The experiments were repeated at least three
times. Differences between two groups were analyzed by unpaired
Student's t-test. Differences among three groups were analyzed by
one-way ANOVA followed by Tukey's post hoc test. Linear regression
analysis was performed to analyze the Spearman correlation between
PLEK2 and β-catenin using the GEPIA2021 database (http://gepia.cancer-pku.cn/) (23). P<0.05 was considered to
indicate a statistically significant difference.
Results
APC/β-catenin upregulates PLEK2 in CRC
cells and patients with CRC
To determine whether APC/β-catenin regulates PLEK2
in CRC cells, APC was knocked down in HCT-116 cells. Immunoblotting
results demonstrated that APC was efficiently silenced after
transfection of HCT-116 cells with siRNAs against APC (Fig. 1A). In addition, APC knockdown
stimulated PLEK2 expression at both the mRNA and protein levels
(Fig. 1A-C). Furthermore,
knockdown of APC did not increase the total β-catenin expression
but increased its nuclear translocation (Fig. 1A and B). To investigate whether
APC regulation of PLEK2 was dependent on upregulation of β-catenin,
CTNNB1, which encodes β-catenin, was further knocked down in
APC-silenced HCT-116 cells. β-catenin was effectively knocked down
using siRNA as detected by RT-qPCR (Fig. S1). The western blotting results
suggested that upregulation of PLEK2 was reduced after silencing of
β-catenin (Fig. 1A). Consistent
results were observed in APC-knockdown HCT-116 cells (Fig. 1C). To check whether β-catenin
modulates the transcription activity of PLEK2, a dual-luciferase
activity assay was performed and revealed that downregulation of
β-catenin reduced the luciferase activity compared with the control
group (Fig. 1D). These results
suggested that β-catenin binds to the promoter sequence of PLEK2 to
activate APC/β-catenin and then upregulate PLEK2 at the
transcriptional level.
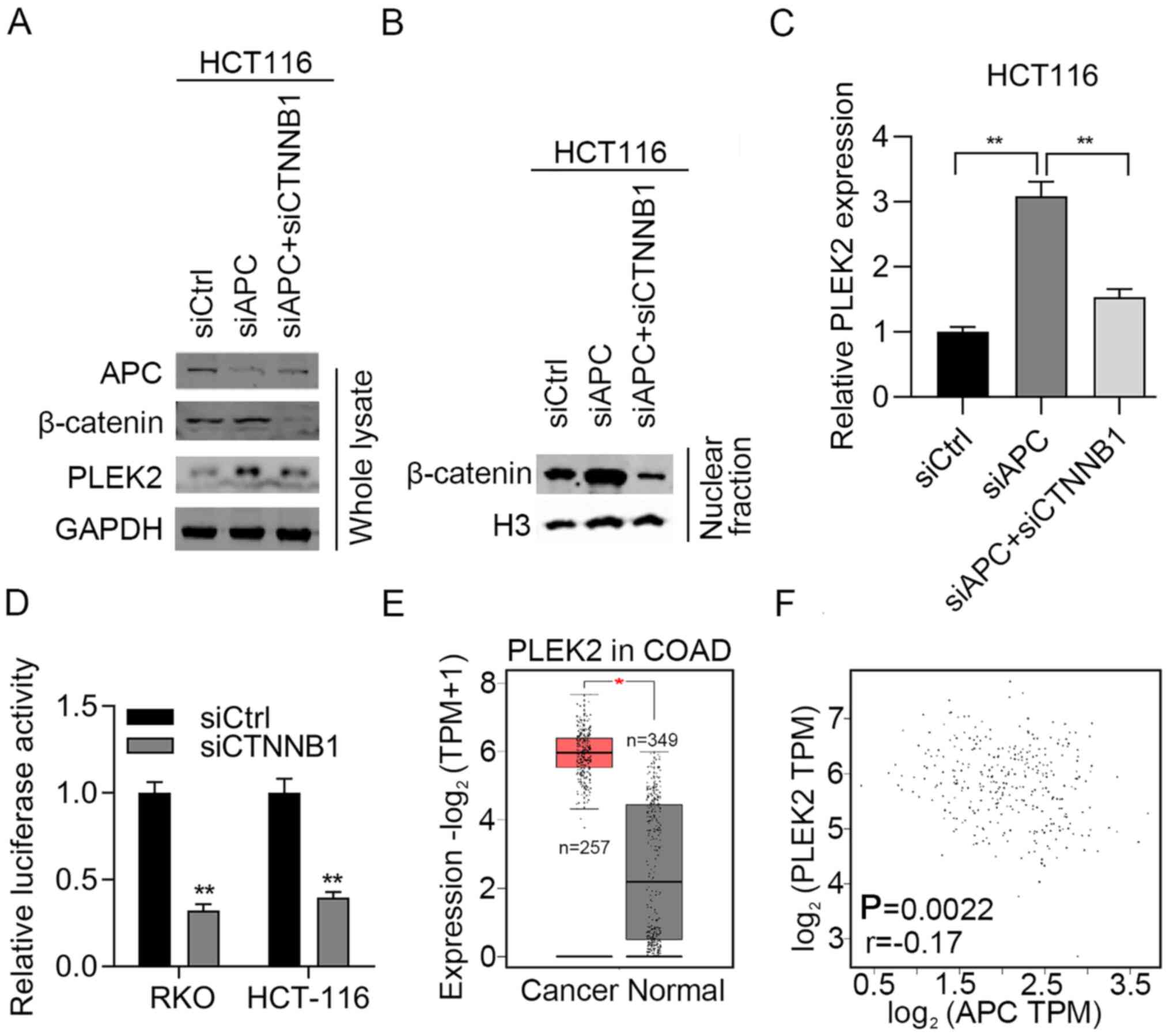 | Figure 1.APC/β-catenin activates PLEK2 in CRC
cells and patients with CRC. (A) HCT-116 cells were transfected
with siCtrl and siAPC. The cells were subjected to immunoblotting
analysis of APC, β-catenin and PLEK2. (B) HCT-116 cells were
transfected with siCtrl and siAPC. The nuclear extract was
subjected to immunoblotting analysis of β-catenin. The expression
levels of indicated genes were normalized to H3. (C) HCT-116 cells
were transfected with siCtrl, siAPC or siAPC + β-catenin. The cells
were subjected to reverse transcription-quantitative PCR analysis
of PLEK2 expression. The expression levels of indicated genes were
normalized to GAPDH. **P<0.01. (D) RKO and HCT-116 cells were
transfected with pGL3. Basic vectors, which contained the PLEK2
promoter sequence, thymidine kinase TK vectors, as well as siCtrl
or siβ-catenin were co-transfected into RKO or HCT-116 cells. Dual
luciferase activity was examined in RKO and HCT-116 cells
transfected with siCtrl and siβ-catenin. **P<0.01 vs. siCtrl.
(E) Expression levels of PLEK2 were analyzed in CRC and normal
tissues from TCGA. *P<0.05. (F) Spearman correlation between APC
and PLEK2 expression in CRC tissues from TCGA. APC, adenomatous
polyposis coli; COAD, colon adenocarcinoma; CRC, colorectal
carcinoma; CTNNB1, catenin β1; PLEK2, pleckstrin 2; si/siRNA, small
interfering RNA; siCtrl, negative control siRNA; TCGA, The Cancer
Genome Atlas; TPM, transcripts per million. |
Subsequently, the present study aimed to explore the
clinical relevance of APC and PLEK2 in CRC. First, the expression
levels of PLEK2 in patients with CRC were explored using TCGA. A
total of 257 CRC tissues and 349 normal tissues were included in
the analysis. The results demonstrated that PLEK2 was significantly
upregulated in CRC tissues compared with normal tissues (Fig. 1E). Furthermore, APC expression was
inversely associated with PLEK2 expression in patients with CRC
(Fig. 1F). Therefore, negative
regulation of PLEK2 by APC may exist in patients with CRC.
PLEK2 promotes CRC cell
proliferation
The present study next assessed whether PLEK2
modulated CRC cell function by assessing cell proliferation and
growth. To this aim, the present study firstly examined the basic
expression levels of PLEK2 in colorectal normal cells (HIEC) and
CRC cell lines (RKO and HCT-116). PLEK2 was highly expressed in RKO
and HCT-116 cells compared with HIEC cells (Fig. 2A). PLEK2 was knocked down and
overexpressed in HCT-116 and RKO cells, respectively.
Immunoblotting results validated the knockdown and overexpression
efficiency (Fig. 2B and C). As
indicated by CCK8 assay results, PLEK2 knockdown reduced the
proliferation of HCT-116 and RKO cells, while PLEK2 overexpression
promoted both HCT-116 and RKO cell proliferation (Fig. 2D and E). In addition, the colony
formation of CRC cells was suppressed by PLEK2 knockdown and
promoted by PLEK2 overexpression (Fig. 2F and G). Therefore, PLEK2
upregulation in CRC may serve tumor-promoting roles.
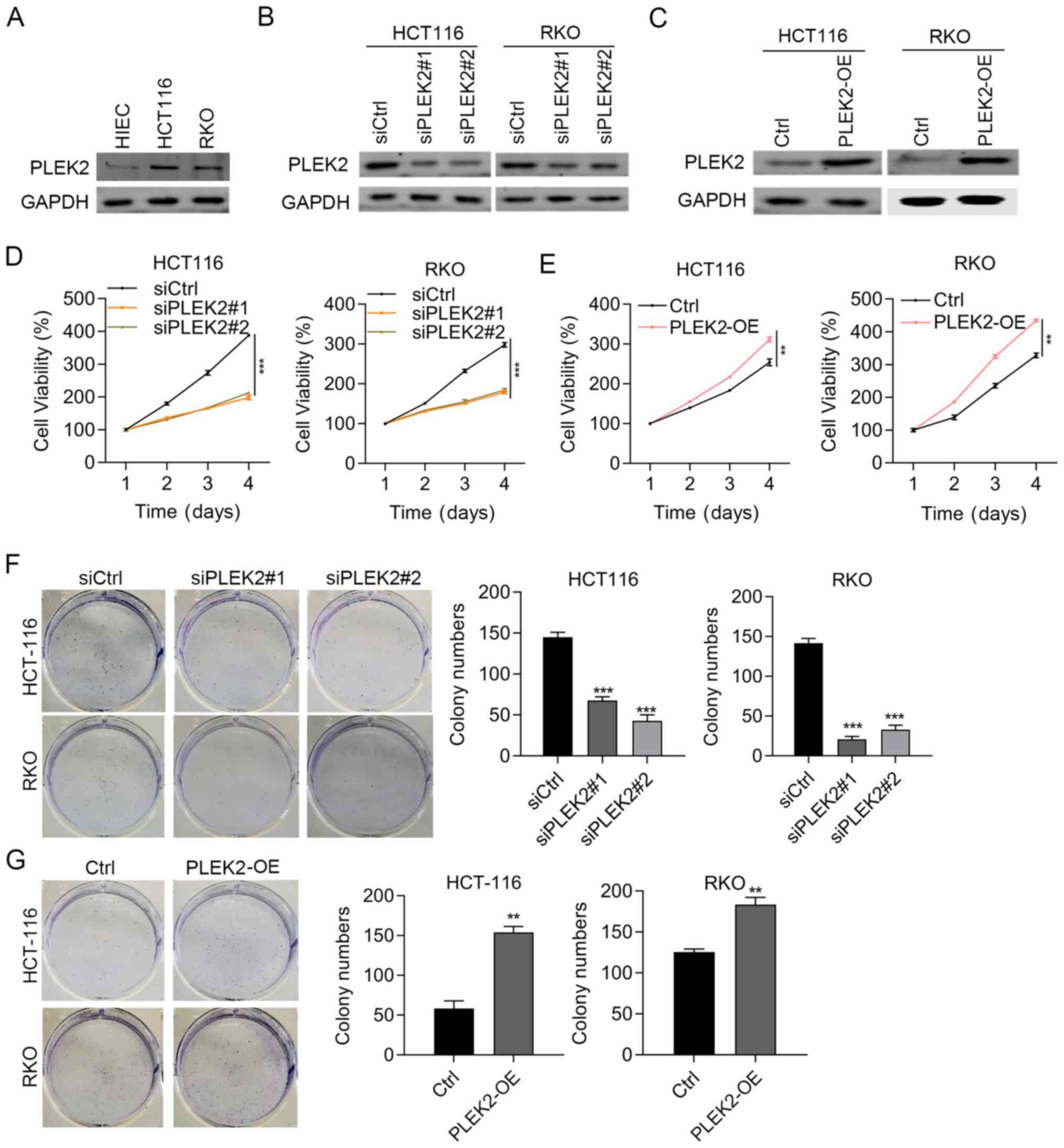 | Figure 2.PLEK2 overexpression reinforces
cellular functions in colorectal carcinoma cells. (A)
Immunoblotting detection of PLEK2 in HIEC, RKO and HCT-116 cells.
GAPDH was used as a loading control. Immunoblotting results of
PLEK2 in (B) HCT116 and RKO cells transfected with siCtrl,
siPLEK2#1 and siPLEK2#2, and (C) in Ctrl- and PLEK2-OE HCT-116 and
RKO cells. GAPDH was used as a loading control. Cell proliferation
was evaluated in (D) HCT116 and RKO cells transfected with siCtrl,
siPLEK2#1 and siPLEK2#2, and (E) in Ctrl- and PLEK2-OE HCT-116 and
RKO cells. **P<0.01 and ***P<0.001. Colony formation assay of
(F) HCT-116 and RKO cells transfected with siCtrl, siPLEK2#1 and
siPLEK2#2, and (G) Ctrl- and PLEK2-OE HCT-116 and RKO cells.
**P<0.01 and ***P<0.001 vs. siCtrl or Ctrl. Ctrl, empty
vector; PLEK2-OE, pleckstrin 2 overexpression; si/siRNA, small
interfering RNA; siCtrl, negative control siRNA. |
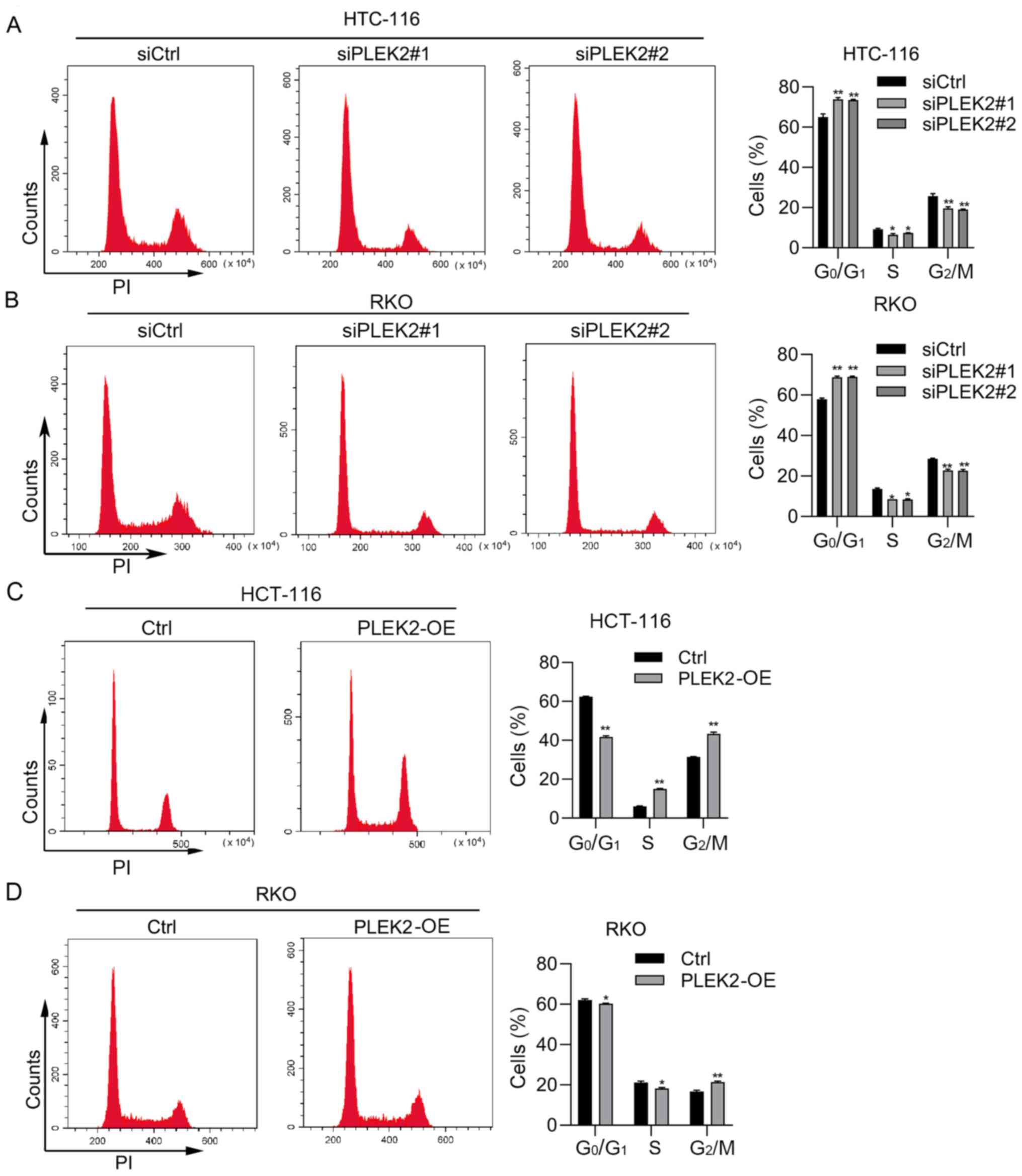
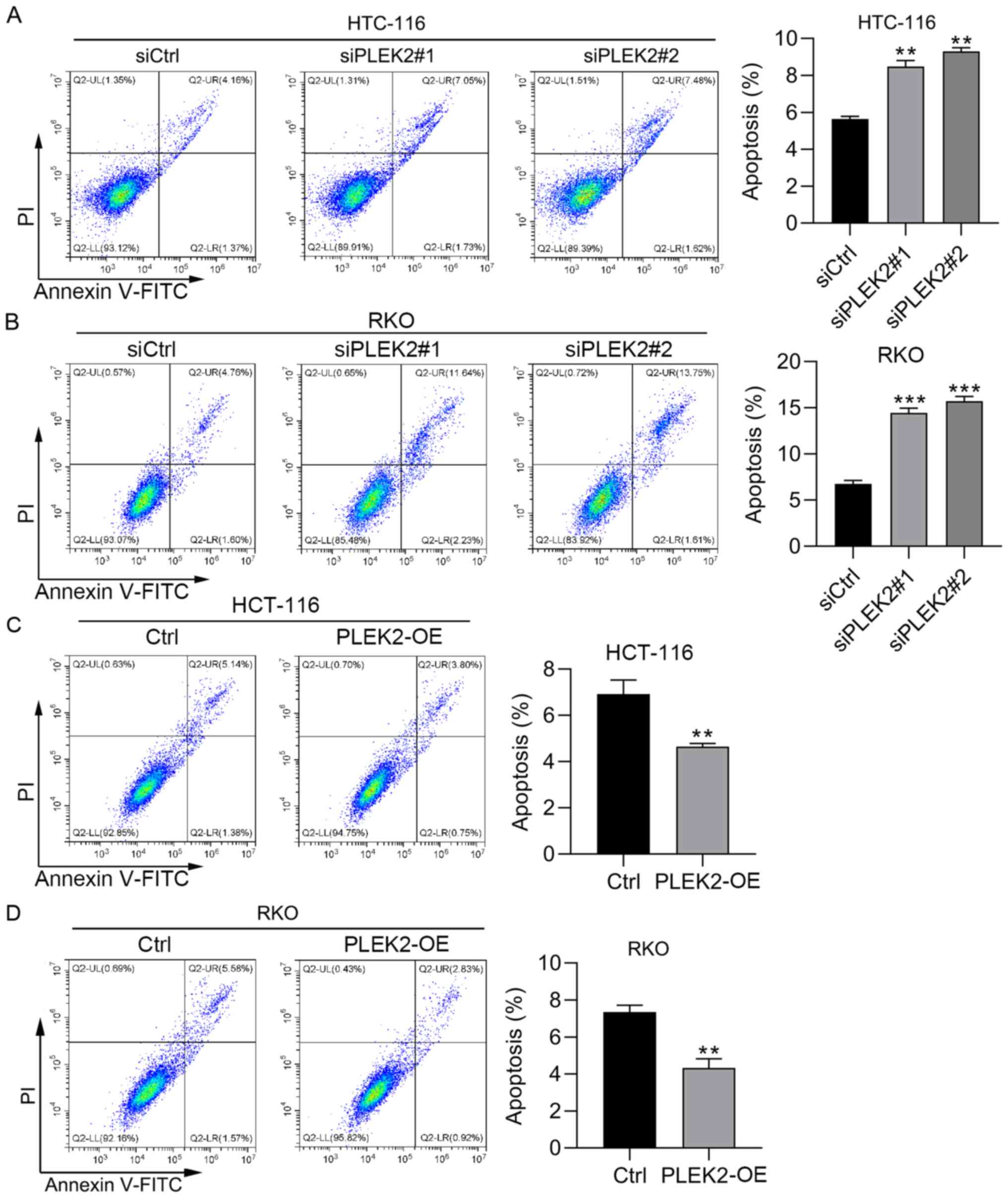
PLEK2 regulates the cell cycle and
apoptosis in CRC cells
To further determine the function of PLEK2, PI
staining and PI/Annexin V staining were performed to detect the
cell cycle distribution and apoptosis in CRC cells with PLEK2
knockdown or overexpression. Based on PI staining analysis of the
cell cycle distribution using flow cytometry, PLEK2 knockdown
promoted HCT-116 and RKO cell cycle arrest at
G0/G1 phase, while overexpression of PLEK2
decreased the CRC cell percentage at G0/G1
phase (Fig. 3A-D). PI/Annexin V
staining results demonstrated that PLEK2 knockdown induced
apoptosis in HCT-116 and RKO cells (Fig. 4A and B). Furthermore,
overexpression of PLEK2 suppressed HCT-116 and RKO cell apoptosis
(Fig. 4C and D). Collectively,
PLEK2 suppressed cell apoptosis and promoted cell cycle progression
in CRC.
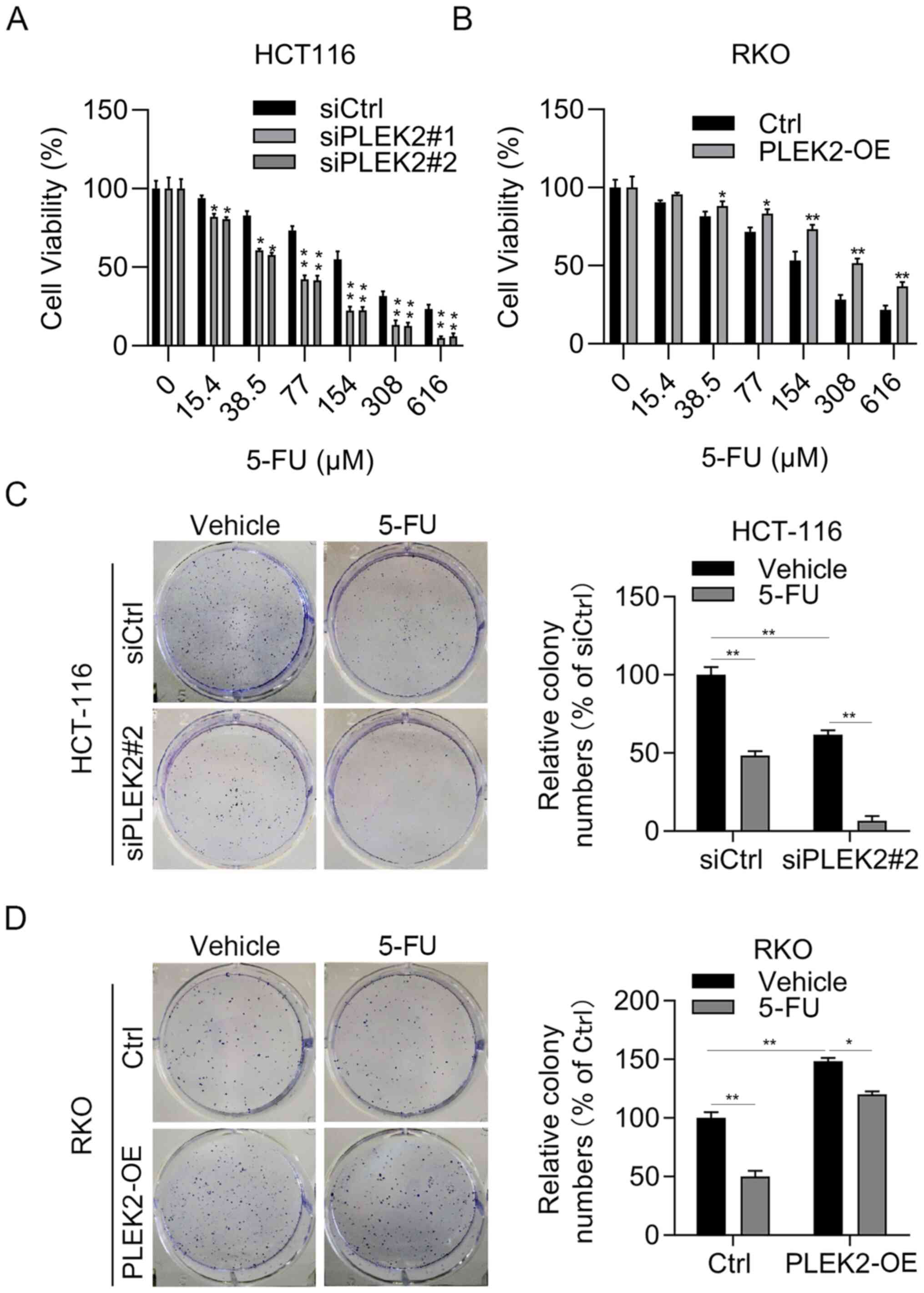
PLEK2 overexpression confers 5-FU
resistance in CRC cells
Chemotherapy resistance is a clinical problem for
patients with CRC. 5-FU represents the most commonly used
chemotherapy drug for patients with CRC (26). Therefore, the present study next
attempted to investigate whether PLEK2 regulated the sensitivity of
CRC cells to 5-FU. Firstly, CRC cells were incubated with different
concentrations of 5-Fu for 48 h. The results demonstrated that
PLEK2 knockdown enhanced the cytotoxicity of 5-FU in HCT-116 cells
(Fig. 5A), while overexpression
of PLEK2 decreased the cytotoxicity of 5-FU in RKO cells (Fig. 5B). Since the cell viability assay
results demonstrated that 5-FU inhibited cell proliferation at 15.4
µM in HCT-116 cells and at 38.5 µM in RKO cells, respectively,
these concentrations were selected for subsequent experiments.
Similarly, a colony formation assay indicated that downregulation
of PLEK2 obviously enhanced the anti-growth effect of 5-FU at 15.4
µM in HCT-116 cells, while PLEK2 overexpression significantly
reduced the toxicity of 5-FU at 38.5 µM in RKO cells (Fig. 5C and D). Overall, PLEK2 promoted
5-FU resistance in CRC cells.
Discussion
The present study demonstrated that upregulation of
PLEK2 not only acted as an oncogene activated by APC/β-catenin
signaling at the transcriptional level but also as a predictor for
5-FU resistance in CRC. Based on knockdown and overexpression
results, PLEK2 contributed to the proliferation of CRC cells. PLEK2
also regulated cell cycle progression and cell apoptosis.
Furthermore, PLEK2 promoted 5-FU resistance in CRC cells. The
present study demonstrated that APC/β-catenin upregulation of PLEK2
contributes to CRC cell proliferation and 5-FU resistance.
Abundant evidence has demonstrated that the tumor
suppressor gene APC is commonly mutated or absent in CRC (27–29). Inactivation of APC promotes the
protein stability of β-catenin by regulating GSK3β-mediated
phosphorylation of β-catenin, resulting in activation of the
Wnt/β-catenin signaling pathway (30). Stable β-catenin then enters into
the cell nucleus and exerts transcription factor functions.
Numerous targets, including guanine nucleotide-binding protein,
axin 2, leucine rich repeat containing G protein-coupled receptor 5
and NKD inhibitor of WNT signaling pathway, are activated by
β-catenin during the development of cancer (31). During the past years, large
amounts of effort have been focused on the exploration of small
molecule inhibitors to specifically target the activity of the
Wnt/β-catenin signaling cascade (14). However, the side effects, such as
tissue homeostasis and regeneration impairment, caused by these
inhibitors limit their application for the treatment of patients
with CRC (26). Therefore, it is
urgent to identify novel downstream effectors of Wnt/β-catenin to
enable the development of effective drugs with limited side
effects. The present study revealed that knockdown of APC promoted
β-catenin and PLEK2 expression. Inhibition of β-catenin
downregulated PLEK2 expression in CRC cells at both the mRNA and
protein levels. Notably, As APC promoted β-catenin through
regulating GSK3β-mediated phosphorylation (25), the present study demonstrated that
β-catenin directly regulated the transcription activity of PLEK2
promoter as demonstrated by a dual luciferase activity assay. Thus,
these results demonstrated that APC suppressed the expression of
PLEK2 and β-catenin.
PLEK2 is a paralog of pleckstrin that can bind to
polyphosphoinositides and regulate actin rearrangement (16,32,33). Dysregulation of PLEK2 is involved
in the development of different cancer types (17,34). For instance, transcriptome
analysis has demonstrated that PLEK2 is highly expressed in the
whole blood cells from patients with melanoma (35). In MPNs, depletion of PLEK2
alleviates JAK2V617F-triggered malignant phenotypes in a transgenic
mouse model (21). PLEK2
expression is upregulated in patients with GBC, whereas
overexpression of PLEK2 promotes GBC carcinogenesis by potentiating
PLEK2/EGFR signaling (17). In
addition, PLEK2 is highly expressed in lung adenocarcinoma samples
and associated with poor prognosis of the patients (34). Upregulation of PLEK2 also serves a
tumor-promoting role in NSCLC via regulation of the
ubiquitination-dependent stabilization of SH2 domain containing
inositol 5-phosphatase 2 (22).
Although PLEK2 is upregulated with activation of APC/β-catenin
signaling in CRC cells and patients with CRC, to the best of our
knowledge, the role of PLEK2 in CRC remains undetermined. The
present study demonstrated that PLEK2 overexpression resulted in
enhanced cell cycle progression, reduced cell apoptosis and
increased cell proliferation. Furthermore, PLEK2 overexpression
conferred resistance to 5-FU in CRC cells. This suggested that
PLEK2 serves an oncogenic role in CRC.
In conclusion, the present study demonstrated that
PLEK2 served as a downstream substrate of the APC/β-catenin
signaling cascade. Overexpression of PLEK2 contributed to the
proliferation of CRC cells. Furthermore, PLEK2 was associated with
the sensitivity to 5-FU treatment in CRC cells. Therefore,
APC/β-catenin activation of PLEK2 may contribute to CRC growth and
chemotherapy resistance.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was financially supported by
Shanxi Natural Science Foundation (grant nos. 201701D221176 and
201901D111413), The Health and Family Planning Commission of Shanxi
Province Scientific Research Project (grant no. 201601032), and The
Doctor Start-up Fund of The First Hospital of Shanxi Medical
University (grant no. YB161705).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YG designed the present study. BC carried out most
of the experiments. NZ and JJ performed quantitative PCR analysis.
ZZ and MP performed western blot analysis. KJ, YS and XC analyzed
and interpretated data. YG and BC confirmed the authenticity of all
the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
No authors listed. Erratum: Global cancer
statistics 2018: GLOBOCAN estimates of incidence and mortality
worldwide for 36 cancers in 185 countries. CA Cancer J Clin.
70:3132020.Erratum for: CA Cancer J Clin 68: 394-424, 2018.
View Article : Google Scholar
|
|
2
|
Dekker E, Tanis PJ, Vleugels JLA, Kasi PM
and Wallace MB: Colorectal cancer. Lancet. 394:1467–1480. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Meyerhardt JA and Mayer RJ: Systemic
therapy for colorectal cancer. N Engl J Med. 352:476–487. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gustavsson B, Carlsson G, Machover D,
Petrelli N, Roth A, Schmoll HJ, Tveit KM and Gibson F: A review of
the evolution of systemic chemotherapy in the management of
colorectal cancer. Clin Colorectal Cancer. 14:1–10. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Van der Jeught K, Xu HC, Li YJ, Lu XB and
Ji G: Drug resistance and new therapies in colorectal cancer. World
J Gastroenterol. 24:3834–3848. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rattray NJW, Charkoftaki G, Rattray Z,
Hansen JE, Vasiliou V and Johnson CH: Environmental influences in
the etiology of colorectal cancer: The premise of metabolomics.
Curr Pharmacol Rep. 3:114–125. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Caspi M, Wittenstein A, Kazelnik M,
Shor-Nareznoy Y and Rosin-Arbesfeld R: Therapeutic targeting of the
oncogenic Wnt signaling pathway for treating colorectal cancer and
other colonic disorders. Adv Drug Deliv Rev. 169:118–136. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carethers JM and Jung BH: Genetics and
Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology.
149:1177–1190.e3. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shang S, Hua F and Hu ZW: The regulation
of β-catenin activity and function in cancer: Therapeutic
opportunities. Oncotarget. 8:33972–33989. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bian J, Dannappel M, Wan C and Firestein
R: Transcriptional Regulation of Wnt/β-Catenin Pathway in
Colorectal Cancer. Cells. 9:21252020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cho YH, Ro EJ, Yoon JS, Mizutani T, Kang
DW, Park JC, Il Kim T, Clevers H and Choi KY: 5-FU promotes
stemness of colorectal cancer via p53-mediated WNT/β-catenin
pathway activation. Nat Commun. 11:53212020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Y, Wang R, Huang D, Ma X, Mo S, Guo Q,
Fu G, Li Y, Xu X, Hu X, et al: A novel human colon signet-ring cell
carcinoma organoid line: Establishment, characterization and
application. Carcinogenesis. 41:993–1004. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jung YS and Park JI: Wnt signaling in
cancer: Therapeutic targeting of Wnt signaling beyond β-catenin and
the destruction complex. Exp Mol Med. 52:183–191. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Krishnamurthy N and Kurzrock R: Targeting
the Wnt/beta-catenin pathway in cancer: Update on effectors and
inhibitors. Cancer Treat Rev. 62:50–60. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhan T, Rindtorff N and Boutros M: Wnt
signaling in cancer. Oncogene. 36:1461–1473. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hu MH, Bauman EM, Roll RL, Yeilding N and
Abrams CS: Pleckstrin 2, a widely expressed paralog of pleckstrin
involved in actin rearrangement. J Biol Chem. 274:21515–21518.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shen H, He M, Lin R, Zhan M, Xu S, Huang
X, Xu C, Chen W, Yao Y, Mohan M and Wang J: PLEK2 promotes
gallbladder cancer invasion and metastasis through EGFR/CCL2
pathway. J Exp Clin Cancer Res. 38:2472019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang J, He Z, Sun B, Huang W, Xiang J,
Chen Z, Li Z and Gu X: Pleckstrin-2 as a Prognostic Factor and
Mediator of Gastric Cancer Progression. Gastroenterol Res Pract.
2021:55273872021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang J, Sun Z, Wang J, Tian Q, Huang R,
Wang H, Wang X and Han F: Expression and prognostic potential of
PLEK2 in head and neck squamous cell carcinoma based on
bioinformatics analysis. Cancer Med. 10:6515–6533. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cacemiro MDC, Cominal JG, Tognon R, Nunes
NS, Simões BP, Figueiredo-Pontes LL, Catto LFB, Traina F, Souto EX,
Zambuzi FA, et al: Philadelphia-negative myeloproliferative
neoplasms as disorders marked by cytokine modulation. Hematol
Transfus Cell Ther. 40:120–131. 2018.Erratum in: Hematol Transfus
Cell Ther 43: 117, 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao B, Mei Y, Cao L, Zhang J, Sumagin R,
Yang J, Gao J, Schipma MJ, Wang Y, Thorsheim C, et al: Loss of
pleckstrin-2 reverts lethality and vascular occlusions in
JAK2V617F-positive myeloproliferative neoplasms. J Clin Invest.
128:125–140. 2018. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu DM, Deng SH, Zhou J, Han R, Liu T,
Zhang T, Li J, Chen JP and Xu Y: PLEK2 mediates metastasis and
vascular invasion via the ubiquitin-dependent degradation of SHIP2
in non-small cell lung cancer. Int J Cancer. 146:2563–2575.
2020.Erratum in: Int J Cancer 149: E11, 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weinstein JN, Collisson EA, Mills GB, Shaw
KR, Ozenberger BA, Ellrott K, Shmulevich I, Sander C and Stuart JM;
Cancer Genome Atlas Research Network, : The Cancer Genome Atlas
Pan-Cancer analysis project. Nat Genet. 45:1113–1120. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chai B, Guo Y, Cui X, Liu J, Suo Y, Dou Z
and Li N: MiR-223-3p promotes the proliferation, invasion and
migration of colon cancer cells by negative regulating PRDM1. Am J
Transl Res. 11:4516–4523. 2019.PubMed/NCBI
|
|
26
|
Xie YH, Chen YX and Fang JY: Comprehensive
review of targeted therapy for colorectal cancer. Signal Transduct
Target Ther. 5:222020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Michels BE, Mosa MH, Streibl BI, Zhan T,
Menche C, Abou-El-Ardat K, Darvishi T, Członka E, Wagner S, Winter
J, et al: Pooled In Vitro and In Vivo CRISPR-Cas9 Screening
Identifies Tumor Suppressors in Human Colon Organoids. Cell Stem
Cell. 26:782–792.e7. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ragusa S, Prat-Luri B, González-Loyola A,
Nassiri S, Squadrito ML, Guichard A, Cavin S, Gjorevski N, Barras
D, Marra G, et al: Antiangiogenic immunotherapy suppresses
desmoplastic and chemoresistant intestinal tumors in mice. J Clin
Invest. 130:1199–1216. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang L and Shay J: Multiple Roles of APC
and its Therapeutic Implications in Colorectal Cancer. J Natl
Cancer Inst. 109:djw3322017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gao C, Xiao G and Hu J: Regulation of
Wnt/β-catenin signaling by posttranslational modifications. Cell
Biosci. 4:132014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang B, Mao L, Li Y, Li Q, Li X, Zhang Y
and Zhai Z: β-catenin, leucine-rich repeat-containing G
protein-coupled receptor 5 and GATA-binding factor 6 are associated
with the normal mucosa-adenoma-adenocarcinoma sequence of
colorectal tumorigenesis. Oncol Lett. 15:2287–2295. 2018.PubMed/NCBI
|
|
32
|
Inazu T, Yamada K and Miyamoto K: Cloning
and expression of pleckstrin 2, a novel member of the pleckstrin
family. Biochem Biophys Res Commun. 265:87–93. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hamaguchi N, Ihara S, Ohdaira T, Nagano H,
Iwamatsu A, Tachikawa H and Fukui Y: Pleckstrin-2 selectively
interacts with phosphatidylinositol 3-kinase lipid products and
regulates actin organization and cell spreading. Biochem Biophys
Res Commun. 361:270–275. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang W, Li T, Hu B and Li H: PLEK2 Gene
Upregulation Might Independently Predict Shorter Progression-Free
Survival in Lung Adenocarcinoma. Technol Cancer Res Treat.
19:15330338209570302020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Luo Y, Robinson S, Fujita J, Siconolfi L,
Magidson J, Edwards CK, Wassmann K, Storm K, Norris DA,
Bankaitis-Davis D, et al: Transcriptome profiling of whole blood
cells identifies PLEK2 and C1QB in human melanoma. PLoS One.
6:e209712011. View Article : Google Scholar : PubMed/NCBI
|