Introduction
Next-generation sequencing (NGS) technologies with
massively parallel sequencing are widely used for medical genomics
studies (1). NGS techniques, such
as whole-exome sequencing (WES) and whole-genome sequencing (WGS),
are more desirable than individual gene sequencing due to high
coverage sequencing at a lower cost (2). However, data processing and analysis
remain a limitation in WES and WGS (1). Specifically, identifying all single
nucleotide variants (SNVs) and short insertion/deletions (indels)
from the protein-coding region is a challenge with WES data
analysis (3). Confounding factors
such as DNA quality and numerous potential errors during the
library preparation, DNA sequencing, alignment and mapping steps
affect the accuracy of the variants called (3). Multiple quality control steps are
employed in a standard bioinformatics pipeline; however, false
positive and negative variants still occur (4). Therefore, visual inspection of read
alignments is key to accurately identify the variants from NGS data
(4,5).
Autosomal dominant polycystic kidney disease (ADPKD)
is the most common type of inherited cystic kidney disease, with an
estimated prevalence of 9.3 per 10,000 people worldwide (6) and is characterized by development of
multiple cysts in both kidneys. Due to the enlargement of the
kidneys and progressive loss of renal function, ~50% of patients
with ADPKD suffer from end-stage renal disease (ESRD) by age 60
(7). ADPKD is primarily caused by
mutations in the polycystin 1, transient receptor potential channel
interacting (PKD)1 and PKD2 genes (7). PKD1 is composed of 46 exons
with a coding length of 12,912 bp (NM_001009944.3) (8), whereas PKD2 is composed of 15
exons with a coding length of 2,907 bp (NM_000297.4) (9). Besides being a large gene,
sequencing PKD1 is complicated by the presence of six
pseudogenes (PKD1P1-PKD1P6) that share >97%
sequence similarity with exons 1–33 of PKD1 (10).
Molecular analysis of ADPKD is performed using
several techniques such as long-range PCR followed by direct Sanger
sequencing (11), multiple
ligation probe assay (12), NGS
techniques (13,14) or a mixture of the aforementioned
approaches (15). Conventionally,
long-range PCR is used to exclude pseudogenes (11). However, the subsequent Sanger
sequencing for PKD1 is laborious, expensive and
time-consuming (16). Due to
these factors, NGS technologies such as WGS (13) and WES (14), have increasingly been utilized to
genotype PKD1 and PKD2 in patients with ADPKD.
Compared with Sanger sequencing, a recent study reported that WGS
has 100% sensitivity and specificity in detecting variants
associated with ADPKD (13).
However, the utility of WES for ADPKD remains in unknown, as recent
study reported the sensitivity to be limited at 50% (14).
Our previous studies reported successful use of WES
to identify genetic mutations for several types of monogenic
disease that belong to the group of inborn errors of immunity
(17,18). In the present study, the
application of WES to ADPKD was evaluated. Key visual inspection
steps in identifying a novel insertion mutation in PKD1,
which was not identified by the variant caller, are described.
Subjects and methods
Study subject
A 50-year-old woman was diagnosed with advanced
chronic kidney disease in September 2013 during health screening in
Kuala Lumpur Hospital, Malaysia. Ultrasound was performed because
the patient had abnormal kidney function. Ultrasonography of her
kidneys demonstrated bilateral polycystic kidneys with features
which were highly suggestive of ADPKD. Diagnosis of ADPKD was made
clinically based on the ultrasound result in September 2013. The
patient was then recruited into the research cohort before her
kidney transplant in August 2015.
WES
Genomic DNA was extracted from whole blood in EDTA
tubes using QIAsymphony DSP DNA Midi kit (cat. no. 937255; Qiagen
GmbH) on a QIAsymphony SP instrument (Qiagen GmbH). The DNA
concentration and purity were evaluated through optical density
measurement at 260 nm and 260/280 ratio respectively, using the
QIAxpert Slide-40 (cat. no. 990700; Qiagen GmbH) on a QIAxpert
System (Qiagen, GmbH). Two µg of genomic DNA was fragmented into
150–200 bp using Covaris LE220-plus focused-ultrasonicator
(Covaris, Inc.). A genomic library with fragment size of ~330 bp
was constructed using SureSelectXT Reagent kit (cat. no. G9641C;
Agilent Technologies, Inc.). Exome enrichment was performed using
the SureSelect Human All Exon V6 kit (cat. no. 5190-8864; Agilent
Technologies, Inc.) with a target size of 60 Mb. The size of PCR
enriched fragments was verified using Agilent DNA 1000 kit (cat.
no. 5067-1504; Agilent Technologies, Inc.) on a 2100 Bioanalyzer
instrument (Agilent Technologies, Inc.). The final library was
quantified using qPCR according to the Illumina qPCR Quantification
Protocol Guide (KAPA SYBR FAST qPCR Master Mix (2X) Universal; cat.
no. KK4602; Kapa Biosystems, Inc.). The loading concentration of
the final library was 300 pM. Paired-end reads of 2×101 bp were
sequenced using the HiSeq 3000/4000 SBS kit (300 cycles) (cat. no.
FC-410-1003; Illumina, Inc.) on a HiSeq 4000 System (Illumina,
Inc.) with a minimum coverage of 100×. The raw data were converted
into FASTQ format.
Bioinformatics analysis
The bioinformatics processing pipeline for germline
short variant discovery was modified from the Genome Analysis
Toolkit (GATK) Best Practices Workflows (version 4.1.2.0) (19). Briefly, pre-processing of the
FASTQ file began with addition of specific read group information
and tagging of Illumina adapters using Picard (version 2.20.1)
(20). Next, the reads were
aligned and mapped to the human reference genome GRCh38
(GCA_000001405.15) using the Burrows-Wheeler Aligner-maximal exact
matches (BWA-MEM) (version 0.7.17-r1188) (21). Additional quality control step in
BAM file processing was included to unmap contaminant reads using
Picard (version 2.20.1) (20).
Following alignment, duplicate reads were marked using Picard
(version 2.20.1) (20) and base
quality score recalibration (BQSR) was performed using GATK
(version 4.1.2.0) (19). Variants
including SNVs and indels were called using HaplotypeCaller
(version 4.1.2.0) (22). Finally,
the resulting variant call format file was annotated using the
web-based wANNOVAR tool (accessed in March 2020) (23).
The alignment of reads in the binary alignment and
map (BAM) file was visualized using the Integrative Genomics Viewer
(IGV) (version 2.8.10) from The Eli and Edythe L. Broad Institute
of MIT and Harvard (24).
Sequence similarity analysis was performed using the Basic Local
Alignment Search Tool (BLAST) (25). The pathogenicity of variants was
computationally evaluated using in silico prediction tools,
namely Sorting Intolerant from Tolerant (version 2.3) (26), MutationTaster (version 2)
(27) and PolyPhen-2 (version
2.2.2) (28). Nucleotide and
protein changes were identified by comparison with National Center
for Biotechnology Information (NCBI) reference sequences of
PKD1 (NM_001009944.3) (8)
and PKD2 (NM_000297.4) (9). The detected mutation sites were
compared with the ADPKD Variant Database (PKDB) (29) and NCBI Single Nucleotide
Polymorphism Database (dbSNP) human build 155 (30). Allelic frequency of the variants
was checked using the genome Aggregation Database (gnomAD)
(31). The final decision on
pathogenicity of the detected mutations was based on the American
College of Medical Genetics and Genomics (ACMG) classification
(32). Changes to protein
sequences following an indel event were determined using the ExPASy
translation tool (33).
Mutation validation
Primers were designed to flank the targeted region
using Primer3 (version 0.4.0) (34) and validated using Primer-BLAST
(35). Using genomic DNA
extracted from whole blood of patient, primer sequences (forward,
5′-CTGCTCTTCCTGCTTTTGGT-3′ and reverse, 5′-CCGTACCCACCTCCTTGAC-3′)
were used to amplify a product of 633 bp from the PKD1 gene
using MyFi™ Mix kit (Meridian Bioscience, Inc.). Genomic DNA from a
healthy unrelated individual was used as the control. The PCR
cycling conditions were initial denaturation at 95°C for 3 min,
followed by 33 cycles of denaturation at 95°C for 30 sec, annealing
at 60°C for 30 sec and extension at 72°C for 40 sec and final
extension at 72°C for 8 min. PCR products were purified and
subjected to bidirectional Sanger sequencing using BigDye™
Terminator v3.1 Cycle Sequencing kit (Applied Biosystems; Thermo
Fisher Scientific, Inc.) on a 3500×l Genetic Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The chromatograms were
viewed and analyzed using FinchTV (version 1.4.0; Geospiza,
Inc.).
Results
Case presentation
The patient was the sixth of eight children. Her
father had ischemic heart disease and her mother had diabetes
mellitus. No family history of renal disease was noted; however,
only one of her brothers was screened for the disease. The patient
developed ESRD in September 2013 and hemodialysis was initiated.
Her kidney ultrasound demonstrated >15 cysts in each kidney with
bipolar lengths of 15 and 17 cm. Diagnosis of ADPKD was made
despite negative family history and genetic testing. Bilateral
nephrectomy was performed in November 2014 and she subsequently
received a living kidney transplant from her younger brother in
August 2015. The kidney transplant was successful with good kidney
function.
Bioinformatics analysis of WES
WES generated 70,808,986 paired-end reads, of which
69,941,464 reads (98.77%) were properly paired and mapped to the
human reference genome GRCh38. A total of 110,294 reads suspected
of cross-species contamination due to extremely short alignments
with clipping on both sides were unmapped. Variant calling
annotated 24,901 SNVs and 703 indels in the exonic and splice site
regions. A total of 13 SNVs was identified in the PKD1 and
PKD2 genes. Of these, eight were synonymous and five were
non-synonymous mutations. The pathogenicity and allelic frequency
for all variants were carefully examined. All five non-synonymous
mutations were assessed to be benign using in silico
prediction tools and were present in >5% of the population. The
workflow for bioinformatics processing and variant prioritization
is presented in Fig. 1.
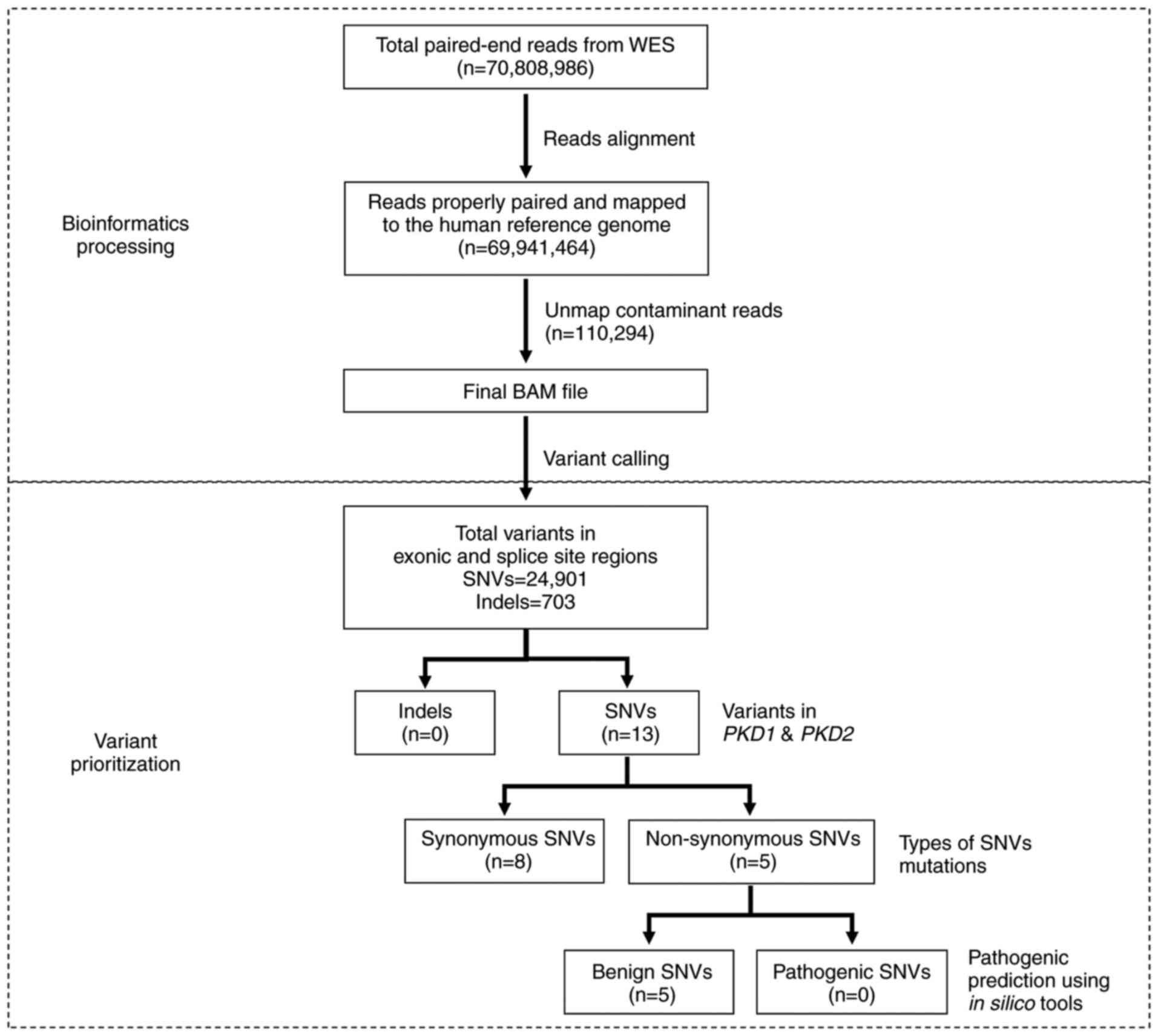 | Figure 1.Workflow of WES for the patient. WES
is divided into bioinformatics processing and variant
prioritization. In bioinformatics processing, a total of 70,808,986
paired-end reads were generated, of which 69,941,464 reads were
properly paired and mapped to the human reference genome.
Subsequently, 110,294 reads suspected as cross-species
contamination were unmapped as part of quality control. The final
BAM file was subjected to variant calling and annotated 24,901 SNVs
and 703 indels. In the variant prioritization step, variants were
filtered against PKD1 and PKD2 genes. A total of 13
SNVs was identified, of which eight were synonymous; five were
non-synonymous mutations. The pathogenicity of these five SNVs was
predicted using in silico prediction tools SIFT,
MutationTaster and PolyPhen-2. All five SNVs were assessed to be
benign. BAM, binary alignment and map; SNV, single nucleotide
variant; indel, insertions/deletion; SIFT, Sorting Intolerant from
Tolerant. |
Identification of atypically aligned
soft-clipped reads
As no pathogenic genetic variants were demonstrated,
the read alignments of PKD1 and PKD2 genes were
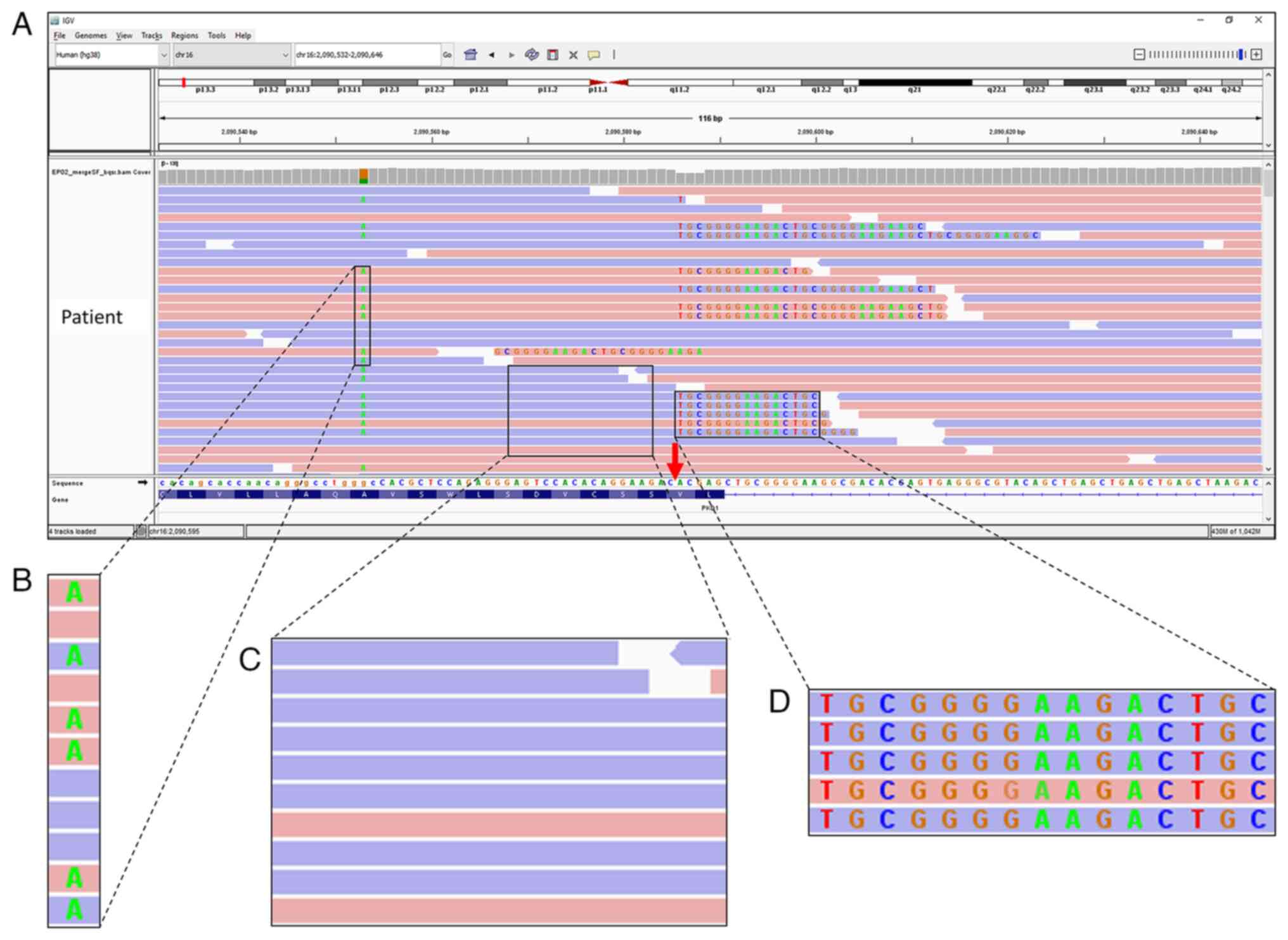
visually inspected using IGV. One position at chr16:2,090,586 of
exon 45 of PKD1 was assessed as having numerous reads
containing long sequences of soft-clipped bases (Fig. 2). This region was unique because
all soft-clipped bases were in alignment with each other, which
prompted further evaluation. This atypical alignment may have
resulted from strand bias during capture, poor mapping, sequencing
errors or DNA template contamination.
Soft-clipped reads are of high-quality
mapping and good base score
The number of reads that spanned the affected site
was counted (Fig. 3A); there was
a total of 91 reads, with 52 reads without any soft-clipped bases
and 39 reads with soft-clipped bases. The forward reads were
colored pink and the reverse reads blue. For 52 reads without
soft-clipped bases, 25 were forward and 27 were reverse reads. For
the 39 reads with soft-clipped bases, 21 were forward and 18 were
reverse reads. The coverage of reads at the site of interest was
sufficiently high to suggest heterozygosity. There was no evidence
of strand bias as the distribution of forward and reverse reads was
balanced.
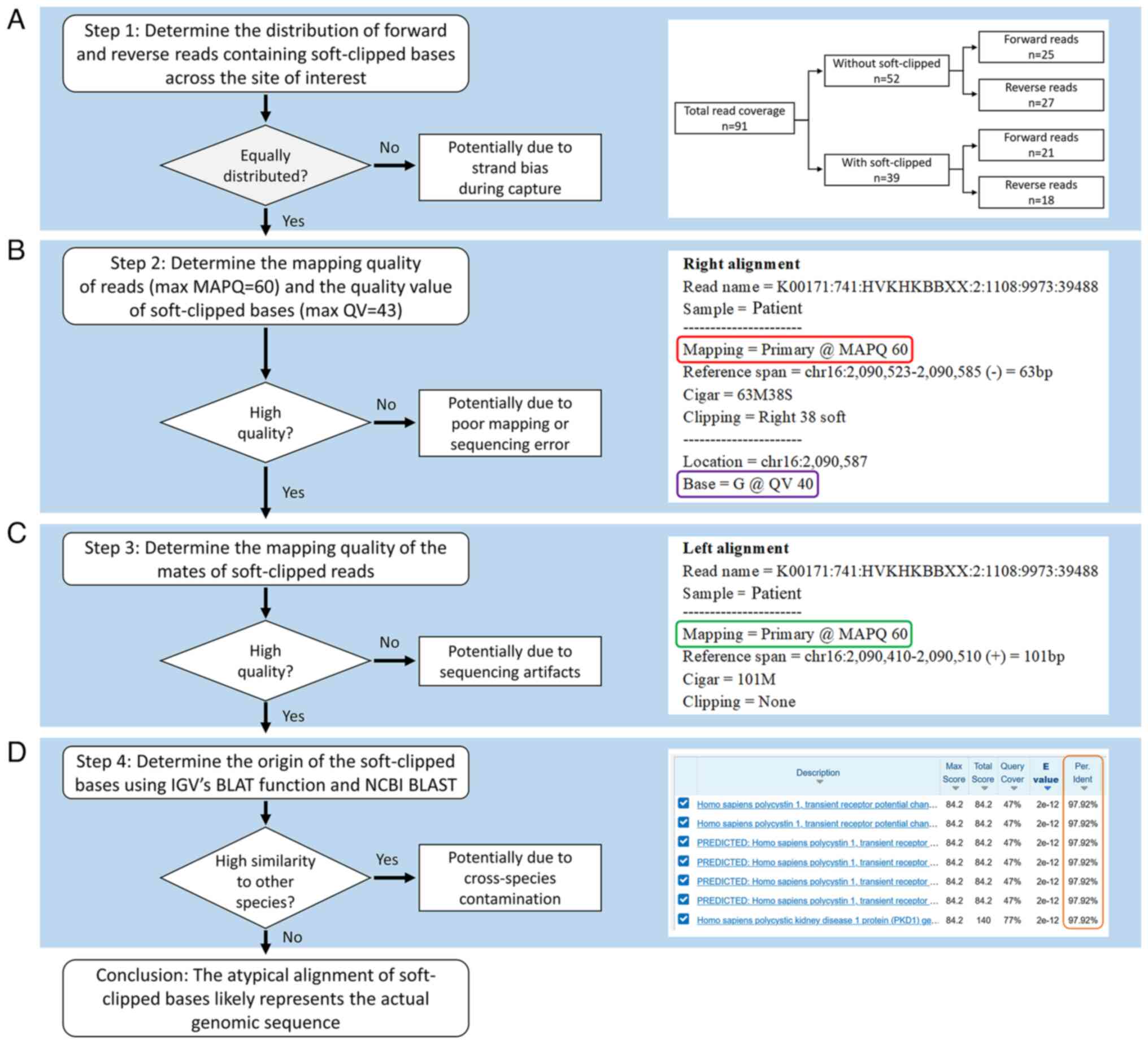 | Figure 3.Flow chart of assessment of atypical
alignment of soft-clipped reads using IGV and BLAST. (A)
Determination of quantity and coverage of reads with and without
soft-clipped bases. The total coverage of 91 was high and equally
distributed between the with and without soft-clipped groups, which
suggested true heterozygosity. (B) Determination of MAPQ of reads
and QV of bases. MAPQ=60 indicated high confidence that the read
was correctly aligned; MAPQ=0 indicated the read could be mapped to
more than one location in the human reference genome. The potential
QV scores for bases ranged from 6 to 43. Higher score indicated
higher probability of correctly calling the base. In the example
presented, the read with soft-clipped bases had MAPQ=60 (red box)
and the soft-clipped base G had QV 40 (purple box). (C) Checking
mapping quality of mates of soft-clipped reads. If mates had
low-quality MAPQ=0, this indicated potential sequencing artifacts
or errors during library preparation. In the example presented, the
mate (left) for the read presented (right) had MAPQ=60 (green box).
(D) Determination of origin of the soft-clipped bases using IGV
built-in BLAT function and BLAST. Atypical alignment with
soft-clipped bases results from non-human DNA contamination, which
can be determined from BLAST results. In the example presented, the
results obtained from BLAST demonstrated that soft-clipped read had
>97% identity (orange box) with Homo sapiens polycystin
1, which indicated no evidence of cross-species contamination. When
all conditions were fulfilled, the atypical alignment likely
represented the actual genomic sequence. IGV, Integrative Genomics
Viewer; BLAST, Basic Local Alignment Search Tool; BLAT, BLAST-like
Alignment Tool; MAPQ, mapping quality; QV, quality value; NCBI,
National Center for Biotechnology Information. |
The mapping quality (MAPQ) of the soft-clipped reads
and the quality value (QV) of the soft-clipped bases were checked.
In the example presented in Fig.
3B, the read was 101 bp in length with 63 bases matched and 38
bases soft-clipped on the right. After BWA-MEM, the MAPQ of reads
ranged from 0 to 60. This read had the highest possible value of
MAPQ=60, which indicated high confidence that the read was
correctly aligned. Read with low-quality mapping (MAPQ=0) appeared
translucent on IGV and indicated that the read could be mapped to
more than one location in the human reference genome. Assessment of
all 91 reads that spanned the affected region demonstrated that all
reads had MAPQ=60. Following BQSR, the QV ranged from a minimum of
6 to a maximum of 43. The base G in the example presented had a QV
score of 40. Generally, soft-clipped bases had high QV scores of
30–42. These findings ruled out poor mapping and sequencing errors
as potential causes for atypical alignment.
Subsequently, the mapping parameters of mates of the
soft-clipped reads were checked using the IGV option to view the
reads as pairs. The mate of the read presented in Fig. 3B was used as the example presented
in Fig. 3C. The right read had 38
bases soft-clipped and the left read was 101 bases and fully
matched with MAPQ=60. Mates with MAPQ=0 may have been due to
sequencing artifacts or errors during the library preparation.
While in ‘view as pairs’ mode, the start and end of insert size
were compared. Read pairs with the same start and end positions may
indicate duplication events that were not removed during quality
control and may have given a false sense of coverage. No evidence
of duplication events was discovered and all mates of soft-clipped
reads had MAPQ=60.
Reads with soft-clipped bases are of
PKD1 origin
The final step was to identify the origin of
soft-clipped bases. Atypical alignment with soft-clipped bases may
result from non-human DNA contamination (36). To evaluate this, BLAST was
performed on five reads with long soft-clipped bases (Fig. 3D); reads had >97% similarity to
PKD1. This was also performed using IGV built-in BLAST-like
Alignment Tool (BLAT) function on the soft-clipped bases to assess
whether the soft-clipped sequence could be mapped to other parts of
the human reference genome. Five results from BLAT analysis
demonstrated that the soft-clipped bases were partially matched to
PKD1. The results demonstrated the atypically aligned
soft-clipped reads were of PKD1 origin.
Sanger sequencing confirms presence of
a 20-bp novel insertion within the soft-clipped region
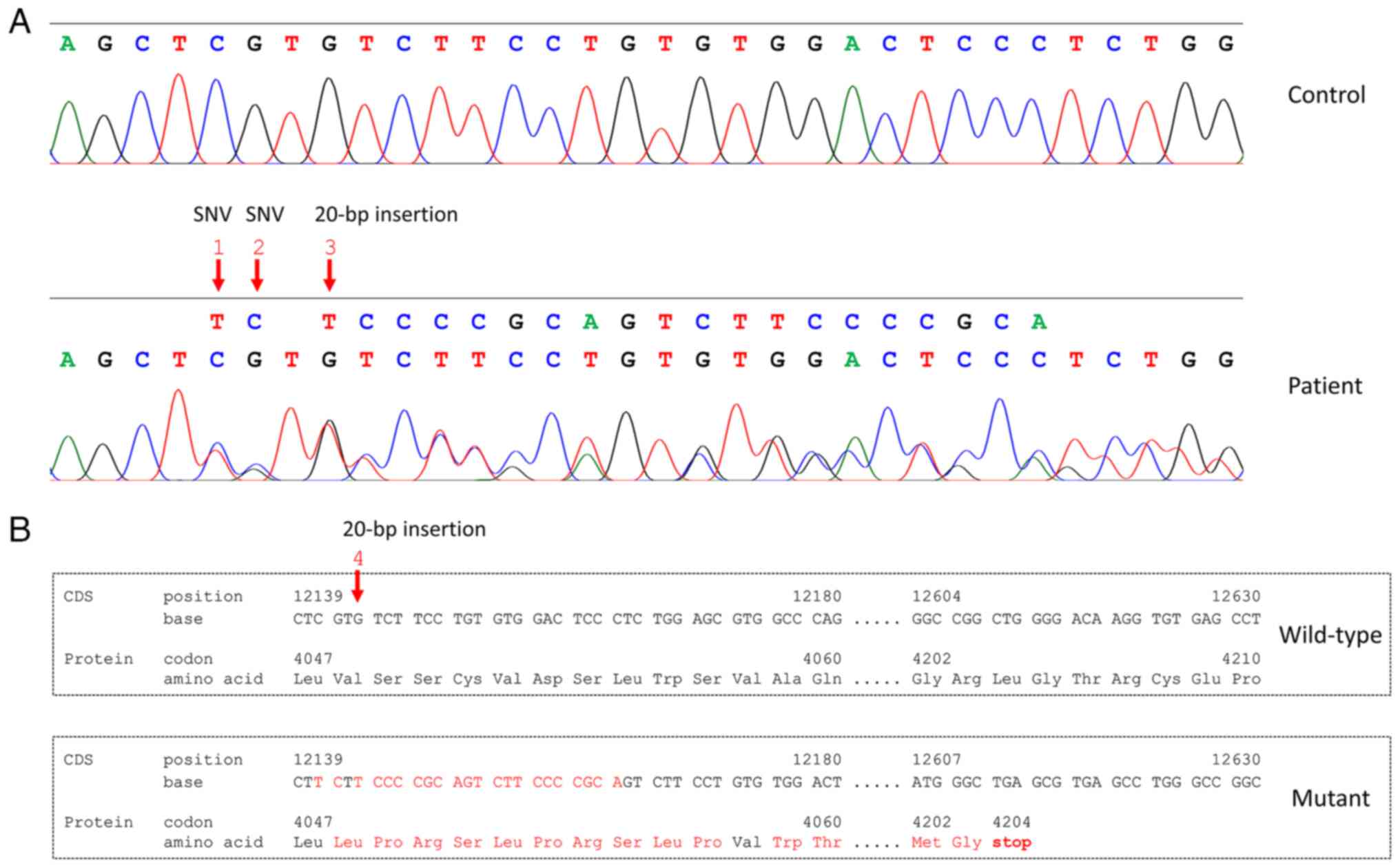
The atypical alignment at PKD1 was
hypothesized to be caused by an indel mutation. Sanger sequencing
confirmed the presence of two SNVs (rs1384786564 and rs1337808849)
and a 20-bp insertion (c.12143_12144insTCCCCGCAGTCTTCCCCGCA;
Fig. 4A). This 20-bp insertion
was considered novel because it has not been previously reported in
commonly used databases including PKDB, dbSNP (build 155) and
gnomAD. This mutation was predicted to change the reading frame
from codon 4,048 for 156 amino acids, followed by introduction of a
premature stop codon at codon 4,204. The prematurely truncated
protein was predicted to be 100 amino acids shorter than the
wild-type protein of 4,303 amino acids (Fig. 4B). Assessment using ACMG
classified this novel 20-bp insertion as pathogenic.
Discussion
A novel 20-bp insertion mutation in PKD1 from
a patient with ADPKD was identified using WES. However, this
mutation was not identified by the variant caller. Instead, a
region with atypical alignment of numerous soft-clipped reads was
identified by visual inspection of read alignments. A total of four
visual inspection steps was outlined using IGV and BLAST to rule
out potential errors that may yield soft-clipped reads. Finally,
the hypothesis that an indel event caused the atypically aligned
soft-clipped reads was confirmed by Sanger sequencing. To the best
of our knowledge, this 20-bp insertion in PKD1 is novel and
predicted to introduce a premature stop codon, acting as a
loss-of-function mutation. Despite the lack of ADPKD in the family
history, assessment using ACMG classified this mutation as
pathogenic, which supported the clinical diagnosis of ADPKD for the
patient.
SNV and indel detection using WES is routine;
however, researchers rely on the output generated by automated
pipelines and inaccurate or false variant calls occur. Visual
inspection of aligned reads is a key step for variant discovery and
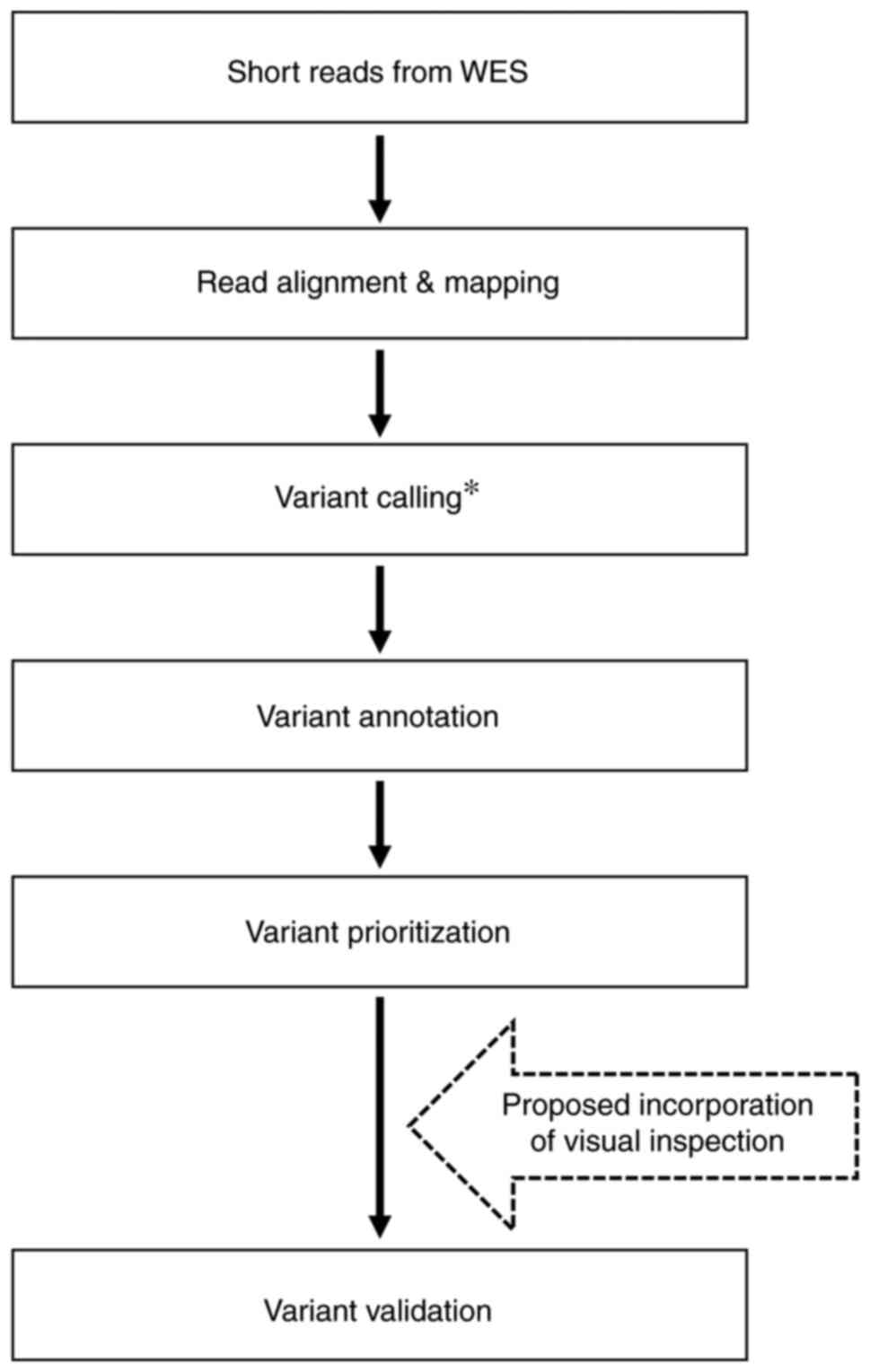
validation (4,5). The proposed visual inspection steps
can be incorporated into WES data analysis workflow following
variant prioritization (Fig. 5).
Visual inspection at the genes of interest may reveal atypical
alignment. It has been reported that atypical alignment with
soft-clipped bases results from contamination with non-human DNA
(36). Samson et al
(36) reported that non-human DNA
originating from the oral microbiome aligns with the human
reference genome, producing atypical alignment and false positive
variants. However, the DNA source of the aforementioned study was
saliva, whereas the DNA source in the present study was peripheral
blood. Visual inspection using BLAST and BLAT confirmed that the
atypical alignment in the present patient was not due to
cross-species contamination.
In the present study, an indel mutation leading to
truncation in PKD1 was found in a patient with ADPKD. Generally,
mutations are more commonly identified in PKD1 (~85%) than
PKD2 (~15%) (37–39). At the time of analysis, a total of
1,225 PKD1 and 196 PKD2 pathogenic variants were
cataloged in the PKDB (29). For
PKD1, indels that lead to a truncated protein are the most
common mutation (36.7%; 449/1,225), followed by missense (22.6%;
277/1,225), nonsense (21.5%; 263/1,225) and other forms of mutation
(19.3%; 236/1,225).
Despite numerous variants reported in PKD1
and PKD2, no pathogenic variant has been reported in these
two genes for 11–39% of patients with ADPKD in large cohort studies
(37–39). For these patients, several other
genes have been reported to cause ADPKD, such as glucosidase II
alpha subunit (40), DnaJ heat
shock protein family 40 member B11 (DNAJB11) (41), ALG8 alpha-1,3-glucosyltransferase
and protein kinase C substrate 80K-H (39). Furthermore, numerous other
candidate genes have been reported to affect progression and
severity of ADPKD. Using WES, Hu et al (42) assessed 313 genes associated with
polycystic kidney disease; demonstrated that molecular analysis of
patients with ADPKD using global approaches such as WES, was more
useful than individual gene sequencing using Sanger sequencing. For
patients without mutations in PKD1 and PKD2, WES data
can be used to screen for other cystic genes that may point to
atypical ADPKD (16).
Sequencing PKD1 is challenging due to the
presence of six pseudogenes (10). However, WGS is reported to
overcome this (43) and has 100%
sensitivity in detecting pathogenic variants in PKD1
(13). To the best of our
knowledge, however, the diagnostic ability of WES for PKD1
is still uncertain. Al-Muhanna et al (44) reported that 100% coverage of
PKD1 is possible with WES, whereas Ali et al
(14) reported poor coverage of
PKD1 at duplicated regions. The difference in reported
sensitivity of WES may be due to different capture kits and data
analysis methods. NGS is becoming more affordable; however, WGS is
considerably more expensive compared with WES (2). WES that specifically targets
protein-coding exomes at 100× coverage generates ~6 GB of data,
whereas WGS that targets the entire genome at 30× coverage
generates ~90 GB of data (1). The
markedly larger data output from WGS demands higher computing power
for data analysis, which is not readily available in many
laboratories. Hence, WES is a cheaper and faster solution than
WGS.
In clinical practice, diagnosis of ADPKD is usually
made using ultrasonography, together with clinical presentations
and family history. However, there is lower sensitivity of
diagnosis using ultrasonography for younger individuals (45). Therefore, genetic testing may be
beneficial to provide a definite ADPKD diagnosis. Genetic testing
may also be used for testing patients with negative family history
and atypical clinical presentation and for the selection of family
members for living kidney transplantation (46).
In the present study, a novel 20-bp insertion in
exon 45 of PKD1 in a patient with ADPKD was demonstrated
using WES. This frameshift mutation was predicted to be pathogenic
due to the introduction of a premature stop codon. Therefore, WES
was demonstrated to be a suitable option for genetic testing for
ADPKD, which is cheaper and faster than WGS or Sanger sequencing.
However, data analysis of WES relying on a standard bioinformatics
pipeline may miss out disease-causing variants. As demonstrated in
the present study, visual inspection of read alignments was key to
identifying the pathogenic variant. The proposed visual inspection
steps can be incorporated into a typical WES data analysis workflow
and may improve diagnostic yield. The limitations of this study
include the requirement of experienced personnel to perform visual
inspection in WES data analysis, and the results were based on a
single case. Therefore, future research is warranted to validate
the use of visual inspection on a larger patient cohort.
Acknowledgements
The authors would like to thank the Director General
of Health, Malaysia for permission to publish this article. The
authors would also thank Mr Muhammad Johari (Allergy and Immunology
Research Centre, Institute for Medical Research) for assistance in
DNA extraction.
Funding
The present study was funded by a grant from the Ministry of
Health, Malaysia (grant no. NMRR-17-892-35929).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are available in the NCBI Sequence Read Archive under
accession number PRJNA798582
(ncbi.nlm.nih.gov/sra/?term=PRJNA798582). The human reference
genome GRCh38 (filename:
GCA_000001405.15_GRCh38_no_alt_plus_hs38d1_analysis_set) can be
downloaded from https://ftp.ncbi.nlm.nih.gov/genomes/all/GCF/000/001/405/GCF_000001405.26_GRCh38/GRCh38_major_release_seqs_for_alignment_pipelines/.
dbSNP (build 155) (filename: GCF_000001405.39.gz) can be downloaded
from https://ftp.ncbi.nlm.nih.gov/snp/archive/b155/VCF/.
Author's contributions
BTK conceived the study, acquired the funding,
performed laboratory experiments and data analysis, interpreted the
data and drafted the manuscript. MYC performed laboratory
experiments and drafted the manuscript. JI and NKF recruited the
patient, interpreted the data and constructed figures. SYY provided
the clinical history of the patient and interpreted the data. NM
and MA conceived the study and acquired the funding. AMR and SBM
designed and supervised the study. BTK, AMR and SBM confirm the
authenticity of all raw data. All authors have read and approved
the final manuscript.
Ethics approval and consent to
participate
Written informed consent for participation was
collected from the patient prior to blood sample collection. The
present study was approved by the Medical Research and Ethics
Committee, Ministry of Health, Malaysia (approval no.
KKM/NIHSEC/P17-1084) and was performed according to the Declaration
of Helsinki.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
NGS
|
next-generation sequencing
|
|
WES
|
whole-exome sequencing
|
|
WGS
|
whole-genome sequencing
|
|
SNV
|
single nucleotide variant
|
|
indel
|
insertion/deletion
|
|
ADPKD
|
autosomal dominant polycystic kidney
disease
|
|
IGV
|
Integrative Genomics Viewer
|
|
MAPQ
|
mapping quality
|
|
QV
|
quality value
|
References
|
1
|
Suwinski P, Ong C, Ling MHT, Poh YM, Khan
AM and Ong HS: Advancing personalized medicine through the
application of whole exome sequencing and big data analytics. Front
Genet. 10:492019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schwarze K, Buchanan J, Taylor JC and
Wordsworth S: Are whole-exome and whole-genome sequencing
approaches cost-effective? A systematic review of the literature.
Genet Med. 20:1122–1130. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ma X, Shao Y, Tian L, Flasch DA, Mulder
HL, Edmonson MN, Liu Y, Chen X, Newman S, Nakitandwe J, et al:
Analysis of error profiles in deep next-generation sequencing data.
Genome Biol. 20:502019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Robinson JT, Thorvaldsdottir H, Wenger AM,
Zehir A and Mesirov JP: Variant review with the integrative
genomics viewer. Cancer Res. 77:e31–e34. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koboldt DC: Best practices for variant
calling in clinical sequencing. Genome Med. 12:912020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lanktree MB, Haghighi A, Guiard E, Iliuta
IA, Song X, Harris PC, Paterson AD and Pei Y: Prevalence estimates
of polycystic kidney and liver disease by population sequencing. J
Am Soc Nephrol. 29:2593–2600. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chebib FT and Torres VE: Autosomal
dominant polycystic kidney disease: Core curriculum 2016. Am J
Kidney Dis. 67:792–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
National Library of Medicine (US),
National Center for Biotechnology Information, . Nucleotide
NM_001009944.3. https://www.ncbi.nlm.nih.gov/nuccore/NM_001009944.3December
30–2021
|
|
9
|
National Library of Medicine (US),
National Center for Biotechnology Information, . Nucleotide
NM_000297.4. https://www.ncbi.nlm.nih.gov/nuccore/NM_000297.4December
30–2021
|
|
10
|
Bogdanova N, Markoff A, Gerke V, McCluskey
M, Horst J and Dworniczak B: Homologues to the first gene for
autosomal dominant polycystic kidney disease are pseudogenes.
Genomics. 74:333–341. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Audrézet MP, Cornec-Le Gall E, Chen JM,
Redon S, Quéré I, Creff J, Bénech C, Maestri S, Le Meur Y and Férec
C: Autosomal dominant polycystic kidney disease: Comprehensive
mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum
Mutat. 33:1239–1250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu G, Qian X, Wu Y, Li X, Chen J, Xu J and
Qi J: Analysis of gene mutations in PKD1/PKD2 by multiplex
ligation-dependent probe amplification: Some new findings. Ren
Fail. 37:366–371. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Mallawaarachchi AC, Lundie B, Hort Y,
Schonrock N, Senum SR, Gayevskiy V, Minoche AE, Hollway G, Ohnesorg
T, Hinchcliffe M, et al: Genomic diagnostics in polycystic kidney
disease: An assessment of real-world use of whole-genome
sequencing. Eur J Hum Genet. 29:760–770. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ali H, Al-Mulla F, Hussain N, Naim M,
Asbeutah AM, AlSahow A, Abu-Farha M, Abubaker J, Al Madhoun A,
Ahmad S and Harris PC: PKD1 duplicated regions limit clinical
utility of whole exome sequencing for genetic diagnosis of
autosomal dominant polycystic kidney disease. Sci Rep. 9:41412019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eisenberger T, Decker C, Hiersche M,
Hamann RC, Decker E, Neuber S, Frank V, Bolz HJ, Fehrenbach H, Pape
L, et al: An efficient and comprehensive strategy for genetic
diagnostics of polycystic kidney disease. PLoS One.
10:e01166802015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cordido A, Besada-Cerecedo L and
García-González MA: The genetic and cellular basis of autosomal
dominant polycystic kidney disease-a primer for clinicians. Front
Pediatr. 5:2792017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ripen AM, Chear CT, Baharin MF, Nallusamy
R, Chan KC, Kassim A, Choo CM, Wong KJ, Fong SM, Tan KK, et al: A
single-center pilot study in Malaysia on the clinical utility of
whole-exome sequencing for inborn errors of immunity. Clin Exp
Immunol. 206:119–128. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ripen AM, Chiow MY, Rama Rao PR and
Mohamad SB: Revealing chronic granulomatous disease in a patient
with Williams-Beuren Syndrome using whole exome sequencing. Front
Immunol. 12:7781332021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Van der Auwera GA, Carneiro MO, Hartl C,
Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high confidence variant
calls: The genome analysis toolkit best practices pipeline. Curr
Protoc Bioinformatics. 43:11.10.1–11.10.33. 2013.PubMed/NCBI
|
|
20
|
Picard. Broad Institute, . GitHub
repository. http://broadinstitute.github.io/picard/February
25–2020
|
|
21
|
Li H: Aligning sequence reads, clone
sequences and assembly contigs with BWA-MEM. https://arxiv.org/abs/1303.3997February 25–2020
|
|
22
|
Poplin R, Ruano-Rubio V, DePristo MA,
Fennell TJ, Carneiro MO, Van der Auwera GA, Kling DE, Gauthier LD,
Levy-Moonshine A, Roazen D, et al: Scaling accurate genetic variant
discovery to tens of thousands of samples. bioRxiv. 201178. 2018
February 27–2020
|
|
23
|
Chang X and Wang K: wANNOVAR: annotating
genetic variants for personal genomes via the web. J Med Genet.
49:433–436. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Robinson JT, Thorvaldsdottir H, Winckler
W, Guttman M, Lander ES, Getz G and Mesirov JP: Integrative
genomics viewer. Nat Biotechnol. 29:24–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
National Library of Medicine (US),
National Center for Biotechnology Information, . BLAST. https://blast.ncbi.nlm.nih.gov/Blast.cgiNovember
24–2021
|
|
26
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
ADPKD Variant Database. https://pkdb.mayo.edu/28–January. 2022
|
|
30
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: dbSNP: The NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Karczewski KJ, Francioli LC, Tiao G,
Cummings BB, Alfoldi J, Wang Q, Collins RL, Laricchia KM, Ganna A,
Birnbaum DP, et al: The mutational constraint spectrum quantified
from variation in 141,456 humans. Nature. 581:434–443. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hedge M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gasteiger E, Gattiker A, Hoogland C,
Ivanyi I, Appel RD and Bairoch A: ExPASy: The proteomics server for
in-depth protein knowledge and analysis. Nucleic Acids Res.
31:3784–3788. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Untergasser A, Cutcutache I, Koressaar T,
Ye J, Faircloth BC, Remm M and Rozen SG: Primer3-new capabilities
and interfaces. Nucleic Acids Res. 40:e1152012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ye J, Coulouris G, Zaretskaya I,
Cutcutache I, Rozen S and Madden TL: Primer-BLAST: A tool to design
target-specific primers for polymerase chain reaction. BMC
Bioinformatics. 13:1342012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Samson CA, Whitford W, Snell RG, Jacobsen
JC and Lehnert K: Contaminating DNA in human saliva alters the
detection of variants from whole genome sequencing. Sci Rep.
10:192552020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rossetti S, Consugar MB, Chapman AB,
Torres VE, Guay-Woodford LM, Grantham JJ, Bennett WM, Meyers CM,
Walker DL, Bae K, et al: Comprehensive molecular diagnostics in
autosomal dominant polycystic kidney disease. J Am Soc Nephrol.
18:2143–2160. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Carrera P, Calzavara S, Magistroni R, den
Dunnen JT, Rigo F, Stenirri S, Testa F, Messa P, Cerutti R, Scolari
F, et al: Deciphering variability of PKD1 and PKD2 in an Italian
cohort of 643 patients with autosomal dominant polycystic kidney
disease (ADPKD). Sci Rep. 6:308502016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mantovani V, Bin S, Graziano C, Capelli I,
Minardi R, Aiello V, Ambrosini E, Cristalli CP, Mattiaccio A,
Pariali M, et al: Gene panel analysis in a large cohort of patients
with autosomal dominant polycystic kidney disease allows the
identification of 80 potentially causative novel variants and the
characterization of a complex genetic architecture in a subset of
families. Front Genet. 11:4642020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Porath B, Gainullin VG, Cornec-Le Gall E,
Dillinger EK, Heyer CM, Hopp K, Edwards ME, Madsen CD, Mauritz SR,
Banks CJ, et al: Mutations in GANAB, encoding the glucosidase IIα
subunit, cause autosomal-dominant polycystic kidney and liver
disease. Am J Hum Genet. 98:1193–1207. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cornec-Le Gall E, Olson RJ, Besse W, Heyer
CM, Gainullin VG, Smith JM, Audrezet MP, Hopp K, Porath B, Shi B,
et al: Monoallelic mutations to DNAJB11 cause atypical
autosomal-dominant polycystic kidney disease. Am J Hum Genet.
102:832–844. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hu HY, Zhang J, Qiu W, Liang C, Li CX, Wei
TY, Feng ZK, Guo Q, Yang K and Liu ZG: Comprehensive strategy
improves the genetic diagnosis of different polycystic kidney
diseases. J Cell Mol Med. 25:6318–6332. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mallawaarachchi AC, Hort Y, Cowley MJ,
McCabe MJ, Minoche A, Dinger ME, Shine J and Furlong TJ:
Whole-genome sequencing overcomes pseudogene homology to diagnose
autosomal dominant polycystic kidney disease. Eur J Hum Genet.
24:1584–1590. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Al-Muhanna FA, Al-Rubaish AM, Vatte C,
Mohiuddin SS, Cyrus C, Ahmad A, Shakil Akhtar M, Albezra MA, Alali
RA, Almuhanna AF, et al: Exome sequencing of Saudi Arabian patients
with ADPKD. Ren Fail. 41:842–849. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Torres VE, Harris PC and Pirson Y:
Autosomal dominant polycystic kidney disease. Lancet.
369:1287–1301. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Khadangi F, Torkamanzehi A and Kerachian
MA: Identification of missense and synonymous variants in Iranian
patients suffering from autosomal dominant polycystic kidney
disease. BMC Nephrol. 21:4082020. View Article : Google Scholar : PubMed/NCBI
|