Introduction
Subarachnoid hemorrhage (SAH) is a serious acute
cerebrovascular disease caused by the flow of blood into the
subarachnoid space of the brain (1). However, at present, there are no
effective drugs available for improving brain tissue damage after
SAH following the prevention of rebleeding (2). Therefore, further studies into the
mechanisms of brain injury following SAH and the exploration of
appropriate treatment options for the improvement of the prognosis
of SAH are urgently required.
Early brain injury (EBI) refers to secondary brain
tissue damage caused by multiple physiological disturbances and a
range of pathological changes that occur within 72 h of the onset
of SAH (3). In the acute response
phase following SAH, the injury begins with the entry of large
amounts of blood into the subarachnoid space and this is followed
by a series of pathological injuries (4). As mitochondria are particularly
susceptible to hypoxia and ischemic injury, mitochondrial
dysfunction is one of the primary pathological injuries suffered
after SAH (5). Large numbers of
reactive oxygen species (ROS) are released, which leads to
enhancement of oxidative stress and activation of the mitochondrial
apoptotic pathway, causing mitochondrial injury and dysfunction
(6). Therefore, therapeutic
modalities that reduce oxidative stress and apoptosis are key for
the improvement of the prognosis of SAH.
Apoptosis signal-regulating kinase 1 (ASK1), a
member of the mitogen-activated protein 3 kinase (MAP3K) family, is
activated by numerous different types of cellular stress and has an
important role in oxidative stress and apoptosis (7). In primary cultures of mouse
hippocampal neurons, the Rho kinase inhibitor fasudil reverses
β-amyloid-induced elevation of phosphorylated (p-)ASK1, which
decreases neuronal apoptosis in Alzheimer's disease (8). To the best of our knowledge, however,
therapeutic options targeting ASK1 following SAH have not been
evaluated.
Ethyl-2,7-dioxo-2,7-dihydro-3H-naphtho(1,2,3-de)quinoline-1-carboxylate
(NQDI-1) has been reported to be a specific inhibitor of ASK1 and
has been validated using in vitro kinase assays (9,10).
Previous studies have reported that NQDI-1 may be effective in the
treatment of numerous diseases through inhibition of ASK1 activity
(11,12). NQDI-1 alleviates
microcystin-LR-induced mitochondrial damage and apoptosis in
ovarian granulosa cells via inhibition of the activation of ASK1
(13). Furthermore, indomethacin
can induce apoptotic cascade responses in glial cells via ASK1
activation of endoplasmic reticulum stress (14). Therefore, the ASK1-specific
inhibitor NQDI-1 may exert neuroprotective effects following SAH
through inhibition of ASK1 activity. p38 and JNK are considered to
form the downstream signaling pathways of ASK1; activated p38 and
JNK are able to further activate a variety of substrates, including
nuclear transcription factors as well as protein kinases and other
functional proteins, which leads to the onset of oxidative stress
and apoptosis (15).
It was hypothesized that ASK1 inhibitor NQDI-1
exerts neuroprotective effects and decreases oxidative stress and
neuroapoptosis via the ASK1/p38 and JNK signaling pathway in EBI
following SAH in rats.
Materials and methods
Experimental animals
The animal experiments were approved by the Ethics
Committee for Experimental Animals of Central South University
(approval no. 2019sydw0104). A total of 191 male Sprague-Dawley
(SD) rats weighing 280–320 g were housed in a constant temperature
(21–23°C) and humidity (45–50%) environment with a 12/12-h
light/dark cycle and had free access to water and food. All animal
experiments were performed according to the Animal Research:
Reporting In Vivo Experiments guidelines (16) and were performed in accordance with
the National Institutes of Health (NIH) Guide for the Care and Use
of Animals guidelines (17). When
rats reached the designated preset time point of the experiment or
met certain criteria for euthanasia [complete loss of appetite for
24 h or poor appetite (50% below normal) for 3 days, inability to
eat or drink, or they were assessed as being close to death
(self-injurious behavior, abnormal posture, respiratory distress,
etc.)], the rats were placed in a carbon dioxide euthanasia device
with CO2 volume displacement rate set at 30%/min
(18). Death was confirmed by
cessation of breathing and heartbeat and pupil dilation. The
carcasses of all rats were bagged and frozen for environmentally
sound disposal.
SAH model
The rat SAH model was induced using monofilament
perforation (19). After
anesthetizing the rats (induction, 3% isoflurane; maintenance, 2%
isoflurane), the left common, internal and external carotid artery
of the rats were exposed. The distal external carotid artery was
clipped using cauterization to form the stump of the external
carotid artery. A 3-cm sharpened model 4–0 prolene wire was
inserted ~2 cm through the external carotid artery until a slight
resistance was felt. The puncture line was slowly advanced by ~3 mm
to puncture the arterial wall. Brain tissue was removed at the
corresponding time points predetermined for the experiment. If a
blood clot was found on the ventral side of the brain tissue, it
indicated that the rat had SAH. Rats with a score >8 by
assessment of the SAH grading score were considered to meet the
experimental requirements and to be a successful model. The
procedure for the sham group was similar to that of the SAH group,
with the exception that the 4–0 prolene wire did not pierce the
arterial wall. The respiration, heart rate, skin color and pedal
reflex were monitored every 5 min throughout the surgical and
anesthetic recovery stages. Of these, 23 rats died and 8 rats were
excluded, and these deaths and exclusions were supplemented by new
SAH modeled rats.
Severity of SAH
The severity of SAH was assessed using SAH grading
score 24 h after SAH (20). The
ventral side of the brain tissue was divided into 6 regions, each
rated 0–3 depending on the amount of blood clot and how well the
blood vessels were obscured: A score of 0 indicates no clot, 1
indicates a small amount of clot, 2 indicates a moderate amount of
clot but large vessels at the skull base are still identifiable,
and 3 indicates a large amount of clot and unidentifiable vessels
at the skull base. The six regional scores were summed to calculate
a total score (0–18), where higher scores indicated more severe
hemorrhage. Rats with a total SAH grade score of ≤8 were considered
to have mild SAH, excluded from subsequent experiments and were
replaced with newly modeled rats.
Drug administration
To enable the drug concentrations in the brain
tissue to be more precisely regulated independently of the liver
and other factors, the siRNAs and NQDI-1 were administered by
intracerebroventricularly injection (21). Following anesthetization
(induction, 3% isoflurane; maintenance, 2% isoflurane), the rats
were fixed on a brain stereotaxic apparatus. The microinjector was
fixed to the bregma at 1.0 posterior, 1.5 right and 3.3 mm deep.
The ASK1 siRNA (cat. no. 4390771; Thermo Fisher Scientific, Inc.)
or Scr siRNA control (cat. no. 4390843; Thermo Fisher Scientific,
Inc.) were dissolved in 5 µl nuclease-free sterile deionized water
at a concentration of 500 pmol and administered 48 h before SAH.
The sequences of ASK1 siRNA were as follows: Sense
5′-CGGCAGACAUUGUUAUCAAtt-3′ and antisense
5′-UUGAUAACAAUGUCUGCCGtc-3′. Drug concentrations and doses reported
in a similar study (22) and the
solubility of NQDI-1 were used to determine the concentrations used
in the present study. Three doses of NQDI-1 (1, 3 and 10 µg/kg;
Selleck Chemicals) or vehicle solution in a total volume of 5
µl/rat was injected 1 h after SAH. The BMS-582949 (100 mg/kg;
Selleck Chemicals), SP600125 (30 mg/kg; Selleck Chemicals) and
vehicle solutions could cross the blood-brain barrier and were
administered intraperitoneally 1 h after SAH (23,24).
Short-term neurological function
Modified Garcia score and the beam balance score
were used to assess the short-term neurological function 24 h after
SAH. The modified Garcia score was divided into six categories as
follows: Voluntary movement, voluntary limb movement, forepaw
extension, climbing ability, somatosensory responses and tentacle
response, each of which was scored individually and summed to give
a total score ranging from 3–18 (25). Higher scores were considered to
indicate better neurological function. For the beam balance score,
rats were placed on a beam for 1 min and a score ranging from 0–4
was assessed based on the distance walked (26). Higher scores were considered to
indicate better neurological function.
Long-term neurological function
Rotarod pre-adaptation experiments were performed on
rats using the same initial velocity and acceleration before SAH
modeling. The rats were placed on a Rotamex rotating bar tester
(Columbus Instruments) for the Rotarod test on days 7, 14 and 21
after SAH (27). First, the
initial Rotarod speed was set to 5 revolutions per min and the
acceleration to 2 revolutions per 5 sec. After putting on the rats,
the fall latency of the rats was recorded. The rats were then
allowed to rest for 1 h. Finally, the fall latency of the rats was
tested again at the initial speed of 5 revolutions per min and an
acceleration of 2 revolutions per 5 sec. Longer fall latency was
considered to indicate better motor coordination of the rat. At
week 4 after SAH modeling, the Morris water maze test was used to
assess learning memory and spatial orientation of the rats
(27). The learning exercise of
swimming and finding a platform located below the water surface was
performed from day 1 to day 5 on week 4 (days 28–33 post-SAH) and
the swimming trajectory, escape latency and swimming distance were
recorded. On the final day, the platform was removed for testing
and swimming trajectory, swimming distance and probe quadrant
duration were recorded for 60 sec.
IF staining
After sequential perfusion of 4°C saline and 4°C 4%
paraformaldehyde from the heart to remove blood from the brain
tissue, intact brain tissue was obtained. After sucrose gradient
dehydration, the brain tissue was then cut into 10-µm sections in a
coronal position and was stored at −20°C. IF staining was used to
assess the co-localization of ASK1 with nerve cells. Frozen tissue
sections were blocked using 5% donkey serum (cat. no. G1217-5ML;
Wuhan Servicebio Technology Co., Ltd.) for 2 h at room temperature
and incubated overnight at 4°C with primary antibodies as follows:
Anti-ASK1 (mouse; 1:200; cat. no. 67072-1-Ig; ProteinTech Group,
Inc.), anti-NeuN (rabbit; 1:200; cat. no. 26975-1-AP; ProteinTech
Group, Inc.), anti-ionized calcium-binding adapter molecule-1
(Iba-1; rabbit; 1:200; cat. no. 10904-1-AP; ProteinTech Group,
Inc.), and anti-glial fibrillary acidic protein (GFAP; chicken;
1:200; cat. no. ab4674; Abcam). The tissue sections were then
incubated for 2 h at room temperature with the corresponding
fluorescent secondary antibodies as follows: Alexa
Fluor® 594 AffiniPure Donkey Anti-Mouse IgG (H+L) (cat.
no. 715-585-150; 1:500; Jackson ImmunoResearch Laboratories, Inc.),
Alexa Fluor 488 AffiniPure Donkey Anti-Rabbit IgG (H+L) (cat. no.
711-545-152; 1:500; Jackson ImmunoResearch Laboratories, Inc.), and
Alexa Fluor 488 AffiniPure Donkey Anti-Chicken IgY (IgG) (H+L)
(cat. no. 703-545-155; 1:500; Jackson ImmunoResearch Laboratories,
Inc.). Subsequently, they were stained for cell nuclei using 2
µg/ml DAPI (cat. no. G1012-100ML; Wuhan Servicebio Technology Co.,
Ltd.) for 10 min at room temperature. The sections were assessed
using an Olympus BX53 fluorescence microscope (Olympus Corporation)
and images were captured.
Dihydroethidium (DHE) staining
DHE staining was performed to assess the brain
oxidative stress (28). Frozen
brain tissue sections were incubated with 2 µmol/l DHE (Thermo
Fisher Scientific, Inc.) at 37°C for 30 min in the dark. After
staining for cell nuclei using 2 µg/ml DAPI for 10 min at room
temperature, the sections were assessed using an Olympus BX53
fluorescence microscope. A total of six sections from each brain
tissue was randomly selected and six areas in each section were
randomly selected for imaging. The number of DHE-positive cells was
counted using ImageJ 1.4 software (National Institutes of Health)
and the mean was calculated and taken as the final percentages for
each brain tissue section.
TUNEL staining
TUNEL staining was used to evaluate the percentage
of apoptotic neurons (29). Frozen
tissue sections were blocked using 5% donkey serum for 2 h at room
temperature and incubated overnight at 4°C with primary antibody
anti-NeuN (rabbit; 1:200; cat. no. 26975-1-AP; ProteinTech Group,
Inc.). The tissue sections were then incubated with the fluorescent
secondary antibody Alexa Fluor 488 AffiniPure Donkey Anti-Rabbit
IgG (H+L) (cat. no. 711-545-152; 1:500; Jackson ImmunoResearch
Laboratories, Inc.) for 2 h at room temperature. TUNEL staining was
performed using the One Step TUNEL Apoptosis Assay kit (cat. no.
C1090; Beyotime Institute of Biotechnology) according to the
manufacturer's protocol. Finally, the tissue sections were stained
for cell nuclei using 2 µg/ml DAPI for 10 min at room temperature.
Random selection of TUNEL counting locations and calculation of
TUNEL-positive neurons for each section were performed using the
aforementioned method for DHE counting.
Western blotting (WB)
WB was used to semi-quantify protein expression
levels. After perfusion of 4°C saline from the heart to remove
blood from the brain tissue, intact brain tissue was obtained.
Previous studies have reported that the SAH model, induced via
left-sided endovascular puncture, results in a degree of bias
towards bleeding events and can be relatively severe in terms of
damage to left-sided brain tissue (30). Therefore, the left-sided brain
tissue was used to assess the pathological damage following SAH
modeling. Brain tissue proteins were extracted using RIPA lysis
solution (cat. no. P0013B; Beyotime Institute of Biotechnology)
according to the manufacturer's protocol. The protein concentration
of the samples was determined by the BCA assay (cat. no. P0012;
Beyotime Institute of Biotechnology) and subsequently adjusted to 5
µg/µl by adding different amounts of double-distilled water. The 5
µl of 5 µg/µl protein samples were added to each lane and these
protein samples were separated on 10% gels by SDS-PAGE
electrophoresis and transferred to PVDF membranes. The membranes
were blocked for 2 h at room temperature using 5% non-fat powdered
milk (cat. no. P0216-300g; Beyotime Institute of Biotechnology).
The membranes were incubated overnight at 4°C with primary
antibodies as follows: Anti-p-ASK1 (Ser-83; 1:1,000; cat. no.
MA5-28020; Thermo Fisher Scientific, Inc.), anti-ASK1 (1:1,000;
cat. no. 8662; Cell Signaling Technology, Inc.), anti-p-p38 MAPK
(Thr180/Tyr182) (1:1,000; cat. no. 9216; Cell Signaling Technology,
Inc.), anti-p38 MAPK (1:1,000; cat. no. 8690; Cell Signaling
Technology, Inc.), anti-phospho-stress-activated protein
kinase/Jun-amino-terminal kinase (phospho-SAPK/JNK) (Thr183/Tyr185;
1,000; cat. no. 9255; Cell Signaling Technology, Inc.),
anti-SAPK/JNK (JNK; 1:1,000; cat. no. 9252; Cell Signaling
Technology, Inc.), anti-4 hydroxynonenal (4-HNE; 1:1,000; cat. no.
ab46545; Abcam), anti-heme oxygenase 1 (HO-1; 1:1,000; cat. no.
82206; Cell Signaling Technology, Inc.), anti-Bcl-2 (1:1,000; cat.
no. 26593-1-AP; ProteinTech Group, Inc.), anti-Bax (1,1000; cat.
no. 60267-1-Ig; ProteinTech Group, Inc.) and anti-β-actin (1:2,000;
cat. no. 4970; Cell Signaling Technology, Inc.). This was followed
by incubation with the appropriate horseradish
peroxidase-conjugated secondary antibodies for 2 h at room
temperature: Goat anti-mouse IgG-HRP (cat. no. sc-2005; 1:5,000;
Santa Cruz Biotechnology, Inc.) or goat anti-rabbit IgG-HRP (cat.
no. sc-2004; 1:5,000; Santa Cruz Biotechnology, Inc.), and the
specific bands were visualized using an ECL kit (cat. no. P0018AS;
Beyotime Institute of Biotechnology) and imaged using a UVP Che
Studio PLUS system (Analytik Jena GmbH). Relative densitometric
analysis of WB bands was performed using ImageJ 1.4 software (NIH)
and the protein expression levels were normalized against
β-actin.
Experimental design
Expression changes and cellular localization of
ASK1 following SAH
A total of 36 rats were randomly assigned to six
groups (n=6/group) as follows: i) Sham-operated (sham), ii) 3 h
post-SAH, iii) 6 h post-SAH, iv) 12 h post-SAH, v) 24 h post-SAH
and vi) 72 h post-SAH. WB was used to assess changes in the protein
expression levels of endogenous ASK1 and p-ASK1. A total of 4
additional rats were divided into sham (n=2) and SAH 24 h (n=2)
groups and IF staining was used to evaluate the co-localization of
ASK1.
Therapeutic effects of ASK1 inhibitor
NQDI-1 on short- and long-term neurological function following
SAH
A total of 30 rats were divided into 5 groups
(n=6/group) as follows: i) Sham, ii) SAH + vehicle, iii) SAH + 1.0
µg/kg NQDI-1, iv) SAH + 3.0 µg/kg NQDI-1 and v) SAH + 10.0 µg/kg
NQDI-1. The modified Garcia and beam balance scores were used to
assess short-term neurological function 24 h following SAH. Based
on the extent of the short-term neurological improvement, 3.0 µg/kg
NQDI-1 was assessed to be the most appropriate dose group and this
dose of NQDI-1 was used in subsequent experiments. A total of 30
rats were divided into 3 groups (n=10) as follows: i) Sham, ii) SAH
+ vehicle and iii) SAH + NQDI-1. A rotarod experiment was used to
assess long-term neurological function on days 7, 14 and 21 after
SAH, and the Morris water maze test was performed at week 4 after
SAH to assess long-term neurological function.
Therapeutic effects of ASK1 inhibitor
NQDI-1 on oxidative stress and apoptosis
A total of 12 rats were divided into 3 groups
(n=4/group) as follows: i) Sham, ii) SAH + vehicle and iii) SAH +
NQDI-1. (DHE) staining was performed to assess oxidative stress and
TUNEL staining was used to assess neuronal apoptosis 24 h after
SAH.
Role of ASK1/p38 and JNK signaling
pathway in the therapeutic effects of NQDI-1
A total of 48 rats were divided into 8 groups
(n=6/group) as follows: i) Sham, ii) SAH, iii) SAH + vehicle, iv)
SAH + NQDI-1, v) SAH + scrambled (Scr) short interfering (si)RNA,
vi) SAH + ASK1 siRNA, vii) SAH + BMS-582949 and viii) SAH +
SP600125 groups. The ASK1 siRNA, the p38 inhibitor BMS-582949 or
the JNK inhibitor SP600125 were injected to assess whether ASK1,
p38 and JNK were involved in the neuroprotective effects of
NQDI-1.
Statistical analysis
The data were presented as the mean ± standard
deviation and the experimental data of WB were obtained in 6
independent replicates, while DHE staining and TUNEL staining were
performed as 4 independent replicates. Data were analyzed using
SPSS version 17 (SPSS, Inc.). Data from the Morris water maze test
were assessed using a mixed ANOVA/two-way repeated measures ANOVA,
followed by the LSD post hoc test. Data from the remaining
experiments were tested for normal distribution using the
Shapiro-Wilk normal distribution test followed by one-way ANOVA
followed by Tukey's post hoc multiple comparison test. Graphs were
plotted using GraphPad Prism 7 (GraphPad Software, Inc.). P<0.05
was considered to indicate a statistically significant
difference.
Results
Mortality and SAH severity
A total of 191 SD rats were used in the present
study, of which 8 rats were excluded due to only mild SAH (SAH
grading score ≤8). Of the SD rats used in the experiments, 34 rats
were in the sham group and 149 rats underwent SAH modeling. No rats
in the sham group died; however, 23 rats died following SAH
modeling (mortality rate 15.4%) (Table SI, Table SII, Table SIII, Table SIV, Table V). Compared with the sham group,
clots in the subarachnoid space were primarily located near the
Willis ring and on both sides of the basilar artery following SAH
modeling (Fig. 1A). The SAH
grading score 24 h post-SAH modeling demonstrated no significant
difference in the severity of hemorrhage between all non-sham
groups, and a significant difference between sham and all non-sham
groups (Fig. 1B).
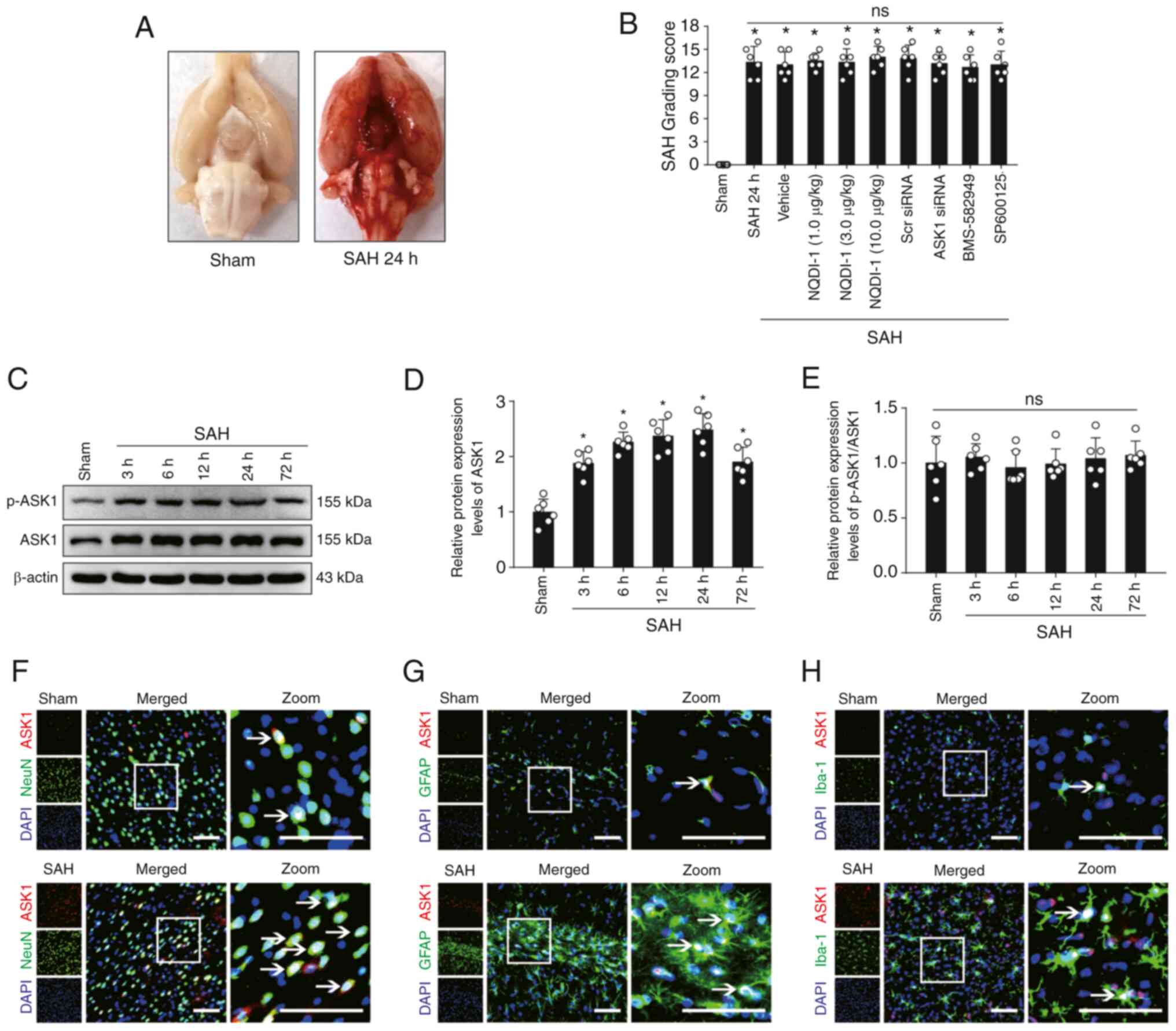 | Figure 1.Expression changes and cellular
localization of ASK1 in rat brain tissue following SAH. (A)
Representative images of rat brain tissue in the sham and SAH 24 h
groups. (B) SAH score of rats 24 h after SAH modeling (n=6; Scr
siRNA was used as the siRNA control). (C) Western blotting images
and semi-quantitative analysis of (D) ASK1 and (E) p-ASK1/ASK1
protein expression levels (n=6). Immunofluorescence of ASK1 with
(F) NeuN-positive neurons, (G) Iba-1-positive microglia and (H)
GFAP-positive astrocytes. White boxes indicate the location of the
zoom plots and white arrows indicate that ASK1 co-localizes with
neuronal cells. Scale bar, 50 µm. All data are presented as mean ±
standard deviation. *P<0.05 vs. sham. ASK1, apoptosis
signal-regulating kinase 1; SAH, subarachnoid hemorrhage; Scr,
scrambled; siRNA, small interfering RNA; NQDI-1,
ethyl-2,7-dioxo-2,7-dihydro-3H-naphtho(1,2,3-de)quinoline-1-carboxylate;
GFAP, glial fibrillary acidic protein; ns, not significant. |
Protein expression changes and
cellular localization of ASK1 following SAH
The protein expression levels of ASK1 increased and
reached a peak at 24 h in the brain following SAH (Fig. 1C and D). However, the ratio of the
protein expression of p-ASK1/ASK1 demonstrated no significant
difference (Fig. 1C and E).
Cellular localization of ASK1 with NeuN-positive neurons,
Iba-1-positive microglia and GFAP-positive astrocytes was
demonstrated in both the sham and SAH 24 h group (Fig. 1F-H).
ASK1 inhibitor NQDI-1 improves
short-term neurological functions 24 h after SAH
NQDI-1 (1, 3 and 10 µg/kg) was injected
intracerebroventricularly 1 h after SAH. Rats undergoing SAH
modeling had significantly lower modified Garcia and beam balance
scores 24 h after SAH modeling; however, all three doses of NQDI-1
led to a significant partial improvement in short-term neurological
function (Fig. 2A and B). The
modified Garcia scores were significantly higher in the SAH +
NQDI-1 (3.0 µg/kg) group compared with the SAH + NQDI-1 (1.0 µg/kg)
group; however, no statistically significant differences were
demonstrated compared with the SAH + NQDI-1 (10.0 µg/kg) group
(Fig. 2A and B), which suggested
that further increasing the concentration of NQDI-1 did not elicit
further improvement in short-term neurological function following
SAH. Therefore, NQDI-1 (3.0 µg/kg) was considered to be the
effective optimal concentration and was used in subsequent
experiments.
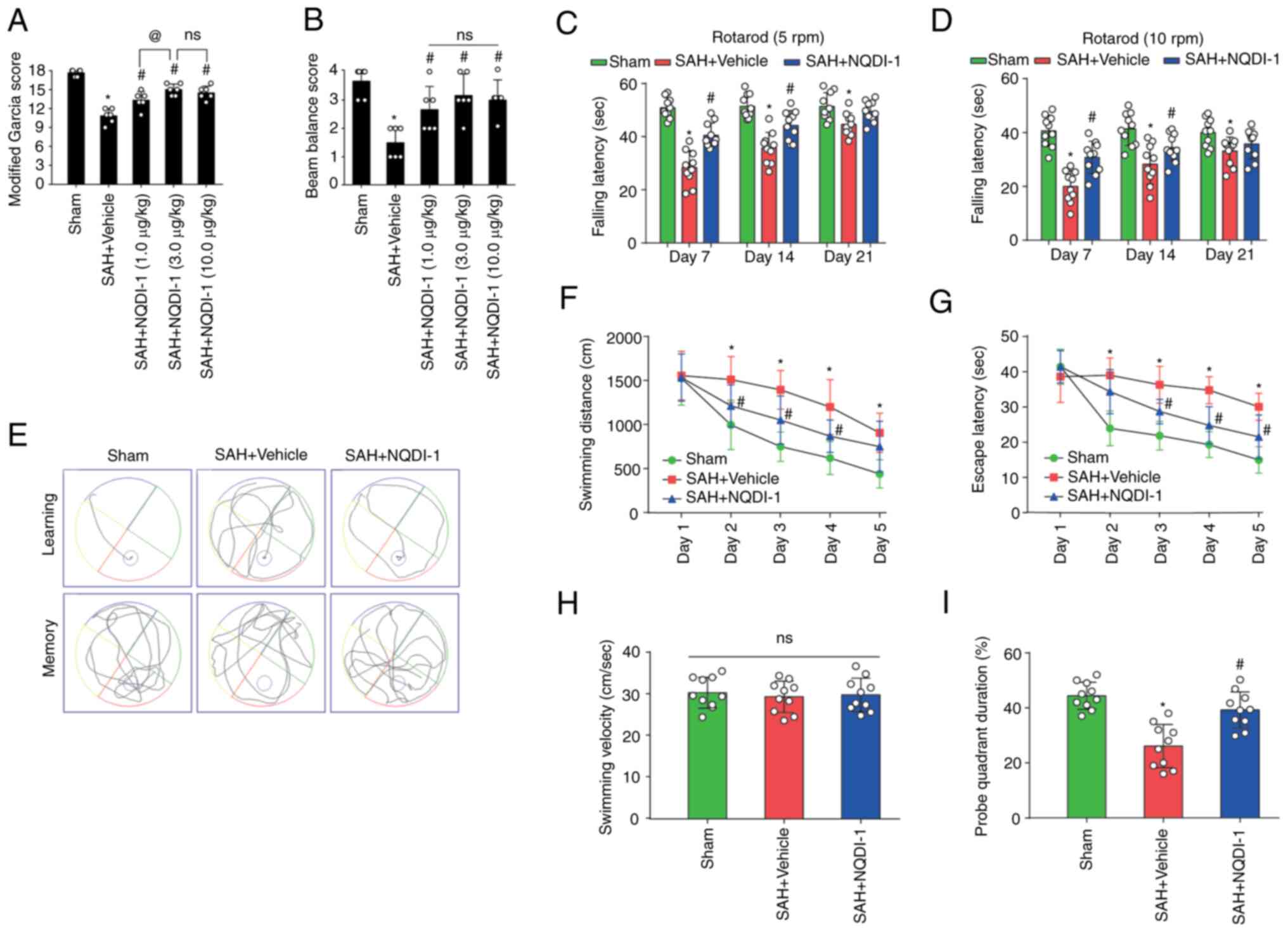 | Figure 2.ASK1 inhibitor NQDI-1 improves short-
and long-term neurological function following SAH. The therapeutic
effects of NQDI-1 on the (A) modified Garcia score and (B) beam
balance score 24 h post-SAH in rats (n=6). Fall latency of rats in
the Rotarod test at initial speeds of (C) 5 and (D) 10 rpm on days
7, 14 and 21 after SAH (n=10). (E) Representative images of the
swimming trajectory in the Morris water maze test. (F) Swimming
distances and (G) escape latency training phase from day 1 to day 5
of the Morris water maze test during week 4 after SAH (n=10). (H)
Mean swimming velocity and (I) probe duration in the target
quadrant during the testing phase of the Morris water maze test
during week 4 post-SAH (n=10) Data are presented as the mean ±
standard deviation. *P<0.05 vs. sham; #P<0.05 vs.
SAH + vehicle; @P<0.05 vs. SAH + NQDI-1 (1.0 µg/kg).
ASK1, apoptosis signal-regulating kinase 1; SAH, subarachnoid
hemorrhage; NQDI-1,
ethyl-2,7-dioxo-2,7-dihydro-3H-naphtho(1,2,3_de)quinoline-1-carboxylate;
GFAP, glial fibrillary acidic protein; ns, not significant; rpm,
revolutions per minute. |
ASK1 inhibitor NQDI-1 improves
long-term neurological functions after SAH
Using starting speeds of 5 or 10 rpm for the Rotarod
test, fall latency was significantly decreased in the SAH + vehicle
group compared with the sham group, whereas the fall latency was
significantly prolonged following intracerebroventricular injection
of NQDI-1 compared with the SAH + vehicle group (Fig. 2C and D). Furthermore, during days
1–5 of the training phase of the Morris water maze test at week 4
post-SAH, the SAH + vehicle group demonstrated significantly longer
escape latency and swimming distance compared with the sham group,
whereas NQDI-1 treatment demonstrated a significant decrease
compared with the SAH + vehicle group (Fig. 2E-G). On the testing phase with
removal of the underwater circular platform, no significant
differences were observed in swimming velocity between the three
groups (Fig. 2H). The probe
quadrant duration was significantly shorter in the SAH + vehicle
group compared with the sham group and NQDI-1 treatment
significantly prolonged the exploration time compared with the SAH
+ vehicle group (Fig. 2I).
ASK1 inhibitor NQDI-1 decreases
oxidative stress and neuronal apoptosis 24 h post-SAH
The level of oxidative stress was measured using DHE
staining and the proportion of apoptotic neurons was assessed using
the percentage of TUNEL-positive neurons 24 h after SAH. The
percentage of DHE-positive cells was significantly higher in the
SAH + vehicle group compared with the sham group and NQDI-1
treatment significantly decreased this compared with the SAH +
vehicle group (Fig. 3A and C). The
percentage of TUNEL-positive neurons was significantly higher in
the SAH + vehicle group compared with the sham group. However, the
percentage of TUNEL-positive neurons decreased significantly
following treatment with NQDI-1 compared with the SAH + vehicle
group (Fig. 3B and D).
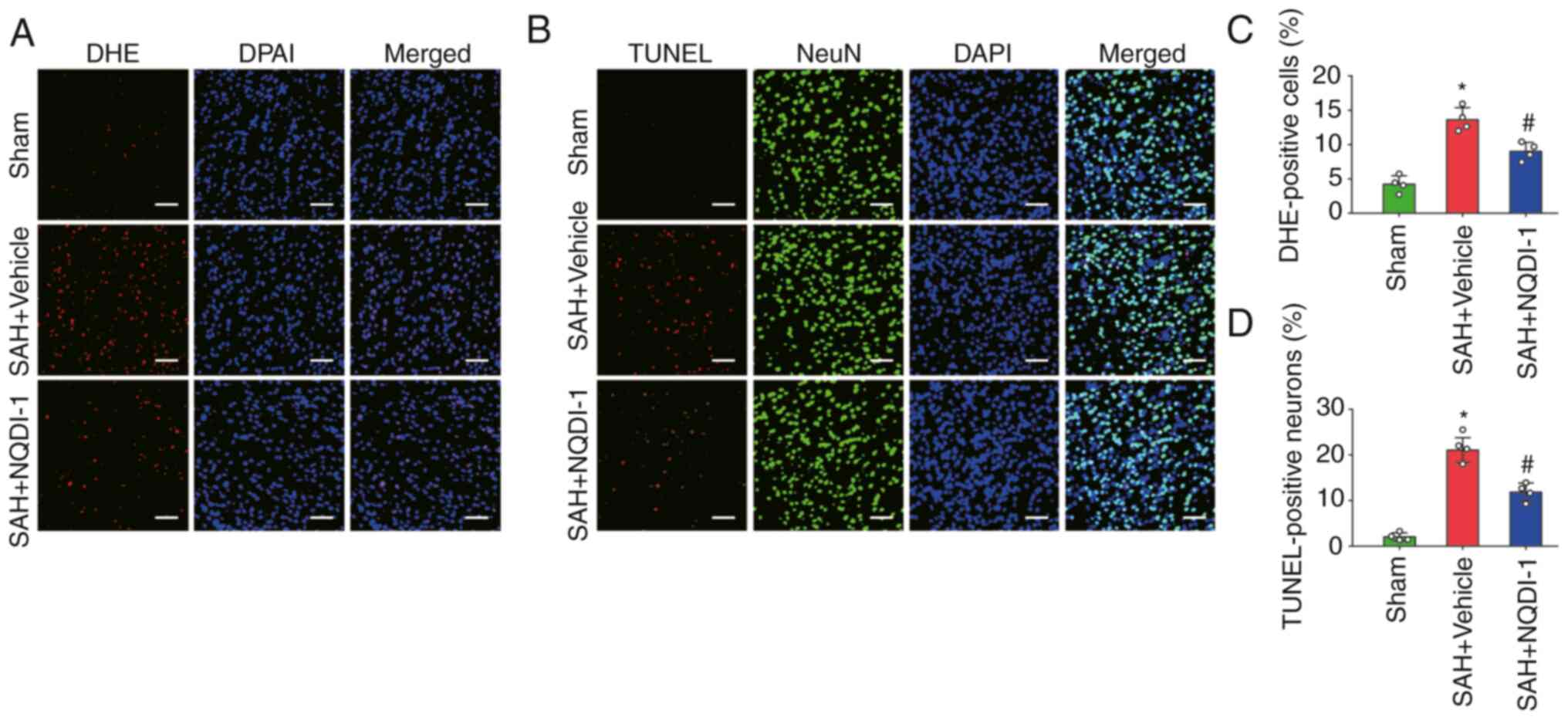 | Figure 3.ASK1 inhibitor NQDI-1 decreases
oxidative stress and neuronal apoptosis 24 h after SAH. (A)
Photomicrographs of DHE staining. Scale bar, 50 µm. (B)
Photomicrographs of TUNEL staining. Scale bar, 50 µm. (C)
Quantitative analysis of DHE staining (n=4). (D) Quantitative
analysis of TUNEL staining (n=4). All data are presented as the
mean ± standard deviation. *P<0.05 vs. sham;
#P<0.05 vs. SAH + vehicle. ASK1, apoptosis
signal-regulating kinase 1; SAH, subarachnoid hemorrhage; NQDI-1,
ethyl-2,7-dioxo-2,7-dihydro-3H-naphtho(1,2,3-de)quinoline-1-carboxylate;
DHE, dihydroethidium. |
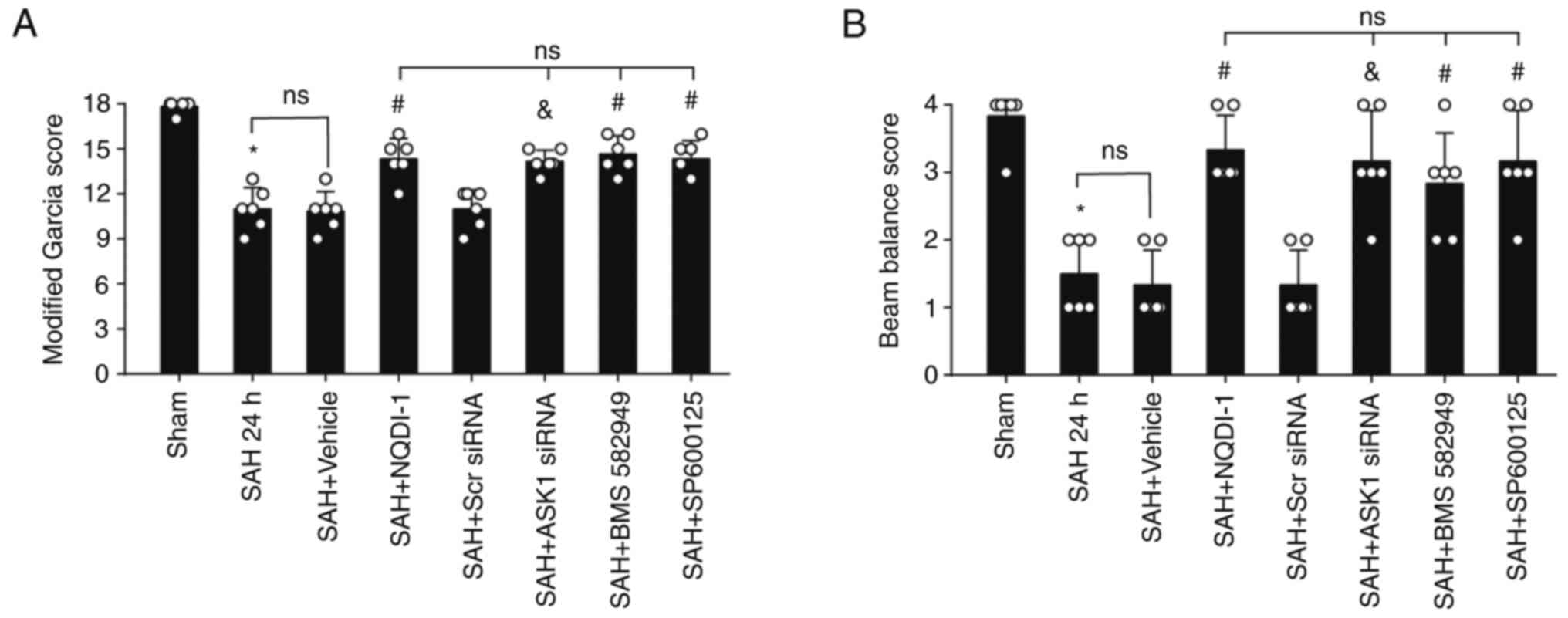
ASK1 siRNA, BMS-582949 and SP600125
improve short-term neurological functions 24 h post-SAH
Compared with the SAH + Scr siRNA group or SAH +
vehicle group, administration of ASK1 siRNA, BMS-582949 or SP600125
demonstrated significant improvement in the modified Garcia and
beam balance score (Fig. 4A and
B).
NQDI-1 attenuates oxidative stress and
apoptosis after SAH via decreased phosphorylation of ASK1, p38 and
JNK
The protein expression levels of ASK1, p-p38, p-JNK,
4-HNE, HO-1, Bax and Bcl-2 were significantly higher after SAH
compared with the sham group; however, the ratio of p-ASK1/ASK1 was
not significantly different (Fig.
5A-M). The injection of vehicle or Scr siRNA did not induce
significant differences in the protein expression levels compared
with the SAH group. Compared with the SAH + vehicle group,
treatment using NQDI-1 caused a significant decrease in the protein
expression levels of p-ASK1/ASK1, p-p38, p-JNK, 4-HNE and Bax,
whereas protein expression levels of HO-1 and Bcl-2 were
significantly increased. Compared with the SAH + Scr siRNA group,
protein expression levels of ASK1 significantly decreased following
injection of ASK1 siRNA. Treatment with ASK1 siRNA also resulted in
a significant decrease in the protein expression levels of p-p38,
p-JNK, 4-HNE and Bax and a significant increase in HO-1 and Bcl-2
expression compared with the SAH + Scr siRNA group.
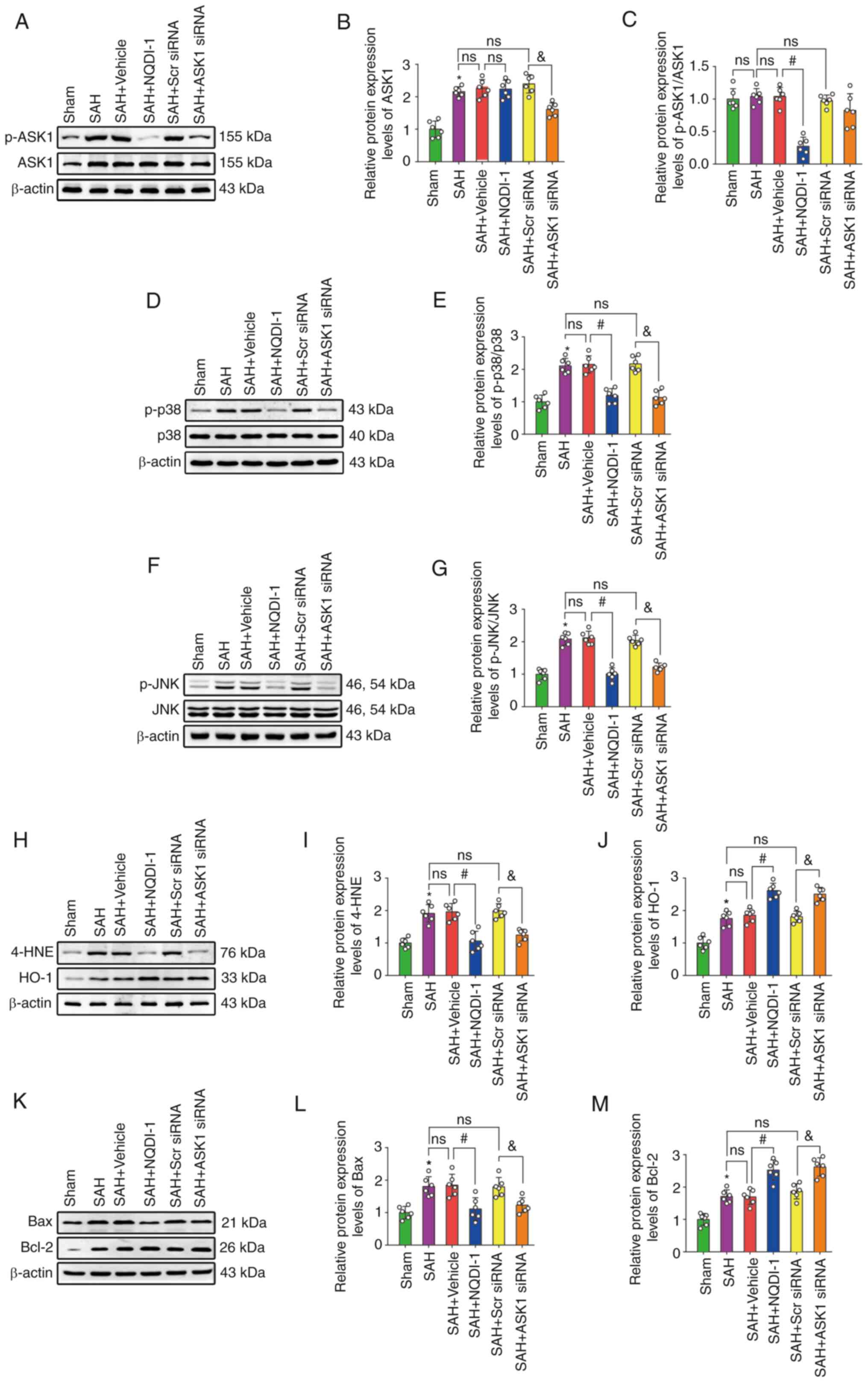 | Figure 5.NQDI-1 attenuates oxidative stress
and apoptosis following SAH via decreased phosphorylation of ASK1,
p38 and JNK. (A) Western blotting images of p-ASK1 and ASK1.
Semi-quantitative analysis of (B) ASK1 protein expression levels
(n=6) and (C) p-ASK1/ASK1 protein expression levels (n=6). (D)
Western blotting images of p-p38 and p38. (E) Semi-quantitative
analysis of p-p38/p38 protein expression levels (n=6). (F) Western
blotting images of p-JNK and JNK. (G) Semi-quantitative analysis of
p-JNK/JNK protein expression levels (n=6). (H) Western blotting
images of 4-HNE and HO-1. Semi-quantitative analysis of (I) 4-HNE
protein expression levels (n=6) and (J) HO-1 protein expression
levels (n=6). (K) Western blotting images of Bax and Bcl-2.
Semi-quantitative analysis of (L) Bax protein expression levels
(n=6) and (M) Bcl-2 protein expression levels (n=6). Scr siRNA was
used as the control. All data are presented as the mean ± standard
deviation. *P<0.05 vs. sham; #P<0.05 vs. SAH +
vehicle; &P<0.05 vs. SAH + Scr siRNA. ASK1,
apoptosis signal-regulating kinase 1; SAH, subarachnoid hemorrhage;
NQDI-1,
ethyl-2,7-dioxo-2,7-dihydro-3H-naphtho(1,2,3-de)quinoline-1-carboxylate;
p, phosphorylated; 4-HNE, 4 hydroxynonenal; HO-1, heme oxygenase 1;
Scr, scrambled; siRNA, small interfering RNA. |
p38 inhibitor BMS-582949 reduces the
protein expression levels of p-p38 but has no effect on p-ASK1,
ASK1 or p-JNK protein expression levels
Treatment with the p38 inhibitor BMS-582949
demonstrated a significant decrease in protein expression levels of
p-p38, 4-HNE and Bax and also significantly increased protein
expression of HO-1 and Bcl-2 compared with the SAH + vehicle group;
however, the protein expression levels of ASK1, p-ASK1/ASK1 and
p-JNK demonstrated no significant differences (Fig. 6A-M).
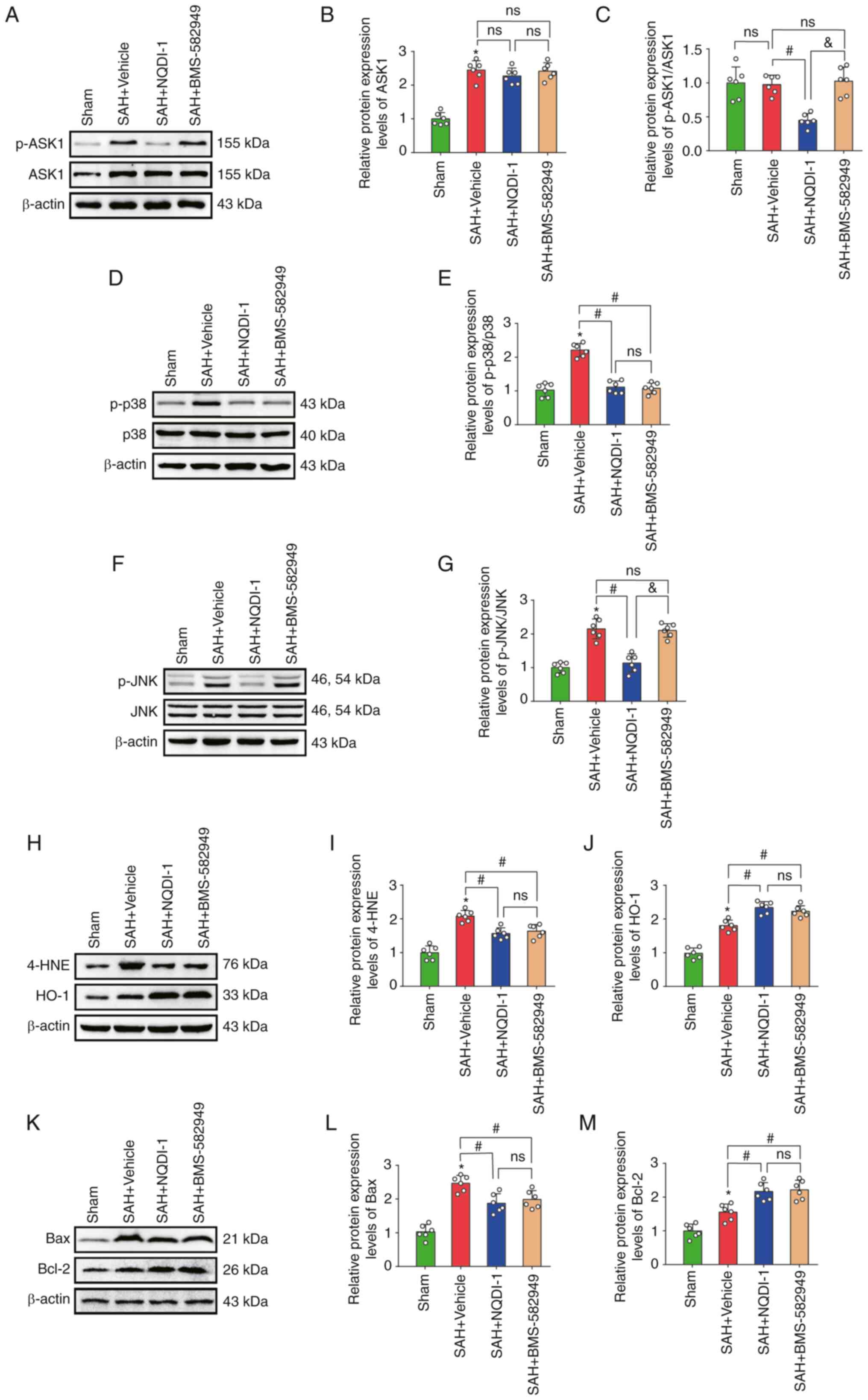 | Figure 6.p38 inhibitor BMS 582949 decreases
protein expression of p-p38 but has no effect on protein expression
levels of p-ASK1, ASK1 or p-JNK. (A) Western blotting images of
p-ASK1 and ASK1. Semi-quantitative analysis of (B) ASK1 protein
expression levels (n=6) and (C) p-ASK1/ASK1 protein expression
levels (n=6). (D) Western blotting images of p-p38 and p38. (E)
Semi-quantitative analysis of p-p38/p38 protein expression levels
(n=6). (F) Western blotting images of p-JNK and JNK. (G)
Semi-quantitative analysis of p-JNK/JNK protein expression levels
(n=6). (H) Western blotting images of 4-HNE and HO-1.
Semi-quantitative analysis of (I) 4-HNE protein expression levels
(n=6) and (J) HO-1 protein expression levels (n=6). (K) Western
blotting images of Bax and Bcl-2. Semi-quantitative analysis of (L)
Bax protein expression levels (n=6) and (M) Bcl-2 protein
expression levels (n=6). Data are presented as the mean ± standard
deviation. *P<0.05 vs. sham; #P<0.05 vs. SAH +
vehicle; &P<0.05 vs. SAH + NQDI-1, apoptosis
signal-regulating kinase 1; SAH, subarachnoid hemorrhage; NQDI-1,
ethyl-2,7-dioxo-2,7-dihydro-3H-naphtho(1,2,3-de)quinoline-1-carboxylate;
p, phosphorylated; 4-HNE, 4 hydroxynonenal; HO-1, heme oxygenase 1;
Scr, scrambled; siRNA, small interfering RNA; ns, not
significant. |
JNK inhibitor SP600125 decreases
protein expression level of p-JNK but has no effect on p-ASK1, ASK1
or p-p38 protein expression levels
Treatment with JNK inhibitor SP600125 caused a
significant decrease in the protein expression levels of p-JNK,
4-HNE and Bax and a significant increase in HO-1 and Bcl-2 protein
expression levels compared with the SAH + vehicle group; however,
protein expression levels of ASK1, p-ASK1/ASK1 and p-p38 were not
significantly altered (Fig.
7A-M).
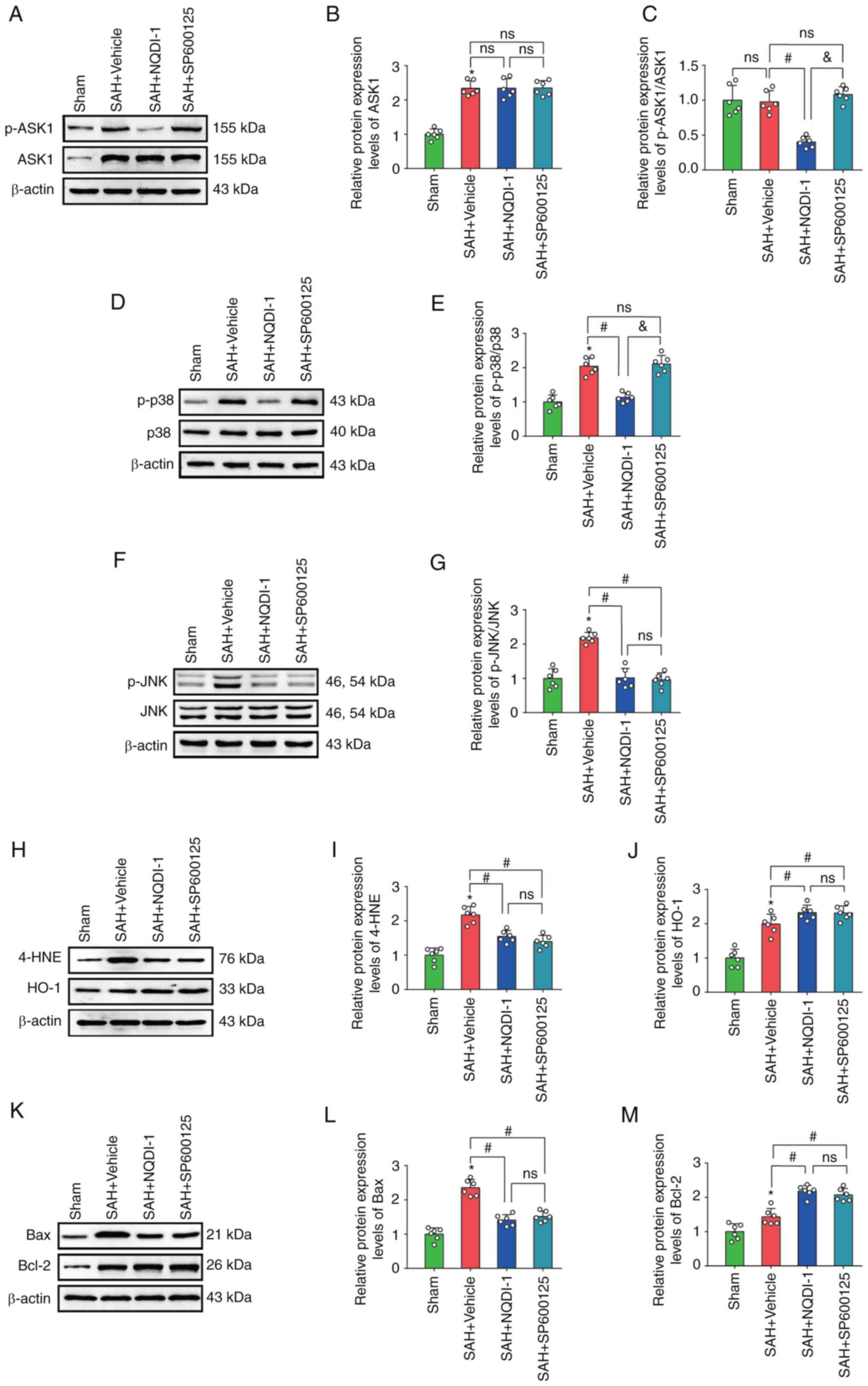 | Figure 7.JNK inhibitor SP600125 decreases
expression of p-JNK, but has no effect on p-ASK1, ASK1 or p-p38.
(A) Western blotting images of p-ASK1 and ASK1. Semi-quantitative
analysis of (B) ASK1 protein expression levels (n=6) and (C)
p-ASK1/ASK1 protein expression levels (n=6). (D) Western blotting
images of p-p38 and p38. (E) Semi-quantitative analysis of
p-p38/p38 protein expression levels (n=6). (F) Western blotting
images of p-JNK and JNK. (G) Semi-quantitative analysis of
p-JNK/JNK protein expression levels (n=6). (H) Western blotting
images of 4-HNE and HO-1. Semi-quantitative analysis of (I) 4-HNE
protein expression levels (n=6) and (J) HO-1 protein expression
levels (n=6). (K) Western blotting images of Bax and Bcl-2.
Semi-quantitative analysis of (L) Bax protein expression levels
(n=6) and (M) Semi Bcl-2 protein expression levels (n=6). All data
are presented as mean ± standard deviation. *P<0.05 vs. sham;
#P<0.05 vs. SAH + vehicle; &P<0.05
vs. SAH + NQDI-1, apoptosis signal-regulating kinase 1; SAH,
subarachnoid hemorrhage; NQDI-1,
ethyl-2,7-dioxo-2,7-dihydro-3H-naphtho(1,2,3-de)quinoline-1-carboxylate;
p, phosphorylated; 4-HNE, 4 hydroxynonenal; HO-1, heme oxygenase 1;
Scr, scrambled; siRNA, small interfering RNA; ns, not
significant. |
Discussion
In the present study, protein expression levels of
ASK1 and p-ASK1 were elevated following SAH. ASK1 inhibitor NQDI-1
improved short- and long-term neurological function after SAH and
decreased oxidative stress and neuronal apoptosis via inhibition of
ASK1 phosphorylation and the ASK1/p38 and JNK signaling pathway in
EBI following SAH.
ASK1 is a member of the MAP3K family and its
activation in response to numerous types of cellular stress
mediates different types of cellular damage (31). For certain diseases involving
oxidative stress and apoptosis, decreased protein expression of
ASK1 or inhibition of its phosphorylation may have a beneficial
role by decreasing oxidative stress and apoptosis (32). To the best of our knowledge,
however, the role and underlying mechanism of action of ASK1 and
its inhibitor NQDI-1 have not previously been reported in SAH. The
present study demonstrated that the protein expression levels of
ASK1 and p-ASK1 were significantly increased following SAH
modeling, which suggested that ASK1 may serve a role in EBI after
SAH. Furthermore, ASK1 was co-expressed in neurons, microglia and
astrocytes, which suggested a wide role for ASK1 in a variety of
neuronal cells following SAH.
NQDI-1 is a specific inhibitor of ASK1 and its
functional role has been previously demonstrated using in
vitro kinase assays (11,33).
In a mouse model of acute pancreatitis, NQDI-1 decreases pancreatic
follicular cell necrosis through decrease of ROS production and
receptor interacting serine/threonine kinase 3 and p-mixed lineage
kinase domain-like pseudokinase protein expression levels (34). In ischemic brain injury, NQDI-1
inhibition of ASK1 decreases matrix metalloproteinase 9 activity
and subsequent neuronal apoptosis in brain endothelial cells
(35). In the present study,
different concentrations of NQDI-1 were injected
intracerebroventricularly, which significantly improved the
modified Garcia and balance beam test score, which suggested that
NQDI-1 led to improvements in short-term neurological function
following SAH.
For long-term neurological function, two methods
were used to assess function over different time periods (36). The Rotarod test was used to assess
the coordinated balance of locomotion on day 7, 14 and 21. At
initial speeds of 5 or 10 rpm, the fall latency of rats in the SAH
+ vehicle group was significantly shorter compared with that in the
sham group and treatment with NQDI-1 led to a significant increase.
The Morris water maze test was used to assess the spatial memory
and learning ability of rats during the fourth week. During the
training phase of days 1–5, the SAH group rats swam for
significantly longer times and for further distances compared with
the sham group and NQDI-1 treatment decreased this phenomenon.
During the testing phase, no significant differences were
demonstrated in the swimming speed between groups of rats. After
removal of the platform, the time spent exploring the target
quadrant was observed and counted for each group of rats. Following
SAH, rats searched for the target quadrant for a shorter time,
which indicated that SAH modeling led to more ambiguous spatial
localization and memory in rats and impaired long-term memory
capacity, whereas NQDI-1 treatment demonstrated improvements in
these outcomes. Rotarod experiment and the Morris water maze test
results suggested that NQDI-1 improved long-term neurological
function after SAH.
Oxidative stress and neuroapoptosis are key
pathological changes of EBI following SAH (37). In steady-state cells, ROS are
primarily byproducts of respiration produced by the mitochondrial
electron respiratory chain and moderate levels of ROS repair
damaged DNA and serve a physiological role in the promotion of cell
survival (38). Upon SAH, due to
the autoxidation of blood in the subarachnoid space, ROS catalysis
by heme and intracellular mitochondrial dysfunction, electrons
escape into the cytoplasm and the antioxidant system is
insufficient to compensate, which results in accumulation of large
amounts of ROS in neuronal cells (39), which leads to oxidative stress
damage. Therefore, therapeutic strategies that target oxidative
stress and apoptosis may be considered effective therapeutic
directions following SAH. In the present study, ROS and oxidative
stress of brain tissues were assessed using DHE, which is oxidized
into ethidium and produces a red fluorescent signal (40). The percentage of DHE-positive cells
significantly increased in the SAH + vehicle group, whereas a
decrease was demonstrated in the percentage of DHE-positive cells
in the SAH + NQDI-1 group, which suggested that NQDI-1 decreased
oxidative stress. Apoptosis was assessed using TUNEL and the
percentage of TUNEL-positive neuronal cells following SAH modeling
significantly increased, whereas NQDI-1 significantly decreased the
percentage of TUNEL-positive neuronal cells. It may be hypothesized
that treatment NQDI-1 decreased oxidative stress and apoptosis in
EBI following SAH.
The effect of NQDI-1 on ASK1 and the potential
underlying molecular mechanism were evaluated. The effects of
NQDI-1 on protein expression levels of ASK1 and p-ASK1 were first
assessed. The ratio of p-ASK1/ASK1 did not change following SAH,
which suggested that changes in the phosphorylation of ASK1 were
similar to those of ASK1 protein expression levels. Treatment with
NQDI-1 demonstrated a significant decrease in p-ASK1 protein
expression levels but demonstrated no effect on ASK1 compared with
the SAH + vehicle group, which suggested that NQDI-1 exerted its
neuroprotective effects primarily via inhibition of ASK1
phosphorylation. Subsequently, protein expression levels of both
p-ASK1 and ASK1 were knocked down using ASK1 siRNA, following which
protein expression levels of p-p38 and p-JNK were significantly
decreased, which suggested that ASK1 was activated primarily via
phosphorylation.
The p38, MAPK and JNK signaling pathways are widely
expressed in brain tissue (41).
Activated p38, MAPK and JNK enhance tumor necrosis factor-induced
apoptosis, participate in the Fas/FasL system, phosphorylate P53
and induce mitochondrial translocation of BAX and other pathways to
promote apoptosis (42).
Inhibition of the phosphorylation activation of p38 MAPK and JNK
decreases oxidative stress and neuronal apoptosis, alleviates EBI
and improves the prognosis of the SAH rat model (43). After injection of the p38 inhibitor
BMS-582949, the WB results demonstrated that it significantly
inhibited phosphorylation of p38 and significantly downregulated
expression levels of oxidative stress and apoptosis-associated
proteins. However, BMS-582949 did not cause a significant
difference in the protein expression levels of p-ASK1, ASK1 or
p-JNK. Similarly, the JNK inhibitor SP600125 significantly
inhibited phosphorylation of JNK, but demonstrated no significant
effect on p-ASK1, ASK1 or p-p38 protein expression levels. These
results suggested that p38 and JNK were downstream of p-ASK1 and
that ASK1 caused oxidative stress and apoptosis following SAH via
phosphorylation-activated p38 and JNK.
There were certain limitations associated with the
present study. Firstly, NQDI-1 was only administered once via
intracerebroventricular injection 1 h after SAH; therefore, the
current study was not suitable to determine the optimal therapeutic
window for NQDI-1 treatment and future studies are required to
address this issue. Secondly, the present study was a pilot study
to evaluate the effect of inhibition of ASK1 on neurological
function and to explore whether NQDI-1 had a therapeutic effect
following SAH. The role of ASK1 in astrocytes or microglia and the
pharmacokinetics of NQDI-1 require further elucidation in future
studies. The Morris water maze test was only performed during the
fourth week post-SAH; therefore, potential changes in early ability
of spatial memory and learning ability may not have been assessed.
Finally, IF was used to assess ASK1 co-localization; however, only
2 animals/experimental group was assessed. The small sample size is
a limitation of the present study that may result in inappropriate
conclusions and should only be used for qualitative, rather than
quantitative, assessment.
In conclusion, ASK1 inhibitor NQDI-1 decreased
oxidative stress and apoptosis and improved short and long-term
neurological function following SAH via inhibition of ASK1
phosphorylation and the p38 and JNK signaling pathways. NQDI-1 may
be a potential therapeutic agent for treatment of SAH.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by The National Natural Science
Foundation of China (grant no. 81870944), The Beijing Science and
Technology Plan Subject: Beijing-Tianjin-Hebei Collaborative
Innovation Promotion Project (grant no. Z181100009618035), The
National Natural Science Foundation of China (grant no. 81771233),
Beijing Municipal Administration of Hospitals' Ascent Plan (grant
no. DFL20190501) and Research and Promotion Program of Appropriate
Techniques for Intervention of Chinese High-risk Stroke People
(grant no. GN-2020R0007).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JD, WY, JJ, FL and AL participated in the
experimental design. JD, JJ, JW and XY performed the experiments
and collected and analyzed the data. XY, FL and AL interpreted the
data. JD and WY drafted the manuscript. XY, FL and AL revised the
manuscript and proofread the language. All authors have read and
approved the final manuscript. JD, JW and FL confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
All experimental procedures were approved by the
Institutional Animal Care and Use Committee of Central South
University (approval. no. 2019sydw0104).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lawton MT and Vates GE: Subarachnoid
Hemorrhage. N Engl J Med. 377:257–266. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
GBD 2019 Stroke Collaborators, . Global,
regional, and national burden of stroke and its risk factors,
1990–2019: A systematic analysis for the Global Burden of Disease
Study 2019. Lancet Neurol. 20:795–820. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Osgood ML: Aneurysmal subarachnoid
hemorrhage: Review of the pathophysiology and management
strategies. Curr Neurol Neurosci Rep. 21:502021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Topkoru B, Egemen E, Solaroglu I and Zhang
JH: Early brain injury or vasospasm? An overview of common
mechanisms. Curr Drug Targets. 18:1424–1429. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Merlini E, Coleman MP and Loreto A:
Mitochondrial dysfunction as a trigger of programmed axon death.
Trends Neurosci. 45:53–63. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kowalczyk P, Sulejczak D, Kleczkowska P,
Bukowska-Ośko I, Kucia M, Popiel M, Wietrak E, Kramkowski K,
Wrzosek K and Kaczyńska K: Mitochondrial oxidative stress-A
causative factor and therapeutic target in many diseases. Int J Mol
Sci. 22:133842021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsushita M, Nakamura T, Moriizumi H,
Miki H and Takekawa M: Stress-responsive MTK1 SAPKKK serves as a
redox sensor that mediates delayed and sustained activation of
SAPKs by oxidative stress. Sci Adv. 6:eaay97782020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao Y, Yan Y, Fang Q, Zhang N, Kumar G,
Zhang J, Song LJ, Yu J, Zhao L, Zhang HT and Ma CG: The Rho kinase
inhibitor fasudil attenuates Aβ1-42-induced apoptosis
via the ASK1/JNK signal pathway in primary cultures of hippocampal
neurons. Metab Brain Dis. 34:1787–1801. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Volynets GP, Chekanov MO, Synyugin AR,
Golub AG, Kukharenko OP, Bdzhola VG and Yarmoluk SM: Identification
of 3H-naphtho[1,2,3-de]quinoline-2,7-diones as inhibitors of
apoptosis signal-regulating kinase 1 (ASK1). J Med Chem.
54:2680–2686. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang QS, Kurpad DS, Mahoney MG, Steinbeck
MJ and Freeman TA: Inhibition of apoptosis signal-regulating kinase
1 alters the wound epidermis and enhances auricular cartilage
regeneration. PLoS One. 12:e01858032017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hao H, Li S, Tang H, Liu B, Cai Y, Shi C
and Xiao X: NQDI-1, an inhibitor of ASK1 attenuates acute perinatal
hypoxic-ischemic cerebral injury by modulating cell death. Mol Med
Rep. 13:4585–4592. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen S, Zuo Y, Huang L, Sherchan P, Zhang
J, Yu Z, Peng J, Zhang J, Zhao L, Doycheva D, et al: The MC
receptor agonist RO27-3225 inhibits NLRP1-dependent neuronal
pyroptosis via the ASK1/JNK/p38 MAPK pathway in a mouse model of
intracerebral haemorrhage. Br J Pharmacol. 176:1341–1356. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Du X, Liu H, Liu X, Chen X, Yuan L, Ma Y,
Huang H, Wang Y, Wang R, Zhang S, et al: Microcystin-LR induces
ovarian injury and apoptosis in mice via activating apoptosis
signal-regulating kinase 1-mediated P38/JNK pathway. Ecotoxicol
Environ Saf. 213:1120662021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chang CY, Li JR, Wu CC, Wang JD, Liao SL,
Chen WY, Wang WY and Chen CJ: Endoplasmic reticulum stress
contributes to indomethacin-induced glioma apoptosis. Int J Mol
Sci. 21:5572020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Win S, Than TA, Zhang J, Oo C, Min RWM and
Kaplowitz N: New insights into the role and mechanism of
c-Jun-N-terminal kinase signaling in the pathobiology of liver
diseases. Hepatology. 67:2013–2024. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Percie du Sert N, Ahluwalia A, Alam S,
Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U,
Emerson M, et al: Reporting animal research: Explanation and
elaboration for the ARRIVE guidelines 2.0. PLoS Biol.
18:e30004112020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
McPherson C: Regulation of animal care and
research? NIH's opinion. J Anim Sci. 51:492–496. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Boivin GP, Hickman DL, Creamer-Hente MA,
Pritchett-Corning KR and Bratcher NA: Review of CO2 as a
euthanasia agent for laboratory rats and mice. J Am Assoc Lab Anim
Sci. 56:491–499. 2017.PubMed/NCBI
|
|
19
|
Luo K, Wang Z, Zhuang K, Yuan S, Liu F and
Liu A: Suberoylanilide hydroxamic acid suppresses axonal damage and
neurological dysfunction after subarachnoid hemorrhage via the
HDAC1/HSP70/TDP-43 axis. Exp Mol Med. 54:1423–1433. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xie Z, Enkhjargal B, Nathanael M, Wu L,
Zhu Q, Zhang T, Tang J and Zhang JH: viaExendin-4 preserves
blood-brain barrier integrity glucagon-like peptide 1
receptor/activated protein kinase-dependent nuclear Factor-Kappa
B/Matrix Metalloproteinase-9 inhibition after subarachnoid
hemorrhage in rat. Front Mol Neurosci. 14:7507262021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Craft TK and DeVries AC: Role of IL-1 in
poststroke depressive-like behavior in mice. Biol Psychiatry.
60:812–818. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xie Z, Enkhjargal B, Wu L, Zhou K, Sun C,
Hu X, Gospodarev V, Tang J, You C and Zhang JH: Exendin-4
attenuates neuronal death via GLP-1R/PI3K/Akt pathway in early
brain injury after subarachnoid hemorrhage in rats.
Neuropharmacology. 128:142–151. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu C, Lin J, Wrobleski ST, Lin S, Hynes
J, Wu H, Dyckman AJ, Li T, Wityak J, Gillooly KM, et al: Discovery
of
4-(5-(cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide
(BMS-582949), a clinical p38α MAP kinase inhibitor for the
treatment of inflammatory diseases. J Med Chem. 53:6629–6639. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bennett BL, Sasaki DT, Murray BW, O'Leary
EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et
al: SP600125, an anthrapyrazolone inhibitor of Jun N-terminal
kinase. Proc Natl Acad Sci USA. 98:13681–13686. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dai J, Xu S, Okada T, Liu Y, Zuo G, Tang
J, Zhang JH and Shi H: T0901317, an agonist of liver X receptors,
attenuates neuronal apoptosis in early brain injury after
subarachnoid hemorrhage in rats via liver X receptors/interferon
regulatory factor/P53 upregulated modulator of
apoptosis/dynamin-1-like protein pathway. Oxid Med Cell Longev.
2021:88491312021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xiao ZP, Lv T, Hou PP, Manaenko A, Liu Y,
Jin Y, Gao L, Jia F, Tian Y, Li P, et al: Sirtuin 5-Mediated lysine
desuccinylation protects mitochondrial metabolism following
subarachnoid hemorrhage in mice. Stroke. 52:4043–4053. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xu W, Yan J, Ocak U, Lenahan C, Shao A,
Tang J, Zhang J and Zhang JH: viaMelanocortin 1 receptor attenuates
early brain injury following subarachnoid hemorrhage by controlling
mitochondrial metabolism AMPK/SIRT1/PGC-1α pathway in rats.
Theranostics. 11:522–539. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mo J, Enkhjargal B, Travis ZD, Zhou K, Wu
P, Zhang G, Zhu Q, Zhang T, Peng J, Xu W, et al: AVE 0991
attenuates oxidative stress and neuronal apoptosis via
Mas/PKA/CREB/UCP-2 pathway after subarachnoid hemorrhage in rats.
Redox Biol. 20:75–86. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
He Y, Zheng Z, Liu C, Li W, Zhao L, Nie G
and Li H: viaInhibiting DNA methylation alleviates
cisplatin-induced hearing loss by decreasing oxidative
stress-induced mitochondria-dependent apoptosis the LRP1-PI3K/AKT
pathway. Acta Pharm Sin B. 12:1305–1321. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
van Lieshout JH, Marbacher S, Muhammad S,
Boogaarts HD, Bartels RHMA, Dibué M, Steiger HJ, Hänggi D and Kamp
MA: Proposed definition of experimental secondary ischemia for
mouse subarachnoid hemorrhage. Transl Stroke Res. 11:1165–1170.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ogier JM, Nayagam BA and Lockhart PJ: ASK1
inhibition: A therapeutic strategy with multi-system benefits. J
Mol Med (Berl). 98:335–348. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang XS, Lu Y, Li W, Tao T, Peng L, Wang
WH, Gao S, Liu C, Zhuang Z, Xia DY, et al: Astaxanthin ameliorates
oxidative stress and neuronal apoptosis via
SIRT1/NRF2/Prx2/ASK1/p38 after traumatic brain injury in mice. Br J
Pharmacol. 178:1114–1132. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen S, Yu Q, Song Y, Cui Z, Li M, Mei C,
Cui H, Cao S and Zhu C: Inhibition of macrophage migration
inhibitory factor (MIF) suppresses apoptosis signal-regulating
kinase 1 to protect against liver ischemia/reperfusion injury.
Front Pharmacol. 13:9519062022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xie X, Yuan C, Yin L, Zhu Q, Ma N, Chen W,
Ding Y, Xiao W, Gong W, Lu G, et al: NQDI-1 protects against acinar
cell necrosis in three experimental mouse models of acute
pancreatitis. Biochem Biophys Res Commun. 520:211–217. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cheon SY, Cho KJ, Kim SY, Kam EH, Lee JE
and Koo BN: Blockade of apoptosis signal-regulating kinase 1
attenuates matrix metalloproteinase 9 activity in brain endothelial
cells and the subsequent apoptosis in neurons after ischemic
injury. Front Cell Neurosci. 10:2132016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhu Q, Enkhjargal B, Huang L, Zhang T, Sun
C, Xie Z, Wu P, Mo J, Tang J, Xie Z and Zhang JH: Aggf1 attenuates
neuroinflammation and BBB disruption via PI3K/Akt/NF-κB pathway
after subarachnoid hemorrhage in rats. J Neuroinflammation.
15:1782018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wu Y, Liu Y, Zhou C, Wu Y, Sun J, Gao X
and Huang Y: Biological effects and mechanisms of caspases in early
brain injury after subarachnoid hemorrhage. Oxid Med Cell Longev.
2022:33456372022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Checa J and Aran JM: Reactive oxygen
species: Drivers of Physiological and pathological processes. J
Inflamm Res. 13:1057–1073. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Z, Zhang A, Liu Y, Hu X, Fang Y,
Wang X, Luo Y, Lenahan C and Chen S: New Mechanisms and Targets of
Subarachnoid Hemorrhage: A Focus on Mitochondria. Curr
Neuropharmacol. 20:1278–1296. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu B, Tian Y, Li Y, Wu P, Zhang Y, Zheng
J and Shi H: ACEA Attenuates Oxidative Stress by Promoting
Mitophagy via CB1R/Nrf1/PINK1 Pathway after Subarachnoid Hemorrhage
in Rats. Oxid Med Cell Longev. 2022:10242792022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Iroegbu JD, Ijomone OK, Femi-Akinlosotu OM
and Ijomone OM: ERK/MAPK signalling in the developing brain:
Perturbations and consequences. Neurosci Biobehav Rev. 131:792–805.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Anjum J, Mitra S, Das R, Alam R, Mojumder
A, Emran TB, Islam F, Rauf A, Hossain MJ, Aljohani ASM, et al: A
renewed concept on the MAPK signaling pathway in cancers:
Polyphenols as a choice of therapeutics. Pharmacol Res.
184:1063982022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wei YX, Zhang DD, Gao YY, Hang CH and Shi
JX: Inhibition of the myeloid differentiation primary response
protein 88 reducres neuron injury in the early stages of
subarachnoid hemorrhage in an in vitro experimental model. J
Physiol Pharmacol. 73:2022.PubMed/NCBI
|