Introduction
Metformin, a biguanide derivative, is a first-line
oral medication for type 2 diabetes mellitus. The best known
effects of metformin include the suppression of hepatic glucose
production and reduction of insulin resistance in peripheral
tissues (1). In addition, the drug
enhances both peripheral glucose uptake and fatty acid oxidation
(1). The Diabetes Prevention Trial
demonstrated that metformin decreased the incidence of diabetes
development in at risk populations by ∼30% (2).
The key mechanism of metformin action is the
activation of its major effector molecule, the adenosine
monophosphate-activated protein kinase (AMPK), a sensor of cellular
energy that is normally activated under conditions of starvation
(3–5). The upregulation of AMPK inhibits
several anabolic/mitogenic pathways activated by growth factors and
nutrients, including the phosphatidylinositol-3-kinase (PI-3K)
pathway, the mammalian target of rapamycin (mTOR) and extracellular
signal-regulated kinases 1 and 2 (ERK1/2) (6). Further downstream, the consequences of
AMPK stimulation may include the inhibition of the cell cycle
regulator cyclin D1, the downregulation of critical transcriptional
regulators such as hypoxia inducible factor 1α, nuclear factor κB
and the c-myc protein, and depletion of other mitosis-related
proteins (7). On a cellular level,
in addition to its insulin-sensitizing effects, the drug is known
to inhibit cell growth, migration, invasion and angiogenesis
(7).
The above effects are particularly attractive in the
context of the potential use of metformin in restricting
premalignant or malignant cell growth. Indeed, in cellular models,
metformin inhibited the growth of breast, colorectal, pancreatic,
lung, ovarian and prostate cancer cells (7). The drug also suppressed the expression
of human epidermal growth factor receptor 2 (HER2) in certain
breast cancer cell lines. In animal models, metformin reduced
chemically-induced carcinogenesis in various organs (mammary gland,
intestine, endometrium, skin, lung and pancreas), and inhibited the
growth of breast cancer xenografts and mammary tumors in HER2
transgenic mice (7–12).
Epidemiological studies in diabetic patients have
suggested that metformin significantly reduces the risk of
pancreatic, liver, colorectal, breast, endometrial and bladder
cancer development, and decreases cancer-related mortality
(7). Moreover, the drug improves
the response to neoadjuvant breast cancer therapy in diabetic women
(7). At present, several
prospective clinical trials are evaluating metformin as a single or
combined treatment for solid tumors (7) and demonstrate its potential value for
cancer prevention (13) (www.clinicaltrials.gov).
Studies in vivo demonstrated that metformin
can, at least to some extent, cross the blood-brain barrier (BBB)
through an organic cation transporter-dependent mechanism and exert
pharmacological effects, including AMPK activation, in intact brain
(8,14) and glioma cells in vitro
(4). Notably, the AMPK pathway
appears to be critical for the growth of epidermal growth factor
receptor-dependent glioblastoma multiforme (GBM), and the
activation of AMPK by its agonist significantly reduces GBM
proliferation (15). However, only
a few studies addressed the effects of the leading AMPK-inducing
pharmaceutical agent, metformin, on brain tumor biology. The
results suggest that the drug reduces the growth and/or migration
of different rat or human glioma cell lines that have a mutation in
the phosphatase and tensin homolog (PTEN) gene and lack expression
of the PTEN tumor suppressor protein (4,16,17).
Here, we analyzed the effects of metformin on basal
and leptin-induced growth and migration of PTEN-positive LN18 and
LN229 GBM cell lines. Leptin is a multifunctional cytokine that has
been shown to regulate metabolic and neoplastic activities in many
cell types (18,19). We reported previously that leptin
and its receptor (ObR) are overexpressed in different human brain
tumors and that their levels correlate with the degree of
malignancy, being the most abundant in GBM (20). In ObR-positive LN18 and LN229 cells,
leptin acts as a mitogen/survival factor and its effects coincide
with the stimulation of the PI-3K/Akt, signal transducer and
activator of transcription 3 (STAT3) pathways as well as the
modulation of ERK1/2 signaling and retinoblastoma protein (pRb)
phosphorylation (20).
Materials and methods
Cell lines and growth conditions
ObR-positive LN18 and LN229 glioblastoma cell lines
were obtained from ATCC (Manassas, VA, USA). Both cell lines were
cultured in low-glucose Dulbecco’s modified Eagle’s medium (DMEM)
(Cellgro Mediatech, Manassas, VA, USA) supplemented with 5% fetal
bovine serum (Cellgro Mediatech) as described in a previous study
(20). The study was approved by
the Biosafety Committee at Temple University, PA, USA.
Cell proliferation assay
LN18 (5x104) and LN229 (3x104)
cells were seeded in 24-well plates in growth medium. After 24 h,
the cells were placed in serum-free medium (SFM; high-glucose DMEM
supplemented with 0.42 g/ml bovine serum albumin, 1 μM
FeSO4 and 2 mM L-glutamine) for a further 24 h. Next,
the cells were treated for 48 h with 2, 4, 8 and 16 mM metformin
(Sigma Aldrich, St. Louis, MO, USA) in the presence or absence of
200 ng/ml leptin, or were left untreated. At the conclusion of the
experiment, the cells were counted under the microscope using the
trypan-blue exclusion method. Each experiment was repeated a
minimum of 3 times.
Cell migration assay
LN18 and LN229 cells (1.5x104 cells)
suspended in modified SFM (high-glucose DMEM supplemented with 0.42
g/ml bovine serum albumin, 1 μM FeSO4 and 2 mM
L-glutamine, and 0.5 and 1% of FBS for LN229 and LN18,
respectively) were seeded in the upper chambers of a Transwell
system (24-well format, polycarbonate filters, 8-μM pore
size, Corning, Costar, NY, USA). The medium in the lower chambers
was supplemented with leptin (200 ng/ml) acting as a
chemoattractant, whereas metformin (8 and 16 mM) was added in the
upper chambers and the cells were incubated for 16 h. Subsequently,
the cells on the upper surface of the filters were removed, while
the cells that migrated to the underside of the filters were
stained with Giemsa stain for 20 min and counted (5 fields/well)
using a contrast phase microscope (Olympus CKX FA), at
magnification x10. The average number of cells/field was determined
± SD. Each experiment was repeated a minimum of 3 times.
Western blot (WB) analysis
LN18 and LN229 at a concentration of
1.2x106 and 1.0x106 cells/100 mm plate,
respectively, were grown for 16 h and then placed in SFM for 24 h.
The cells were subsequently pretreated with 16 mM metformin for 24
h then exposed to 200 ng/ml leptin for 30 and 60 min. Untreated
cells were used as controls. The cells were then lysed in 1% NP40,
50 mM HEPES pH 7.5, 250 mM NaCl, 5 mM EDTA pH 8.0, 0.1% SDS, 1X
protease inhibitors (Complete Mini EDTA-free protease inhibitors,
Hoffmann-La Roche, Nutley, NJ, USA) and phosphatase inhibitors (10
mM Na3VO4 and 50 mM NaF). The expression of
proteins was analyzed in 100 mg total cell lysates. The following
antibodies (Ab) from Cell Signaling Technology (Danvers, MA, USA)
were used for WB: for phospho-AMPKα, AMPKα Thr172 D79.5E monoclonal
(m)Ab 1:1000; total AMPKα, AMPKα polyclonal (p)Ab 1:1000;
phospho-Akt, Akt Ser473 pAb, 1:1000; total Akt, Akt pAb, 1:1000;
phospho-STAT3, STAT3 Tyr705, D3A7 mAb, 1:1000; and for total STAT3,
STAT3 79D7 mAb, 1:1000. The glyceraldehyde-3-phosphate
dehydrogenase 6C5 (GAPDH) Ab 1:1000 was obtained from Santa Cruz
Biotechnology, CA, USA. The intensity of WB bands corresponding to
studied proteins was analyzed by the ImageJ 1.44 program.
Statistical analysis
The results of the growth and migration experiments
were analyzed by a two-tailed distribution paired Student’s t-test.
P-values ≤0.05 were considered to indicate a statistically
significant result.
Results
Leptin stimulates the growth of GBM
cells, while metformin counteracts this effect
The effects of metformin in PTEN-positive,
leptin-responsive brain cancer cells have never been examined. We
assessed how the drug affects basal and leptin-dependent
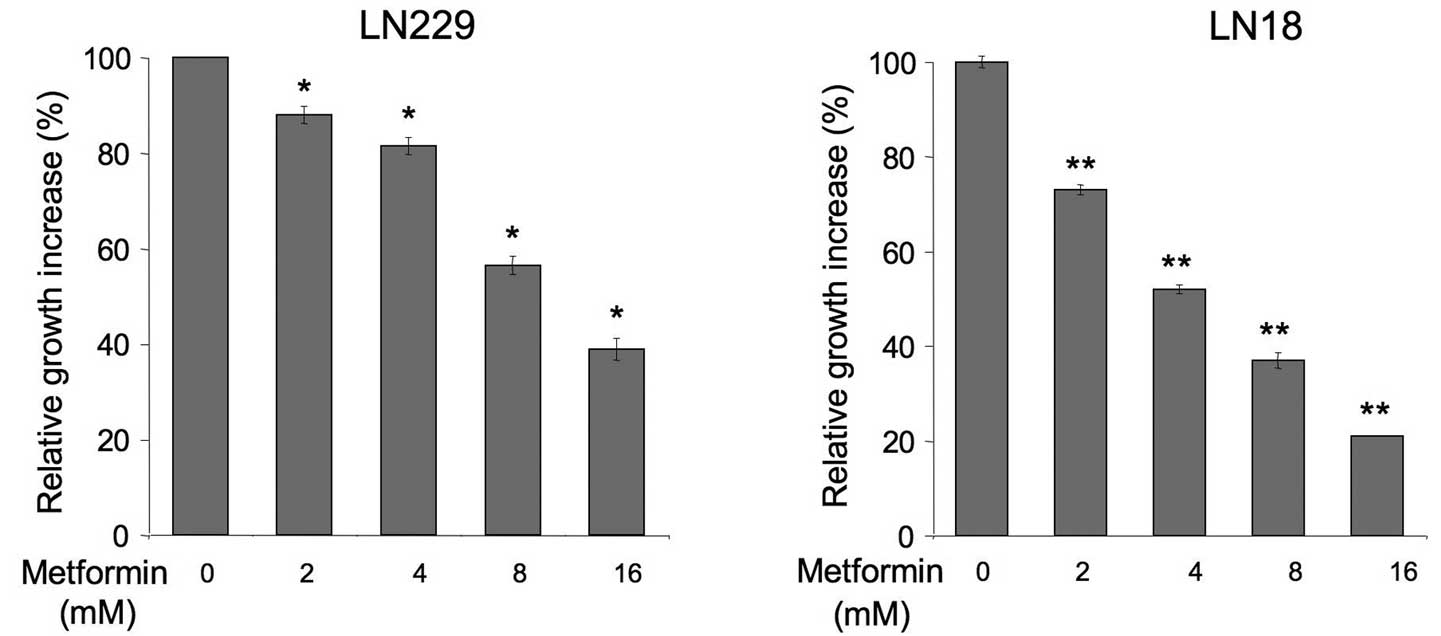
proliferation of LN229 and LN18 cells (Fig. 1). We found that metformin at 2–16 mM
significantly decreased normal cell growth in both cell lines
relative to untreated cells. The activity of the drug was
dose-dependent and the best cytostatic result was observed with 16
mM, while lower concentrations produced lesser effects. Notably,
LN18 cells appeared to be more sensitive to metformin than LN229
cells (Fig. 1).
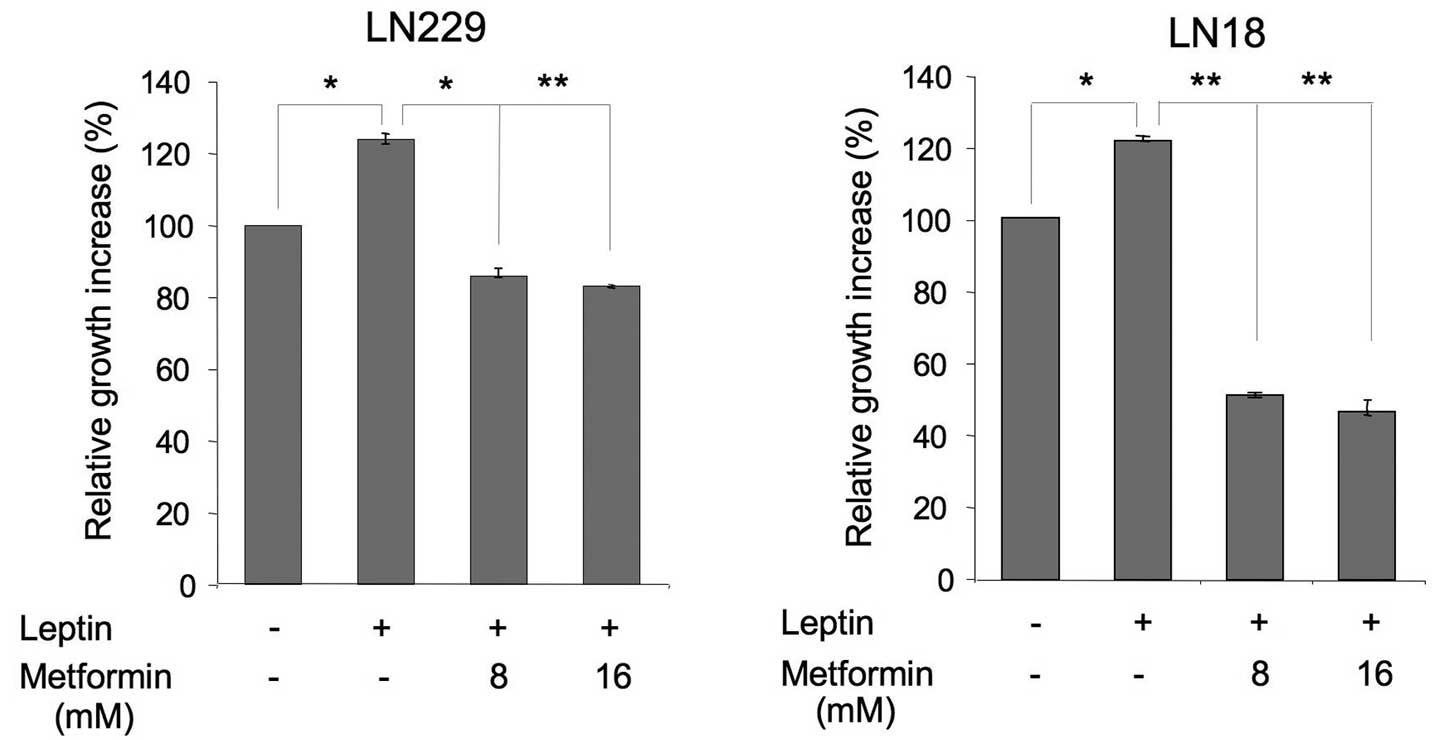
Next, we evaluated the effects of 8 and 16 mM
metformin on leptin-induced proliferation of LN229 and LN18 cells
(Fig. 2). In both cell lines,
leptin increased cell growth by ∼25 and 20%, respectively, whereas
metformin counteracted these effects. In particular, in LN229
cells, 8 and 16 mM metformin reduced cell proliferation to basal
levels. In LN18 cells, both concentrations inhibited the growth to
levels significantly below basal, suggesting that some activity of
the drug is mediated through a leptin-independent mechanism
(Fig. 2).
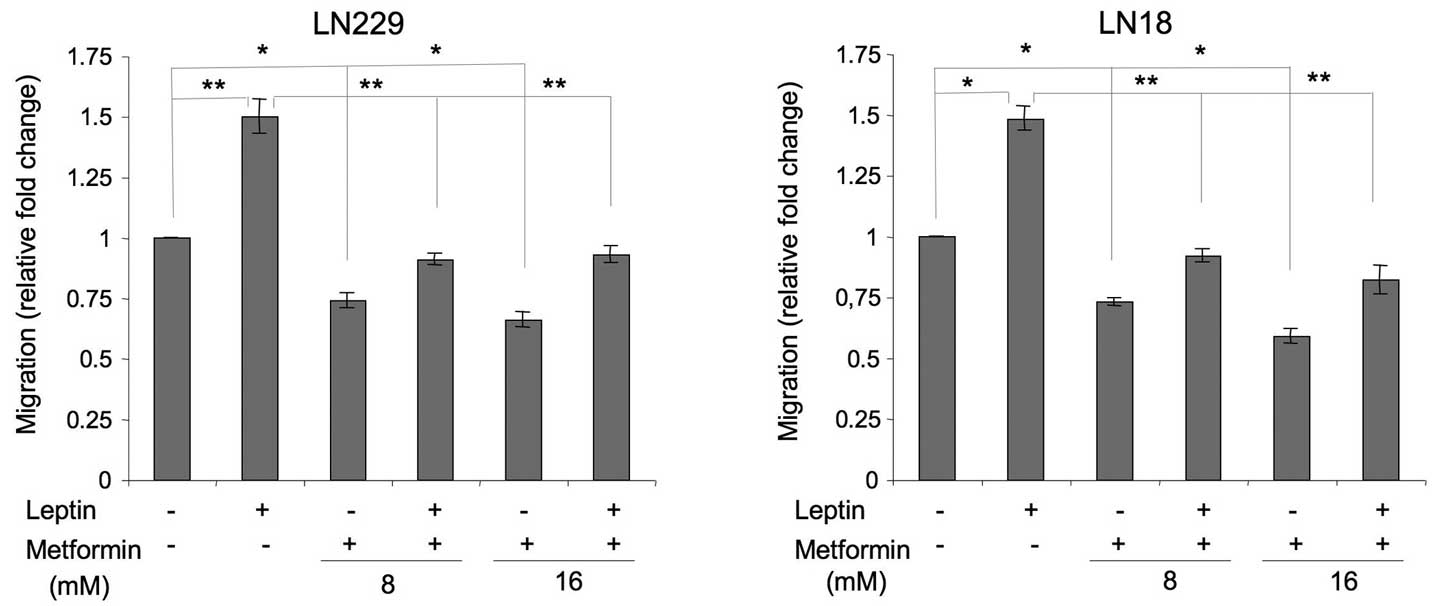
Leptin stimulates the migration of GBM
cells, while metformin counteracts this effect
Leptin is a recognized motogenic and angiogenic
factor in cancer models (21) and
induces migration in rat glioma cells (22); however, its role in human GBM
migration has never been studied. We found that in LN229 and LN18
cells, leptin increased cell migration by ∼50% (Fig. 3). In the presence of metformin,
leptin-dependent migration was reduced to basal or slightly below
basal levels (Fig. 3).
In addition, we observed that metformin alone
significantly suppressed basal cell migration. In LN229 cells, the
drug at 8 and 16 mM inhibited basal cell migration by 26 and 34%,
respectively. In LN18 cells, the decrease of cell migration
observed in the presence of metformin 8 and 16 mM was 27 and 41%,
relative to the control (Fig.
3).
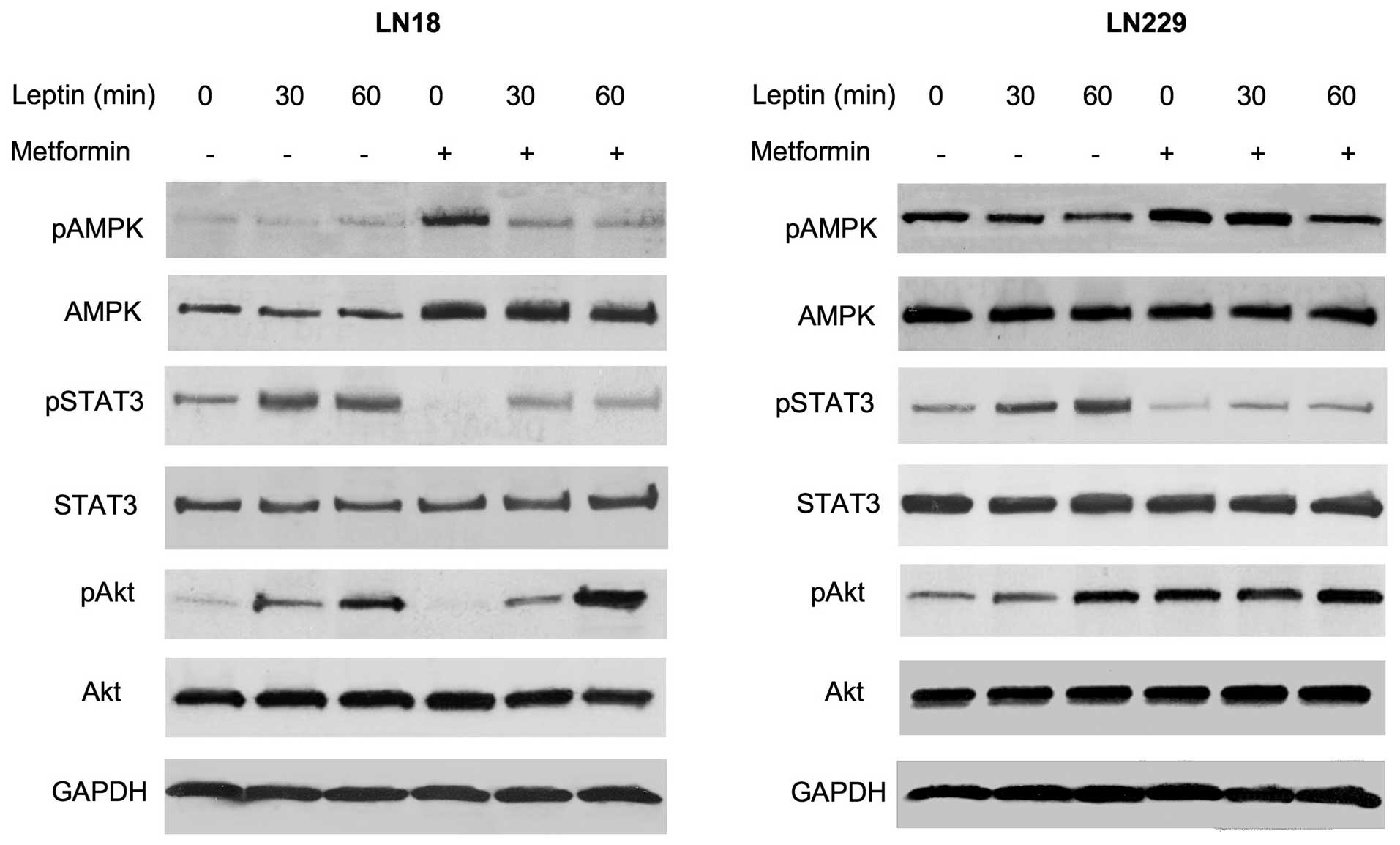
Effects of metformin on leptin signaling
pathways in GBM cells
We previously demonstrated that leptin activates the
STAT3 and Akt pathways, and downregulates ERK1/2 signaling in
ObR-positive GBM cells (20). In
addition, leptin is known to modulate AMPK in a cell
context-dependent manner (19,23),
although its effects on this enzyme in brain tumor cells have never
been studied. Here, we assessed how metformin modulates several
signaling pathways in GBM cells cultured in the presence or absence
of leptin (Fig. 4).
In LN18 cells, the basal levels of activated AMPK
were low and leptin treatment did not modify its phosphorylation
status. However, metformin pretreatment increased total AMPK levels
by ∼90%. The addition of leptin to metformin-treated cultures did
not modulate AMPK abundance, but it decreased AMPK phosphorylation
by 90–120% (Fig. 4). In LN229
cells, leptin treatment for 30 or 60 min reduced the levels of
phosphorylated AMPK by ∼40 and 60%, respectively, without affecting
the basal expression of the enzyme. Metformin did not increase
total AMPK levels in LN229 cells, but it moderately (∼40%) elevated
AMPK phosphorylation. The latter effect was effectively
counteracted by leptin treatment, particularly at 60 min (Fig. 4).
In both cell lines, neither metformin nor leptin
affected total STAT3 levels. Leptin treatment induced STAT3
phosphorylation at 30 and 60 min by ∼80–200%, while metformin
pretreatment reduced STAT3 activation to below basal levels. This
effect of metformin was partially reversed by leptin addition
(Fig. 4).
In LN18 and LN229 cells, leptin significantly
stimulated Akt phosphorylation, particularly at 60 min. Notably,
metformin produced differential effects of Akt signaling in our
cell models. In LN18 cells, metformin inhibited basal Akt
phosphorylation below basal levels, but this effect was totally
negated by leptin treatment. By contrast, in LN229 cells, metformin
increased Akt phosphorylation by ∼250%, and the addition of leptin
did not significantly alter this response. Total Akt levels were
not modified by either leptin or metformin treatment (Fig. 4).
Discussion
There is significant experimental evidence showing
that cellular mechanisms controlling the metabolism converge with
those implicated in the development and progression of neoplastic
diseases (7). In particular, energy
excess appears to act as a tumor promoter in the course of many
common cancers (7). AMPK is a
critical pathway regulating cellular response to energy imbalance
and has also been shown to control cancer progression (24). A well-characterized and extensively
used pharmaceutical agent that activates AMPK, thereby mimicking a
state of energy depletion, is metformin. Metformin has proven
efficacy in the treatment of diabetes and other metabolic diseases
(1,25). The drug has also shown value in
diabetes prevention, and its potential in the treatment and
prevention of cancer is currently being evaluated (7–11).
In this study, we analyzed the effects of metformin
on the growth and migration of human GBM cells cultured either
under basal conditions or exposed to leptin, a hormone known to
regulate various metabolic, mitogenic and motogenic functions in
normal and neoplastic cells (21).
The results of this study may be summarized as follows: i)
metformin restricts basal and leptin-stimulated growth of LN18 and
LN229 GBM cells; ii) metformin inhibits basal and leptin-induced
migration of LN18 and LN229 cells; iii) the action of metformin in
these cells is mediated through the AMPK, STAT3 and Akt pathways;
iv) metformin counteracts leptin effects on the AMPK and STAT3
pathways, but modulates Akt status in a cell-dependent manner.
This study confirmed our previous findings that
leptin stimulates several growth-related intracellular pathways and
acts as a mitogen in GBM cells (20). Furthermore, we demonstrated for the
first time that leptin activates migration in human GBM cells,
which is consistent with the observation that the hormone induces
migration of rat C6 glioma cells (22).
In human LN18 and LN229 cells, metformin inhibited
motogenic as well as mitogenic leptin activity. These findings are
original as no prior study has addressed metformin interference
with leptin activity in cancer models.
Significantly, we observed that metformin not only
inhibited cell growth, migration and signaling induced by leptin
but also restricted cell functions in the absence of the hormone.
This suggests that the drug suppresses the constitutive activation
of several mitogenic and motogenic pathways in GBM cells, which are
frequently induced due to either lack of tumor suppressors, such as
PTEN, or overexpression of activated oncogenic proteins e.g., the
activated epidermal growth factor receptor mutant EGFRvIII, the
insulin-like growth factor receptor, HER2, or focal adhesion kinase
(FAK) (26–30). In fact, previous studies
demonstrated that metformin inhibits growth and/or migration of
PTEN-deficient rat or human glioma cell lines (4,16,17).
Notably, despite the differential genetic
backgrounds of our PTEN-positive cell models (LN18: EGFR+++, HER2+,
IGF-IR+, FAK++; LN229: EGFR+, HER2+++, EGFRvIII+, IGF-IR+++, FAK+),
metformin activated AMPK in both cell lines, either through
increased levels or elevated phosphorylation of the enzyme. This is
consistent with the original observation of Guo et al that
the proliferation of at least some GBM cells is significantly
suppressed by AMPK activation (15,27).
Metformin also inhibited STAT3 activation in both our cell models,
which confirms the importance of STAT3 signaling in GBM (31).
In contrast, we observed differential effects of
metformin on Akt in LN18 and LN229 cells. In LN18 cells, the drug
reduced basal and leptin-induced Akt phosphorylation, which
confirms reports of metformin activity in other cancer models
(32,33). Conversely, in LN229 cells, metformin
significantly increased basal Akt phosphorylation, and this process
was not affected by leptin treatment. The reason for this
difference is unclear, but it may be related to cell-specific
upregulation of Akt by chronic metformin treatment, as noted in
certain models (34,35).
In summary, our results suggest that metformin or
similar AMPK-targeting agents with optimized BBB penetrability
could be developed as potential treatments of GBM. Such modalities
could be used in conjunction with other target drugs, for example
those which inhibit angiogenic and mitogenic pathways stimulated by
leptin or other cytokines/growth factors. However, the analysis of
metformin interaction with conventional anti-neoplastic treatments
is necessary as the drug may decrease the efficacy of some
chemotherapeutic agents (4).
Acknowledgements
This study was supported by funds from
the Sbarro Health Research Organization, PA, USA.
References
|
1.
|
E BosiMetformin - the gold standard in
type 2 diabetes: what does the evidence tell us?Diabetes Obes
Metab11Suppl 238200910.1111/j.1463-1326.2008.01031.x19385978
|
|
2.
|
A RamachandranC SnehalathaDiabetes
prevention programsMed Clin North
Am95353372viii201110.1016/j.mcna.2010.11.006
|
|
3.
|
JG BoyleIP SaltGA McKayMetformin action on
AMP-activated protein kinase: a translational research approach to
understanding a potential new therapeutic targetDiabet
Med2710971106201010.1111/j.1464-5491.2010.03098.x20854376
|
|
4.
|
K JanjetovicL VucicevicM MisirkicMetformin
reduces cisplatin-mediated apoptotic death of cancer cells through
AMPK-independent activation of AktEur J
Pharmacol6514150201110.1016/j.ejphar.2010.11.00521114978
|
|
5.
|
RA MillerMJ BirnbaumAn energetic tale of
AMPK-independent effects of metforminJ Clin
Invest12022672270201010.1172/JCI4366120577046
|
|
6.
|
DG HardieThe AMP-activated protein kinase
pathway - new players upstream and downstreamJ Cell
Sci11754795487200410.1242/jcs.0154015509864
|
|
7.
|
M JalvingJA GietemaJD LefrandtMetformin:
taking away the candy for cancer?Eur J
Cancer4623692380201010.1016/j.ejca.2010.06.01220656475
|
|
8.
|
VN AnisimovMetformin for aging and cancer
preventionAging (Albany NY)2760774201021084729
|
|
9.
|
I Ben SahraY Le Marchand-BrustelJF TantiF
BostMetformin in cancer therapy: a new perspective for an old
anti-diabetic drug?Mol Cancer Ther9109210992010
|
|
10.
|
AM Gonzalez-AnguloF
Meric-BernstamMetformin: a therapeutic opportunity in breast
cancerClin Cancer
Res1616951700201010.1158/1078-0432.CCR-09-180520215559
|
|
11.
|
TV KourelisRD SiegelMetformin and cancer:
new applications for an old drugMed
Oncol2913141327201210.1007/s12032-011-9846-721301998
|
|
12.
|
M PollakMetformin and other biguanides in
oncology: advancing the research agendaCancer Prev Res
(Phila)310601065201010.1158/1940-6207.CAPR-10-017520810670
|
|
13.
|
K HosonoH EndoH TakahashiMetformin
suppresses colorectal aberrant crypt foci in a short-term clinical
trialCancer Prev Res
(Phila)310771083201010.1158/1940-6207.CAPR-10-018620810669
|
|
14.
|
S EyalP HsiaoJD UnadkatDrug interactions
at the blood-brain barrier: fact or fantasy?Pharmacol
Ther12380104200910.1016/j.pharmthera.2009.03.01719393264
|
|
15.
|
D GuoTF CloughesyCG RaduPS MischelAMPK: A
metabolic checkpoint that regulates the growth of EGFR activated
glioblastomasCell Cycle9211212201010.4161/cc.9.2.1054020023392
|
|
16.
|
A IsakovicL HarhajiD StevanovicDual
antiglioma action of metformin: cell cycle arrest and
mitochondria-dependent apoptosisCell Mol Life
Sci6412901302200710.1007/s00018-007-7080-417447005
|
|
17.
|
ME BecknerGT GobbelR AbounaderGlycolytic
glioma cells with active glycogen synthase are sensitive to PTEN
and inhibitors of PI3K and gluconeogenesisLab
Invest8514571470200516170333
|
|
18.
|
F ZhangY ChenM HeimanR DimarchiLeptin:
structure, function and biologyVitam
Horm71345372200510.1016/S0083-6729(05)71012-816112274
|
|
19.
|
L ScolaroM CassoneJW KolaczynskiL Otvos
JrE SurmaczLeptin-based therapeuticsExpert Rev Endocrinol
Metab5875889201010.1586/eem.10.61
|
|
20.
|
M RiolfiR FerlaL Del ValleLeptin and its
receptor are overexpressed in brain tumors and correlate with the
degree of malignancyBrain
Pathol20481489201010.1111/j.1750-3639.2009.00323.x19775291
|
|
21.
|
C GarofaloE SurmaczLeptin and cancerJ Cell
Physiol2071222200610.1002/jcp.20472
|
|
22.
|
WL YehDY LuMJ LeeWM FuLeptin induces
migration and invasion of glioma cells through MMP-13
productionGlia57454464200910.1002/glia.2077318814267
|
|
23.
|
DG HardieAMPK: a key regulator of energy
balance in the single cell and the whole organismInt J Obes
(Lond)32Suppl 4S7S12200810.1038/ijo.2008.11618719601
|
|
24.
|
Z LuoM ZangW GuoAMPK as a metabolic tumor
suppressor: control of metabolism and cell growthFuture
Oncol6457470201010.2217/fon.09.17420222801
|
|
25.
|
T TangJM LordRJ NormanE YasminAH
BalenInsulin-sensitising drugs (metformin, rosiglitazone,
pioglitazone, D-chiro-inositol) for women with polycystic ovary
syndrome, oligo amenorrhoea and subfertilityCochrane Database Syst
RevCD0030532009
|
|
26.
|
S BerezowskaS Diermeier-DaucherG
BrockhoffEffect of additional inhibition of human epidermal growth
factor receptor 2 with the bispecific tyrosine kinase inhibitor
AEE788 on the resistance to specific EGFR inhibition in glioma
cellsInt J Mol Med26713721201010.3892/ijmm_00000518
|
|
27.
|
D GuoIJ HildebrandtRM PrinsThe AMPK
agonist AICAR inhibits the growth of EGFRvIII-expressing
glioblastomas by inhibiting lipogenesisProc Natl Acad Sci
USA1061293212937200910.1073/pnas.090660610619625624
|
|
28.
|
C AngMC GuiotAV RamanakumarD RobergeP
KavanClinical significance of molecular biomarkers in
glioblastomaCan J Neurol
Sci37625630201010.1017/S031716710001080521059509
|
|
29.
|
TJ LiuT LaFortuneT HondaInhibition of both
focal adhesion kinase and insulin-like growth factor-I receptor
kinase suppresses glioma proliferation in vitro and in vivoMol
Cancer Ther613571367200710.1158/1535-7163.MCT-06-047617431114
|
|
30.
|
J SchlegelG PiontekB BuddeF NeffA KrausThe
Akt/protein kinase B-dependent anti-apoptotic pathway and the
mitogen-activated protein kinase cascade are alternatively
activated in human glioblastoma multiformeCancer
Lett158103108200010.1016/S0304-3835(00)00515-210940516
|
|
31.
|
N de la IglesiaSV PuramA BonniSTAT3
regulation of glioblastoma pathogenesisCurr Mol Med95805902009
|
|
32.
|
M ZakikhaniMJ BlouinE PiuraMN
PollakMetformin and rapamycin have distinct effects on the AKT
pathway and proliferation in breast cancer cellsBreast Cancer Res
Treat123271279201010.1007/s10549-010-0763-920135346
|
|
33.
|
IN AlimovaB LiuZ FanMetformin inhibits
breast cancer cell growth, colony formation and induces cell cycle
arrest in vitroCell Cycle8909915200910.4161/cc.8.6.793319221498
|
|
34.
|
J YangGD HolmanLong-term metformin
treatment stimulates cardiomyocyte glucose transport through an
AMP-activated protein kinase-dependent reduction in GLUT4
endocytosisEndocrinology14727282736200610.1210/en.2005-1433
|
|
35.
|
B SonntagM GotteP WulfingAN SchuringL
KieselRR GrebMetformin alters insulin signaling and viability of
human granulosa cellsFertil Steril84Suppl
211731179200510.1016/j.fertnstert.2005.04.04316210009
|