Introduction
Ovarian cancer is the fifth leading cause of
cancer-related mortality in females in the USA. Although
substantial improvements have been made in ovarian cancer research,
the five-year survival rate remains extremely low and the overall
cure rate remains poor. Metastasis continues to be a major clinical
challenge in the treatment of ovarian cancer (1,2), and
an improved understanding of the molecular mechanisms underlying
ovarian cancer invasion and metastasis may lead to the development
of more effective therapeutic strategies. Metastasis is regulated
by a number of factors and diverse mechanisms.
Metastasis-associated in colon cancer 1 (MACC1) (3) was initially shown to promote the
metastatic capacities of colorectal cancer, and clinical studies
have indicated that it may be an independent prognostic indicator
of recurrence and disease-free survival (DFS). Further studies have
shown that MACC1 is overexpressed not only in colon cancer
(4), but also in other carcinomas,
including hepatic and lung cancer (5,6).
Various studies have also revealed that the hepatocyte growth
factor (HGF)/c-MET signaling pathway is key in carcinogenesis
(7,8). Stein et al (3,9)
demonstrated that MACC1-induced tumorigenesis correlates with
HGF/c-MET signaling. MACC1 is a transcription factor that binds to
the promoter of c-MET to stimulate its transcription, ultimately
leading to the activation of the HGF/c-MET signaling pathway. In
the present study, a MACC1-specific small interfering RNA (siRNA)
was constructed, and its effects on adhesion, proliferation,
migration, invasion and angiogenesis were assessed. Finally, a
luciferase reporter assay and western blot analysis were used to
confirm whether MACC1 functions as a metastatic promoter in ovarian
cancer by targeting c-MET.
Materials and methods
Cell culture and siRNA transfection
The human ovarian cancer OVCAR3 cell line was
purchased from the American Type Culture Collection (Manassas, VA,
USA) and grown in Dulbecco’s modified Eagle’s medium (DMEM;
Gibco-BRL, Carlsbad, CA, USA) supplemented with 10% fetal bovine
serum (FBS; Gibco-BRL) and antibiotics (100 U/ml penicillin and 100
μg/ml streptomycin) at 37°C in a humidified incubator containing 5%
CO2. Human umbilical venous endothelial cells (HUVECs)
were obtained from the Institute of Biochemistry and Cell Biology
of the Chinese Academy of Science (GenePharma Co., Shanghai, China)
and cultured in Kaighn’s modified Ham’s F-12K medium (Mediatech,
Inc., Manassas, VA, USA) supplemented with endothelial cell growth
supplement (BD Biosciences, Mississauga, ON, Canada) and 10% FBS.
MACC1-siRNA or a non-specific siRNA (Shanghai, China) was
transfected into cells using Lipofectamine 2000 (Invitrogen Life
Technologies, Carlsbad, CA, USA) according to the manufacturer’s
instructions. Based on design principles and the MACC1 mRNA
sequence, three siRNA sequences that targeted MACC1 and one siRNA
sequence for use as a negative control were designed. The first
sequence used to target MACC1 was 5′-GCCACCAUUUGGGAUUAUATT-3′, the
second sequence was 5′-CACCCUUCGUGGUAAUAAUTT-3′ and the third
sequence was 5′-GCCCGUUGUUGGAAAUCAUTT-3′. The negative control
sequence used was 5′-UUCUCCGAACGUGUCACGUTT-3′.
Quantitative polymerase chain reaction
(qPCR)
Total RNA was extracted from the cells using TRIzol
reagent (Takara Bio, Inc., Shiga, Japan) and reverse-transcribed
into cDNA using the Prime Script RT reagent kit (Takara Bio, Inc.)
according to the manufacturer’s instructions. The RNA was then
analyzed by qPCR using SYBR Premix Ex Taq™ (Takara Bio, Inc.). The
sequences of the primers used were as follows: MACC1 forward,
5′-GGCATTGTCCTGGTGTGGT-3′ and reverse,
5′-CACTCCTTCACCCCTGCTATCT-3′; and GAPDH forward,
5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse, 5′-TGGTGAAGACGCCAGTGGA-3′.
The GAPDH gene was used as an internal control for standardization
in triplicate. The PCR conditions were as follows: 95°C for 30 sec;
40 cycles of 95°C for 5 sec, 60°C for 20 sec and 95°C for 15 sec;
and 60°C for 1 min. PCR amplification was performed using the
Mx3000P qPCR System (Stratagene California, La Jolla, CA, USA) and
the comparative Ct (ΔΔCT) method was used to determine the fold
change in expression.
Western blot analysis
The cells were lysed in radioimmunoprecipitation
buffer, and protein quantification was performed using the
bicinchoninic acid assay (Sigma-Aldrich, St. Louis, MO, USA). A
total of 30 μg of protein was separated using electrophoresis with
a 12% SDS-PAGE gel. Following electrophoresis, the proteins were
transferred to polyvinylidene fluoride membranes (Millipore,
Billerica, MA, USA). Subsequent to being washed, the membranes were
blocked with 5% skimmed milk for 1 h at 4°C and sequentially
incubated with the following primary antibodies at the
manufacturer’s recommended dilutions: rabbit antihuman polyclonal
MACC1 (1:1,000; Abcam, Cambridge, MA, USA), c-MET (1:100; BioWorld
Products, Inc., Visalia, CA, USA) and GAPDH (1:500; BioWorld
Products, Inc.). The mixtures were then incubated overnight at 4°C
on a rocking platform, followed by incubation with horseradish
peroxidase-conjugated secondary antibodies. The proteins were
detected using enhanced chemiluminescence plus detection reagents
(Amersham Pharmacia Biotech, Tokyo, Japan) according to the
manufacturer’s instructions. GAPDH was used as an endogenous
protein for normalization.
Cell growth assay
The cells were grown in 96-well culture plates,
treated as indicated and cultured for 24, 48, 72 and 96 h. Next,
the cells in each well containing 100 μl medium were incubated with
10 μl cell counting kit-8 (CCK-8; Beyotime Institute of
Biotechnology, Shanghai, China) at 37°C for 2 h. The optical
density (OD) of each well was then measured at 450 nm using a
microplate reader (Thermo Fisher Scientific, Waltham, MA, USA).
Cell adhesion assay
For the cell adhesion assay, 96-well plates were
precoated with 50 μl BD Matrigel™ matrix (40 μg/ml; BD Biosciences,
Heidelberg, Germany) at 4°C overnight. Prior to cell seeding, the
wells were washed with phosphate-buffered saline (PBS) twice and
blocked with 1% bovine serum albumin for 1 h at 37°C to prevent
non-specific binding. The cells (100 μl) were trypsinized and
seeded at a density of 1×104 cells per coated well,
incubated at 37°C for 60 or 90 min and then rinsed three times with
PBS to remove the unattached cells. Fresh medium (100 μl)
containing CCK-8 reagent (10 μl) was added to each well and the
plates were incubated for an additional 2 h. The OD was then
measured at 450 nm using a microplate reader. The OD values were
proportional to the number of adherent cells; five duplicate wells
were set up for each group.
Wound-healing assay
The cells were grown to 95% confluence in DMEM
containing 10% FBS in 24-well plates. A straight ‘wound’ was gently
made by scratching the cells with a plastic pipette tip. The cells
were washed twice with PBS and the dead cells were removed.
Following culture in FBS-free medium for 0 and 24 h, wound healing
was observed and images were captured using inverted phase contrast
microscopy (CKX41, Olympus Corporation, Tokyo, Japan). The width of
the scratch at the same position for 0 and 24 h was measured using
Image Pro-Plus software (Media Cybernetics, Inc., Rockville, MD,
USA).
Cell migration and invasion assays
Tumor cell migration and invasion were assessed
using a Transwell insert (8 μm; Corning, Inc., Corning, NY, USA).
The OVCAR3 cells were grown to ~80% confluence and subsequently
transfected with 120 nM MACC1-siRNA or a negative control.
Subsequent to 24 h, the cells were harvested and washed with PBS.
The cells (4×104) were then resuspended in 200 μl
serum-free medium and seeded into the upper chamber of a Transwell
insert. A total of 600 μl DMEM containing 10% FBS as a
chemoattractant was added to the lower chamber. For the invasion
assay, the inserts were precoated with 30 μl Matrigel and
5×104 cells were added to the upper chamber. Following
incubation at 37°C in a humidified atmosphere of 5% CO2
for 24 h, non-migrating (non-invading) cells were removed from the
upper surface of the filter with a cotton-tipped swab. The cells on
the lower surface of the filter were fixed in 4% paraformaldehyde
and stained using crystal violet staining solution. Five random
fields were counted at ×100 magnification. All the data that are
presented are from at least three independent experiments that were
performed in duplicate.
In vitro tube formation assay
In vitro angiogenesis assays were performed
using HUVECs plated on Matrigel. The OVCAR3 cells were cultured and
treated in six-well plates containing fresh complete medium for 48
h, and 1 ml of conditioned medium was collected. The day prior to
the tube formation assay, the BD Matrigel matrix was incubated
overnight on ice. For the tube formation assay, 48-well plates were
coated with Matrigel (100 μl per well) and allowed to polymerize at
37°C in a humidified atmosphere of 5% CO2 for 1 h. Next,
5×104 HUVECs were suspended in 500 μl conditioned medium
and added to the precoated 48-well plates. Following incubation for
an additional 24 h, images were captured using an inverted phase
contrast microscope and the tubular structures that had formed in
the Matrigel were quantified by counting the number of connected
cells in five random fields.
Luciferase reporter assays
Luciferase reporter vectors were constructed as
previously reported (10). Briefly,
the pGL3-Basic vector (Promega Corporation, Madison, WI, USA) was
used to generate luciferase reporter constructs. The
3′-untranslated region (UTR) of human c-MET was amplified from
human genomic DNA using the following primers:
5′-CGGGGTACCCAGACTGCCTGAGCTGGGGGA-3′ (sense) and
5′-CCCAAGCTTGCGACCAGACTGAGGCGCTC-3′ (antisense). The amplicons were
inserted into the KpnI-HindIII restriction sites in
the 3′-UTR of the hRluc gene in the pGL3-Basic vector. The
constructs were confirmed by DNA sequencing and restriction enzyme
digestion. The recombinant plasmid used was the
pGL3-c-MET-promoter. In each well of a 48-well plate, 120 nM
siRNA-MACC1 or a negative control was cotransfected with 0.3 μg of
the firefly luciferase reporter vector and 0.01 μg pRL-SV40, using
Lipofectamine 2000. Each transfection was performed in three
separate wells. Luciferase assays were performed 24 h after
transfection using the Dual Luciferase Reporter Assay System
(Promega Corporation). Renilla luciferase activity was used to
normalize firefly luciferase activity. Experiments were performed
with each construct in triplicate.
Statistical analysis
Quantitative data are presented as the mean ±
standard deviation. All statistical analyses were performed with a
one-way analysis of variance using SPSS version 17.0 (SPSS, Inc.,
Chicago, IL, USA). All experiments were performed at least in
triplicate. P<0.05 was considered to indicate a statistically
significant difference.
Results
Specific inhibition of MACC1 expression
by MACC1-siRNA
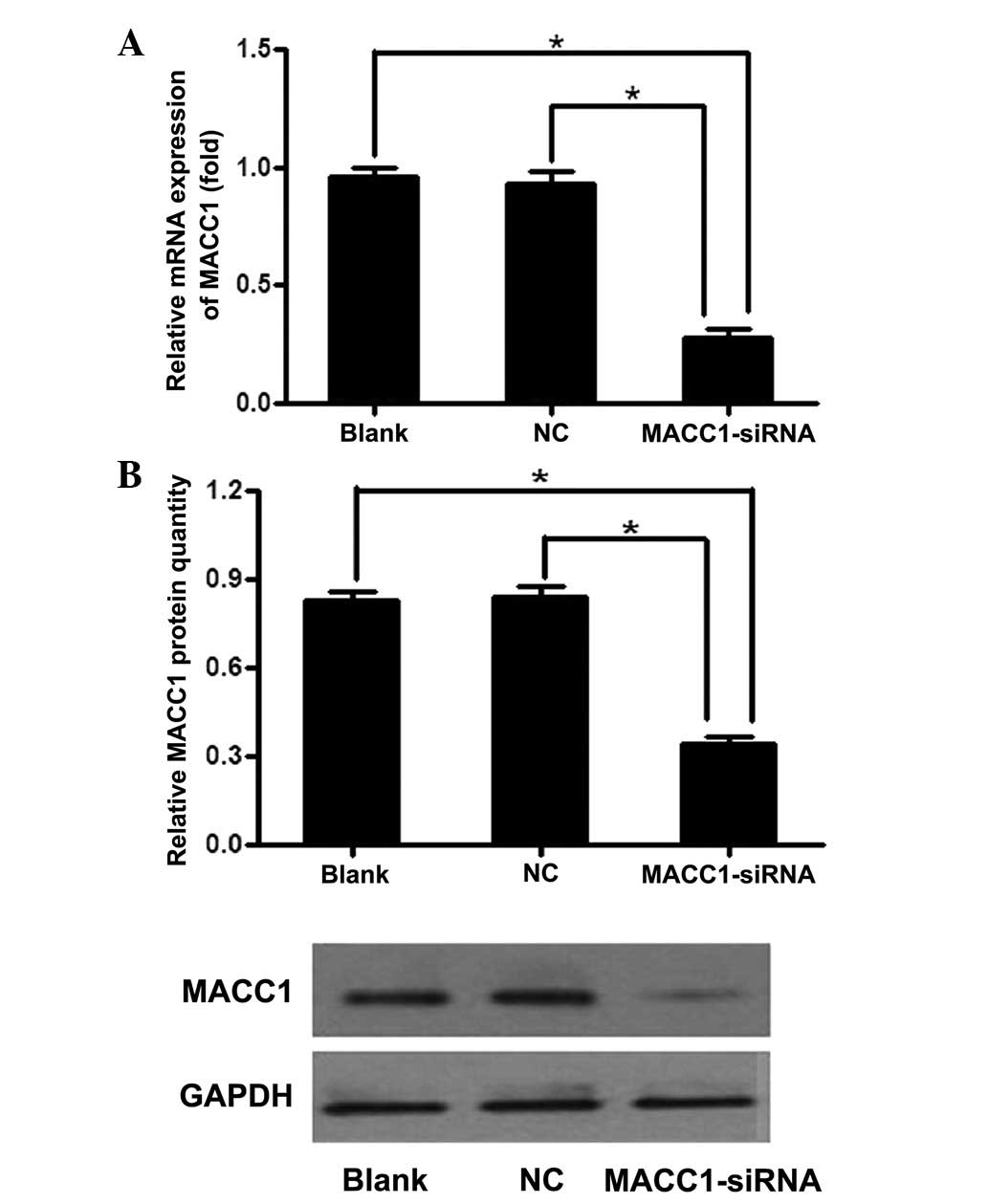
The qPCR results confirmed that the first siRNA
sequence was the most efficient at inhibiting the expression of
MACC1. As shown by qPCR, the expression of MACC1 mRNA was
significantly lower following transfection with MACC1-siRNA at 48
h, with an average inhibition of 75.7% compared with the control
groups. Subsequently, the expression levels decreased (Fig. 1A). MACC1 protein expression was also
decreased, with an average inhibition of 55.3% in the MACC1-siRNA
group (Fig. 1B). These results
indicated that MACC1-siRNA effectively suppresses MACC1 expression
at the mRNA and protein levels in cells.
MACC1 silencing inhibits ovarian cancer
cell proliferation and adhesion
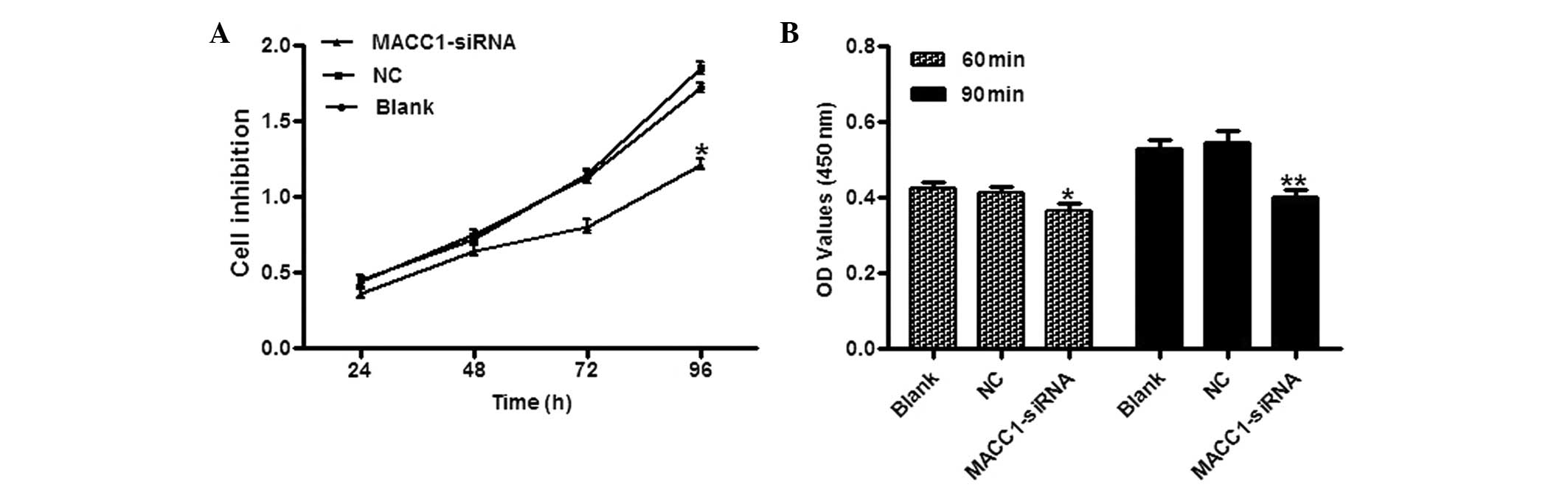
To assess the potential effect of MACC1
downregulation on proliferation and adhesion, cell adhesion and
CCK-8 assays were employed. The results indicated that the
MACC1-siRNA cells showed a time-dependent reduction in cell
proliferation. Compared with the control groups, the inhibition
rates were 10.2, 12.5, 24.0 and 31.7% at 24, 48, 72 and 96 h
post-gene transfection, respectively (Fig. 2A). In the adhesion assay, the
MACC1-siRNA cells exhibited reduced adhesion to the Matrigel
matrix. The adhesion of the MACC1-siRNA-transfected cells was
reduced by 14.3 and 25.9% at 60 and 90 min, respectively, compared
with the control groups (Fig.
2B).
MACC1 silencing suppresses ovarian cancer
cell migration and invasion in vitro
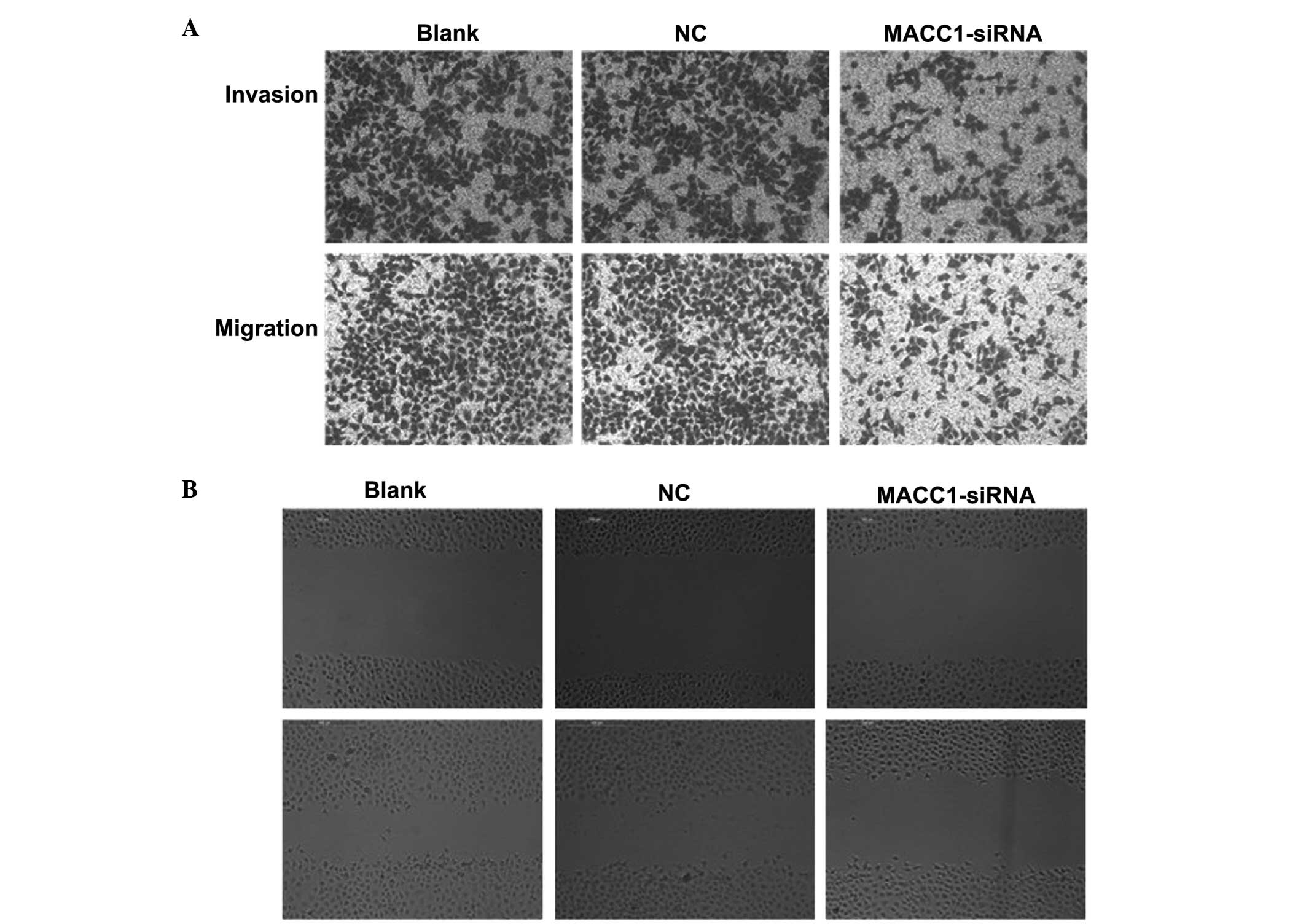
Following MACC1-knockdown using transient
transfection, the Transwell assay results showed that
MACC1-knockdown resulted in a 51.0% reduction in cell migration and
a 55.5% reduction in cell invasion compared with the control groups
(Fig. 3A). In addition, the
wound-healing assay indicated that MACC1-knockdown slowed the
closure of the wounds, whereas the wounds healed more rapidly in
the control groups (Fig. 3B). Taken
together, these results indicated that MACC1 overexpression
promotes the metastasis and invasion of ovarian cancer cells in
vitro.
Effect of MACC1-siRNA transfection on
angiogenesis
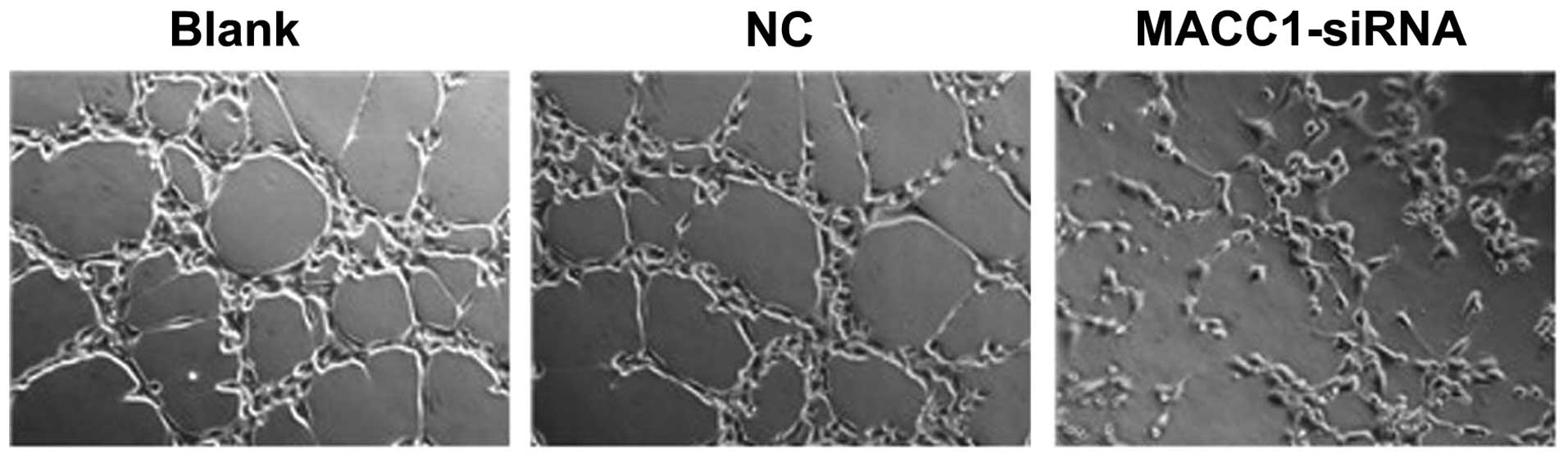
A tube formation assay using HUVECs was employed to
determine the effects of MACC1 on ovarian cancer cell angiogenesis.
As shown in Fig. 4, following
treatment with the different supernatants for 24 h, extensive HUVEC
tube formation was observed in the corresponding controls. However,
when the HUVECs were treated with conditioned media from the
MACC1-siRNA-transfected cells, the average number of complete
tubular structures decreased by 39.5%. These results indicated that
MACC1 inhibition markedly reduces the angiogenic capacity of the
ovarian cancer cells.
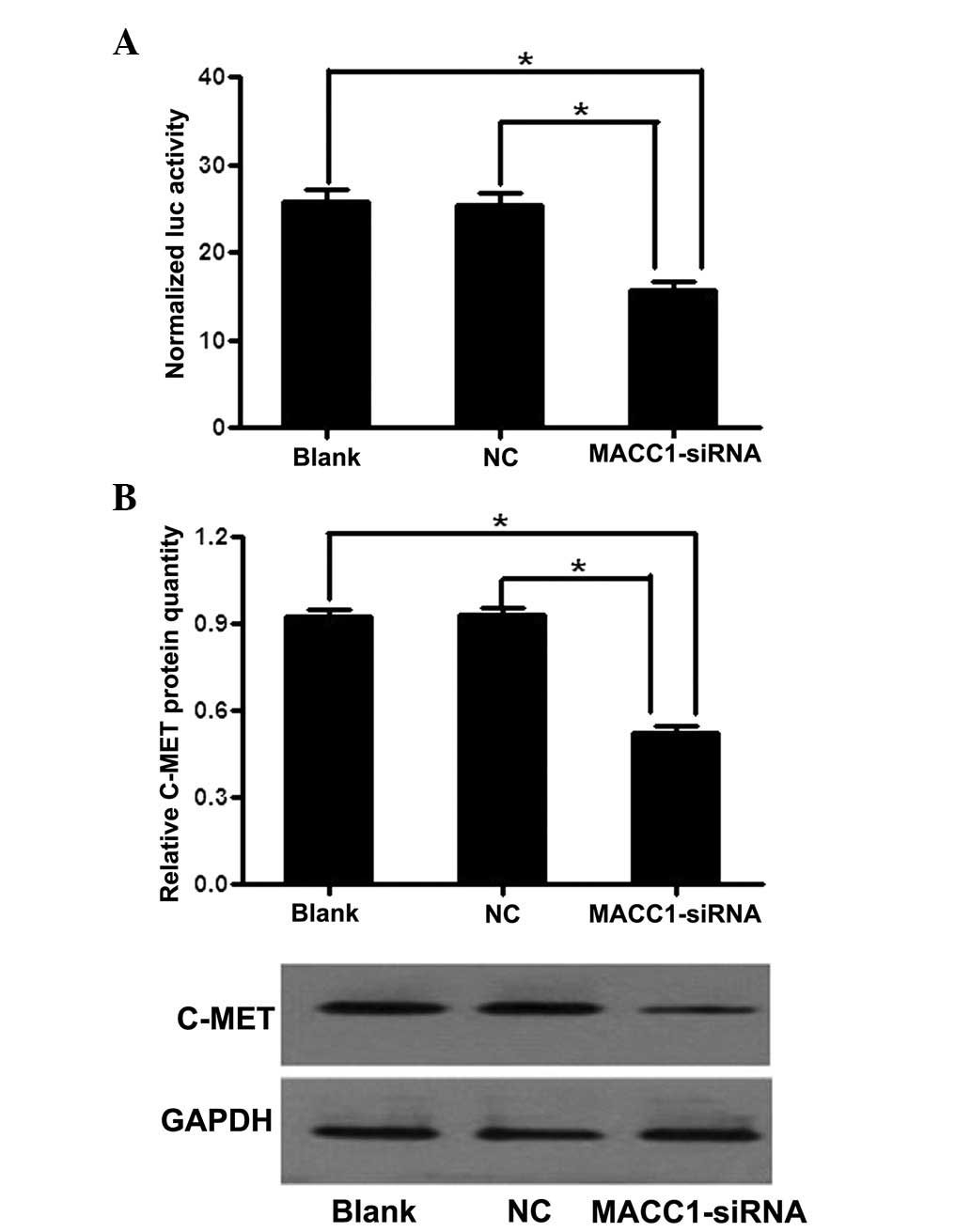
MACC1 activates the c-MET promoter and
upregulates c-MET expression
Based on the evidence that c-MET is a target gene of
MACC1 in colorectal cancer, the c-MET promoter fragments between
−223 and +60 were amplified and cloned into a pGL3-basic vector,
which is a positive regulatory element. Luciferase activity was
analyzed by transiently transfecting the luciferase reporter
cassette (pGL3-c-MET-promoter) into the OVCAR3 cells with or
without MACC1-siRNA. In comparison with the control groups,
transcriptional activity was markedly decreased in the cells that
had been transfected with MACC1-siRNA, with an inhibition rate of
42.9% (Fig. 5A). Furthermore,
MACC1-siRNA was found to significantly reduce c-MET protein
expression levels to 41.5% of those observed in the control groups
(Fig. 5B). These results indicated
that MACC1 may regulate c-MET by activating its promoter between
−223 and +60.
Discussion
Tumor metastasis is characterized by a number of
processes (11). At present, the
primary cause of mortality in patients with solid cancer is tumor
invasion and metastasis, however, the associated mechanisms remain
unknown. The identification of biomarkers that can be used to
monitor tumor invasion and metastasis in clinical practice may aid
clinicians in effectively controlling tumor metastasis, determining
the risk of recurrence and predicting patient survival (12).
MACC1, a recently identified metastasis-related
gene, was identified by differential display qPCR of the normal
colon, primary colon cancer and metastatic tissues (3). MACC1 is located on chromosome 7p21.1
and consists of 2,559 nucleotides encoding a protein containing 852
amino acids. MACC1 functions as a key activator of the HGF/c-MET
signaling pathway, promoting colon cancer cell proliferation,
invasion and metastasis in culture, and the growth and metastasis
of tumors in xenograft models. Stein et al (3) showed that the five-year survival rate
was 80% for colorectal cancer patients with low MACC1 expression,
but 15% for patients with high MACC1 expression. Previous studies
have also demonstrated that c-MET is a prognostic factor for colon
cancer, however, Stein et al showed that the combination of
MACC1 and c-MET expression did not improve the prognosis for
five-year survival or metastasis, indicating that MACC1 may serve
as an independent prognostic factor. In addition, several studies
have also demonstrated the clinical link between MACC1 and tumors.
Shimokawa et al (13) showed
that the expression of MACC1 was significantly higher in recurrent
lung adenocarcinomas than in non-recurrent ones, and that patients
with positive MACC1 staining had poorer DFS. Shirahata et al
(14,15) also observed that MACC1 expression in
hepatocellular and gastric cancers was significantly higher than in
corresponding normal tissues. In addition, MACC1 was found to
correlate with vascular invasion and α-fetoprotein levels in
hepatocellular carcinoma and with peritoneal dissemination in
gastric cancer. In gynecological cancers, studies (16,17)
have shown that compared with normal ovarian tissues and benign
cancer tissues, ovarian cancer tissues exhibit higher MACC1
expression levels. High MACC1 expression was associated with
advanced International Federation of Obstetricians and
Gynecologists stage, poor differentiation and lymph node
metastasis. Furthermore, the transfection of MACC1-siRNA into
OVCAR3 cells was found to significantly reduce invasion and
metastasis in vitro and in vivo. The RNA interference
(RNAi) technique (18), which is
the most effective antisense technique, is important in the study
of gene function and the gene therapy of tumors.
In the present study, RNAi technology was employed
to knock down endogenous MACC1 expression and to analyze the effect
of MACC1 on the metastatic behavior of ovarian cancer cells. qPCR
and western blot analysis confirmed that MACC1-siRNA effectively
suppressed the expression of MACC1 in OVCAR3 cells, with inhibition
rates of 75.7% at the mRNA level and 55.3% at the protein level.
Next, the major malignant characteristics of the ovarian cancer
cells, including adhesion, migration, invasiveness and
angiogenesis, which are essential steps for the establishment of
metastasis, were investigated. Adhesion and CCK-8 assays confirmed
that the MACC1-siRNA sequence altered the adhesion and
proliferation of the cells. A wound-healing assay also demonstrated
the significantly slower migration in the MACC1-silenced group
compared with the control groups, which was further corroborated by
the Transwell assay. Following the transfection of MACC1-siRNA into
the OVCAR3 cells, the invasion and vascularization capacities were
significantly decreased. All the results indicated that MACC1 is
important for the invasion and migration of ovarian cancer.
Previous studies have found that c-MET encodes the
HGF receptor and consists of 21 exons interrupted by 20 introns.
When the complete structural organization and promoter
characterization of c-MET was analyzed in renal epithelial mIMCD3
cells, Liu (19) identified two
positive regulatory elements and one negative regulatory element in
the promoter of the 5′-regulatory region, which were located at
nucleotide positions −2615 to −1621, −223 to −68, and −1621 to
−1093, respectively. Stein et al (3) also identified c-MET as one of the
targets of MACC1 and reported that MACC1 binds between fragments
−223 and −68 of the c-MET promoter, transcriptionally regulating
its expression. MACC1 induces HGF/c-MET signaling pathway
activation, resulting in enhanced cell motility, invasion and
metastasis. c-MET is a metastasis promoter and is overexpressed in
a variety of tumors. Increasing evidence has demonstrated a link
between c-MET overexpression and increased tumor cell metastasis
and invasion. In previous studies (20,21),
c-MET overexpression in ovarian carcinoma has been associated with
advanced tumor stage, and the knockdown of endogenous c-MET
expression using siRNA has been found to greatly reduce the
invasive ability of the cells. Attenuated c-MET expression also
weakens the invasiveness and metastasis of colon cancer and
hepatocellular carcinoma (22,23).
As a result, the current study investigated the potential
association between c-MET and MACC1 in ovarian cancer. MACC1-siRNA
transfected into OVCAR3 cells was found to significantly decrease
the levels of c-MET protein compared with the control cells.
Furthermore, a luciferase reporter assay confirmed that c-MET is a
target of MACC1 in ovarian cancer cells. Consistent with these
results, Stein et al (9) has
also shown that MACC1 can directly bind to the c-MET promoter.
In conclusion, the current study reported that MACC1
downregulation may effectively suppress the malignant biological
behavior of ovarian cancer and confirmed that c-MET is a target of
MACC1. MACC1 may be important in regulating the tumorigenesis and
development of ovarian cancer. In addition, it is indicated that
MACC1 inhibition may be a novel pharmaceutical target for
inhibiting ovarian cancer metastasis.
References
|
1
|
Siegel R, Ward E, Brawley O and Jemal A:
Cancer statistics, 2011: the impact of eliminating socioeconomic
and racial disparities on premature cancer deaths. CA Cancer J
Clin. 61:212–236. 2011.
|
|
2
|
Chen J, Liu X, Zhang J and Zhao Y:
Targeting HMGB1 inhibits ovarian cancer growth and metastasis by
lentivirus-mediated RNA interference. J Cell Physiol.
227:3629–3638. 2012.
|
|
3
|
Stein U, Walther W, Arlt F, et al: MACC1,
a newly identified key regulator of HGF-MET signaling, predicts
colon cancer metastasis. Nat Med. 15:59–67. 2009.
|
|
4
|
Shirahata A, Shinmura K, Kitamura Y, et
al: MACC1 as a marker for advanced colorectal carcinoma. Anticancer
Res. 30:2689–2692. 2010.
|
|
5
|
Qiu J, Huang P, Liu Q, et al:
Identification of MACC1 as a novel prognostic marker in
hepatocellular carcinoma. J Transl Med. 9:1662011.
|
|
6
|
Chundong G, Uramoto H, Onitsuka T, et al:
Molecular diagnosis of MACC1 status in lung adenocarcinoma by
immunohistochemical analysis. Anticancer Res. 31:1141–1145.
2011.
|
|
7
|
Tachibana K, Minami Y, Shiba-Ishii A, et
al: Abnormality of the hepatocyte growth factor/MET pathway in
pulmonary adenocarcinogenesis. Lung Cancer. 75:181–188. 2012.
|
|
8
|
You WK and McDonald DM: The hepatocyte
growth factor/c-Met signaling pathway as a therapeutic target to
inhibit angiogenesis. BMB Rep. 41:833–839. 2008.
|
|
9
|
Stein U, Dahlmann M and Walther W:
MACC1-more than metastasis? Facts and predictions about a novel
gene. J Mol Med. 88:11–18. 2010.
|
|
10
|
Qin X, Yan L, Zhao X, Li C and Fu Y:
microRNA-21 overexpression contributes to cell proliferation by
targeting PTEN in endometrioid endometrial cancer. Oncol Lett.
4:1290–1296. 2012.
|
|
11
|
Ye Q, Yan Z, Liao X, et al: MUC1 induces
metastasis in esophageal squamous cell carcinoma by upregulating
matrix metalloproteinase 13. Lab Invest. 91:778–787. 2011.
|
|
12
|
Wu ZS, Wang CQ, Xiang R, et al: Loss of
miR-133a expression associated with poor survival of breast cancer
and restoration of miR-133a expression inhibited breast cancer cell
growth and invasion. BMC Cancer. 12:512012.
|
|
13
|
Shimokawa H, Uramoto H, Onitsuka T, et al:
Overexpression of MACC1 mRNA in lung adenocarcinoma is associated
with postoperative recurrence. J Thorac Cardiovasc Surg.
141:895–898. 2011.
|
|
14
|
Shirahata A, Fan W, Sakuraba K, et al:
MACC 1 as a marker for vascular invasive hepatocellular carcinoma.
Anticancer Res. 31:777–780. 2011.
|
|
15
|
Shirahata A, Sakata M, Kitamura Y, et al:
MACC 1 as a marker for peritoneal-disseminated gastric carcinoma.
Anticancer Res. 30:3441–3444. 2010.
|
|
16
|
Zhang RT, Shi HR, Huang HL, et al:
Expressions of MACC1, HGF, and C-met protein in epithelial ovarian
cancer and their significance. Nan Fang Yi Ke Da Xue Xue Bao.
31:1551–1555. 2011.(In Chinese).
|
|
17
|
Zhang R, Shi H, Chen Z, et al: Effects of
metastasis-associated in colon cancer 1 inhibition by small hairpin
RNA on ovarian carcinoma OVCAR-3 cells. J Exp Clin Cancer Res.
30:83–107. 2011.
|
|
18
|
Chen XQ, Yang S, Kang MQ, et al: Survivin
expression in human lung cancer and the influence of its
downregulation on the biological behavior of human lung cancer
cells. Exp Ther Med. 3:1010–1014. 2012.
|
|
19
|
Liu Y: The human hepatocyte growth factor
receptor gene: complete structural organization and promoter
characterization. Gene. 215:159–169. 1998.
|
|
20
|
Mitra AK, Sawada K, Tiwari P, et al:
Ligand-independent activation of c-Met by fibronectin and
α(5)β(1)-integrin regulates ovarian cancer invasion and metastasis.
Oncogene. 30:1566–1576. 2011.
|
|
21
|
Bu R, Uddin S, Bavi P, et al: HGF/c-Met
pathway has a prominent role in mediating antiapoptotic signals
through AKT in epithelial ovarian carcinoma. Lab Invest.
91:124–137. 2011.
|
|
22
|
Seiden-Long IM, Brown KR, Shih W, et al:
Transcriptional targets of hepatocyte growth factor signaling and
Ki-ras oncogene activation in colorectal cancer. Oncogene.
25:91–102. 2006.
|
|
23
|
You H, Ding W, Dang H, Jiang Y and
Rountree CB: c-Met represents a potential therapeutic target for
personalized treatment in hepatocellular carcinoma. Hepatology.
54:879–889. 2011.
|