Introduction
Colorectal cancer is the fourth most common cause of
cancer-associated mortality following lung, stomach and liver
cancer (1,2). Treatment methods for colorectal cancer
include chemotherapy, surgery and radiotherapy. Among the available
chemotherapy drugs for treating colorectal cancer, 5-fluorouracil
(5-FU) has been the first-line regimen for the treatment of
colorectal cancer for a number of decades. In cancerous cells, 5-FU
is metabolized into cytotoxic fluorodeoxyuridine monophosphate
(3,4).
However, the clinical benefit of 5-FU is limited because of
resistance of colon tumor cells and adverse side effects (5,6). Previous
studies are consistent with the concept that the combination
therapies are able to improve the management of cancer and decrease
systemic toxicity (7,8). Although colorectal cancer has been
intensely researched, the problem of treatment failure remains a
key obstacle in the improvement of overall patient survival rates,
which remain low at ~50% at 5-year follow-up. Therefore,
combination therapy, including molecular targeting agents and/or
cytotoxic chemotherapy, may delay tumor progression and prolong
survival time in colorectal cancer (9,10).
During apoptosis, procaspase-3 is activated to
caspase-3, which initiates the apoptotic program (11,12). As
procaspase-3 is overexpressed or exhibits increased expression in a
variety of human tumors, drugs that direct active procaspase-3 are
of interest as anticancer agents (13,14). First
procaspase-activing compound (PAC-1) was the first procaspase-3
activator identified. SM-1, a novel PAC-1 derivative, directly
activates procaspase-3 into caspase-3 (15). Our previous study demonstrated that
SM-1 was able to induce cell apoptosis in various cancerous cells
and in vivo murine tumor models (16). SM-1 and 5-FU exert their antitumor
effects by distinct molecular mechanisms, suggesting the potential
for synergistic effects in cancer treatment. In the present study,
the combined effects of SM-1 and 5-FU in the treatment of
colorectal cancer and the potential underlying molecular mechanisms
were investigated.
Materials and methods
Cell culture
The human colorectal cancer cell lines HCT116 and
LoVo were obtained from the American Type Culture Collection
(Manassas, VA, USA). Cells were cultured in McCoy's 5A Modified
Medium (HCT116) or F-12K medium (LoVo) (Gibco; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), supplemented with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a
humidified incubator containing 5% CO2.
Cell proliferation assay
The effects of 5-FU and/or SM-1 on cell
proliferation were determined using an MTT assay. HCT116 and LoVo
cells (5×103 cells/well) were seeded in 96-well plates
and incubated overnight at 37°C, prior to exposure to 5-FU
(KingYork Group Co., Ltd., Tianjin, China) (1.5625, 3.125, 6.25,
12.5, 25, 50, 100, 200, 400 and 800 µmol/l), SM-1 (Xiangya Medical
Research Institute, Changsha, China) (0.25, 0.5, 1, 2, 4, 8, 16,
32, 64 and 128 µmol/l) or 5-FU plus SM-1 at the same doses as
single-agent treatments for 72 h at 37°C. Control cells were
processed identically except omitting the 5-FU or SM-1 treatment.
Subsequently, 20 µl of MTT solution (Sigma-Aldrich; Merck
Millipore, Darmstadt, Germany) (5 mg/ml) was added, and the cells
were incubated at 37°C for an additional 4 h. The culture medium
was discarded and formazan crystals were dissolved in 200 µl DMSO
(Sigma-Aldrich; Merck Millipore). The optical density (OD) of each
well was measured at 570 nm using a microplate reader. The
following formula was used: Cell proliferation inhibition
rate=(1-OD of the experimental sample/OD of the control group)
×100%.
Hoechst staining
Hoechst 33342 staining was used to confirm the
alterations in the nuclear morphology of HCT116 and LoVo cells
following 5-FU and/or SM-1 treatment. Cells were cultured and
treated as described above, prior to staining with 10 µg/ml Hoechst
33342 (Sigma-Aldrich; Merck Millipore) for 15 min at 37°C. Stained
cells were observed using an inverted fluorescence microscope at
magnification, ×400.
Flow cytometry
HCT116 and LoVo cells at 3×105 cells/well
were incubated in 6-well plates overnight at 37°C, then treated
with SM-1 or 5-FU or combinations of SM-1 and 5-FU for 72 h as
aforementioned. Untreated HCT116 and LoVo cells served as the
control. Cells were collected, incubated with Annexin V/propidium
iodide (PI) (BioLegend, Inc., San Diego, CA, USA), and measured
using a Guava EasyCyte 5HT flow cytometer (EMD Millipore,
Billerica, MA, USA). The compound 5,5,
6,6-Tetrachloro-1,1,3,3-tetraethylbenzimidazolylcarbocyanine iodide
(JC-1) (Beyotime Institute of Biotechnology, Haimen, China) was
used to assay the change in mitochondrial membrane potential (MMP).
Following treatment, cells were harvested and stained with JC-1
(0.5 µmol/l) at 37°C for 20 min. The fluorescence intensity was
measured using a Guava EasyCyte 5HT flow cytometer (EMD Millipore).
Guava ExpressPro software (version 5.0, EMD Millipore) was used for
sample analysis.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
HCT116 and LoVo cells were seeded into a 6-well
plate (2×106cells/well) and incubated at 37°C overnight,
prior to treatment with SM-1, 5-FU or combinations of SM-1 with
5-FU for 72 h as described above. Total RNA was extracted using
TRIzol reagent (CWBiotech, Shanghai, China). RNA (1 µg) was used to
synthesize the first-stand cDNA using the PrimeScript RT reagent
kit (TakaraBio, Inc., Otsu, Japan) following the manufacturer's
protocol. qPCR was performed using the SYBR-Green qPCR mixture
(TakaraBio, Inc.) following the manufacturer's protocol. The 20 µl
reaction mixture contained 10 µl 2x SYBR-Green qPCR mixure, 0.5 µl
of the forward and reverse primers each, 1 µl cDNA template and 8
µl RNase-free water. The PCR cycle at which amplification was
detectable above a background threshold (threshold cycle, or Cq)
was calculated using the maximum second derivative method with the
MX3000P qPCR system (Agilent Technologies, Inc., Santa Clara, CA,
USA) (17). All samples were run in
triplicate in each experiment. Primer sequences used to amplify
genes are presented in Table I.
 | Table I.Primer sequences. |
Table I.
Primer sequences.
| Primer name | Sequence |
|---|
| Actin | F:
5′-AGCGGGAAATCGTGCGTG-3′ |
|
| R:
5′-CAGGGTACATGGTGGTGCC-3′ |
| Survivin | F:
5′-TACGCCTGTAATACCAGCAC-3′ |
|
| R:
5′-TCTCCGCAGTTTCCTCAA-3′ |
| XIAP | F:
5′-TGATCGTGCCTGGTCAGAAC-3′ |
|
|
5′-CGCCTTAGCTGCTCTTCAGT-3′ |
| PARP | F:
5′-CATCGAGGTGGCCTACAGTC-3′ |
|
| R:
5′-ACCCATCAGCAACTTAGCGG-3′ |
| Bax | F:
5′-AAGCTGAGCGAGTGTCTCAAG-3′ |
|
| R:
5′-CAAAGTAGAAAAGGGCGACAAC-3′ |
| Bcl-2 | F:
5′-GTTTGATTTCTCCTGGCTGTCTC-3′ |
|
| R:
5′-GAACCTTTTGCATATTTGTTTGG-3′ |
Western blot analysis
HCT116 and LoVo cells at ~1×106
cells/well were harvested following pretreatment with SM-1, 5-FU or
combinations of SM-1 with 5-FU. Cells were incubated in lysis
buffer (Cell Signaling Technology, Inc., Danvers, MA, USA) at 4°C
for 30 min. Lysates were centrifuged at 10,000 × g for 15 min at
4°C. The supernatant obtained was quantified using the Bio-Rad
Protein assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Protein (30 µg) was separated using SDS-PAGE (8, 10 or 12%) and
transferred onto an Immobilon-FL polyvinylidene difluoride membrane
(EMD Millipore). Then the blots were blocked in 5% milk in
TBS-Tween-20 (TBST) for 1 h at room temperature and incubated at
4°C overnight with the following antibodies: anti-caspase-3 (cat.
no. 9662), anti-Survivin (cat. no. 2808), anti-B-cell lymphoma 2
(Bcl-2; cat. no. 15071), Bcl-2-associated X protein (Bax; cat. no.
5023), anti-poly (ADP-ribose) polymerase (PARP; cat. no. 9532) and
anti-β-actin (cat. no. 94970) (all 1:1,000 dilution; Cell Signaling
Technology, Inc.). Following this, blots were washed with TBST and
incubated with a horseradish peroxidase-conjugated goat anti-rabbit
(cat. no. 7074; dilution, 1:10,000; Cell Signaling Technology,
Inc.) or goat anti-mouse (cat. no. 7076; dilution, 1:10,000; Cell
Signaling Technology, Inc.) secondary antibody for 1 h at room
temperature, followed by an additional three washes with TBST. The
immunoreactive bands were visualized using ECL Western Blot kit
(ComWin Biotech, Beijing, China). The experiment was repeated three
times and similar results were obtained.
Mouse xenograft models and
histology
The present study was approved by the Ethics
Committee of the Beijing Medical Experimental Animal Care
Commission (Beijing, China). Female athymic nu/nu mice, between 3
and 4 weeks old, weighing between 18 and 20 g, were purchased from
Vital River Laboratories Co., Ltd. (Beijing, China). Mice were kept
under conditions of constant temperature (21–23°C) and humidity
(40–60%) with a 12 h light/dark cycle. Mice were allowed free
access to an irradiated standard rodent diet and sterilized water.
To generate tumors, viable HCT116 cells (5×106
cells/mouse) and LoVo cells (5×106 cells/mouse) were
subcutaneously injected into the right flanks of the mice. Vernier
calipers were used to measure tumor dimensions, and tumor volume
was calculated as 0.5 × length × width2. When the tumor
volume reached ~100 mm3, 32 mice were divided into four
groups at random, with each group containing 8 mice: i) Control
group treated with saline alone; ii) SM-1 group in which the drug
was given at 50 mg/kg/day via oral gavage (13); iii) 5-FU group in which the drug was
administered intraperitoneally at 30 mg/kg/3 days (13); and iv) combination group of SM-1 and
5-FU at the same dose and schedule as the single-agent groups.
Tumor size and body weight were measured every 3 days. At the
conclusion of the experiment, mice were sacrificed and tumors were
excised. Tumor specimens were stained with hematoxylin and eosin
(H&E) for histological evaluation. Slides were scanned using a
Pannoramic MIDI scanner and analyzed using Pannoramic ViewerRTM
software (version 15.3) (both 3DHistech, Ltd., Budapest,
Hungary).
Statistical analysis
SPSS (version 13.0; SPSS, Inc., Chicago, IL, USA)
was used to perform the statistical analysis. Results are presented
as the mean ± standard deviation. Statistical intergroup
differences were analyzed using one-way analysis of variance
followed by Bonferroni's post hoc test. Differences between two
groups were evaluated using a two-tailed Student's t-test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
SM-1 significantly enhances the
anti-proliferative effect of 5-FU in colorectal cancer cells
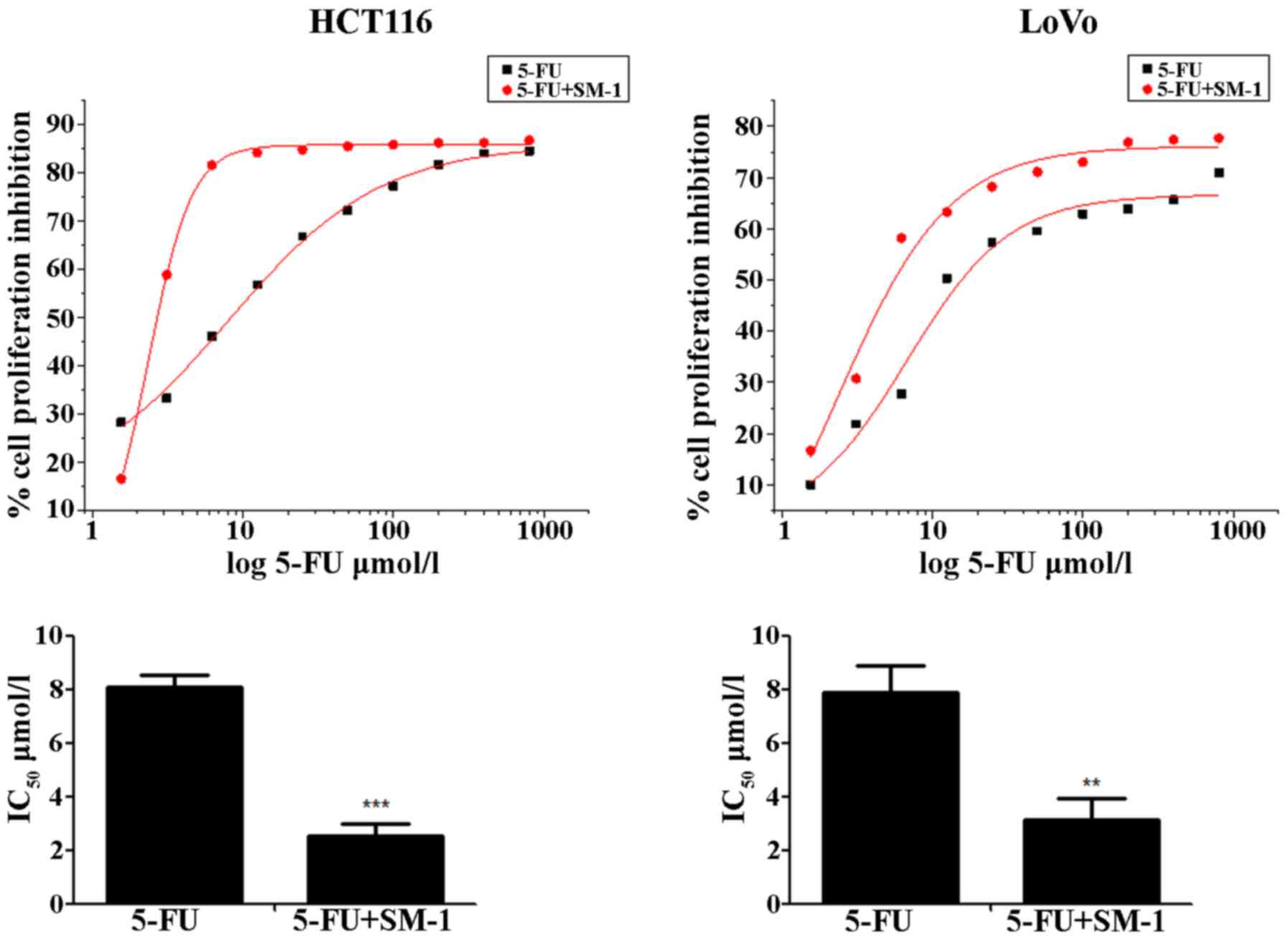
HCT116 and LoVo cells were exposed to increasing
concentrations of SM-1 and/or 5-FU for 72 h, and proliferation
inhibition rates were analyzed using an MTT assay. 5-FU inhibited
the proliferation of HCT116 and LoVo cells in a
concentration-dependent manner. The half-maximal inhibitory
concentration (IC50) values were 8.07±0.49 and 7.90±0.98
µmol/l for HCT116 and LoVo cells, respectively. When cells were
cotreated with 5-FU and SM-1 simultaneously, SM-1 significantly
enhanced the anti-proliferative activity of 5-FU and decreased the
IC50 values to 2.55±0.41 and 3.14±0.81 µmol/l in HCT116
and LoVo cells, respectively (P<0.001 and P<0.01,
respectively; Fig. 1).
SM-1 and 5-FU cotreatment induces
apoptosis in HCT116 and LoVo cells
Combination treatment with SM-1 and 5-FU led to the
appearance of a number of apoptotic biomarkers. During apoptosis,
cells are unable to modulate phospholipid distribution in the cell
membrane, and phosphatidylserine is exposed to the outer membrane
of cells, as assessed using an Annexin V/PI co-staining assay
(18). HCT116 and LoVo cells were
incubated with SM-1 and/or 5-FU. SM-1 markedly increased the
proportion of apoptotic cells from 7.27 to 21.75% in HCT116 cells,
and from 3.07 to 17.05% in LoVo cells. Similarly, 5-FU increased
the apoptotic rate from 7.27 to 19.10% in HCT116 cells and from
3.07 to 12.88% in LoVo cells. SM-1 cotreatment with 5-FU led to
markedly increased proapoptotic effects and an increased proportion
of apoptotic cells to 47.95% in HCT116 and 35.19% in LoVo cells
(Fig. 2A). Condensation of chromatin
is another apoptotic hallmark (19).
Consistent with the flow cytometry data, only the combination of
SM-1 and 5-FU exhibited significant morphological alterations in
Hoechst 33342-stained HCT116 and LoVo cells, including condensed
chromatin and formation of apoptotic bodies (Fig. 2B). A JC-1 assay was used to detect
mitochondrial outer membrane permeabilization. The decreased
fluorescence of JC-1 aggregates indicates a loss of MMP and
primarily appears in the early phase of mitochondrial apoptosis
(20). Significantly decreased
fluorescence intensity was identified in HCT116 and LoVo cells
cotreated with SM-1 and 5-FU, which implied that the combined
treatment mediated the loss of MMP and induced apoptosis (Fig. 2C).
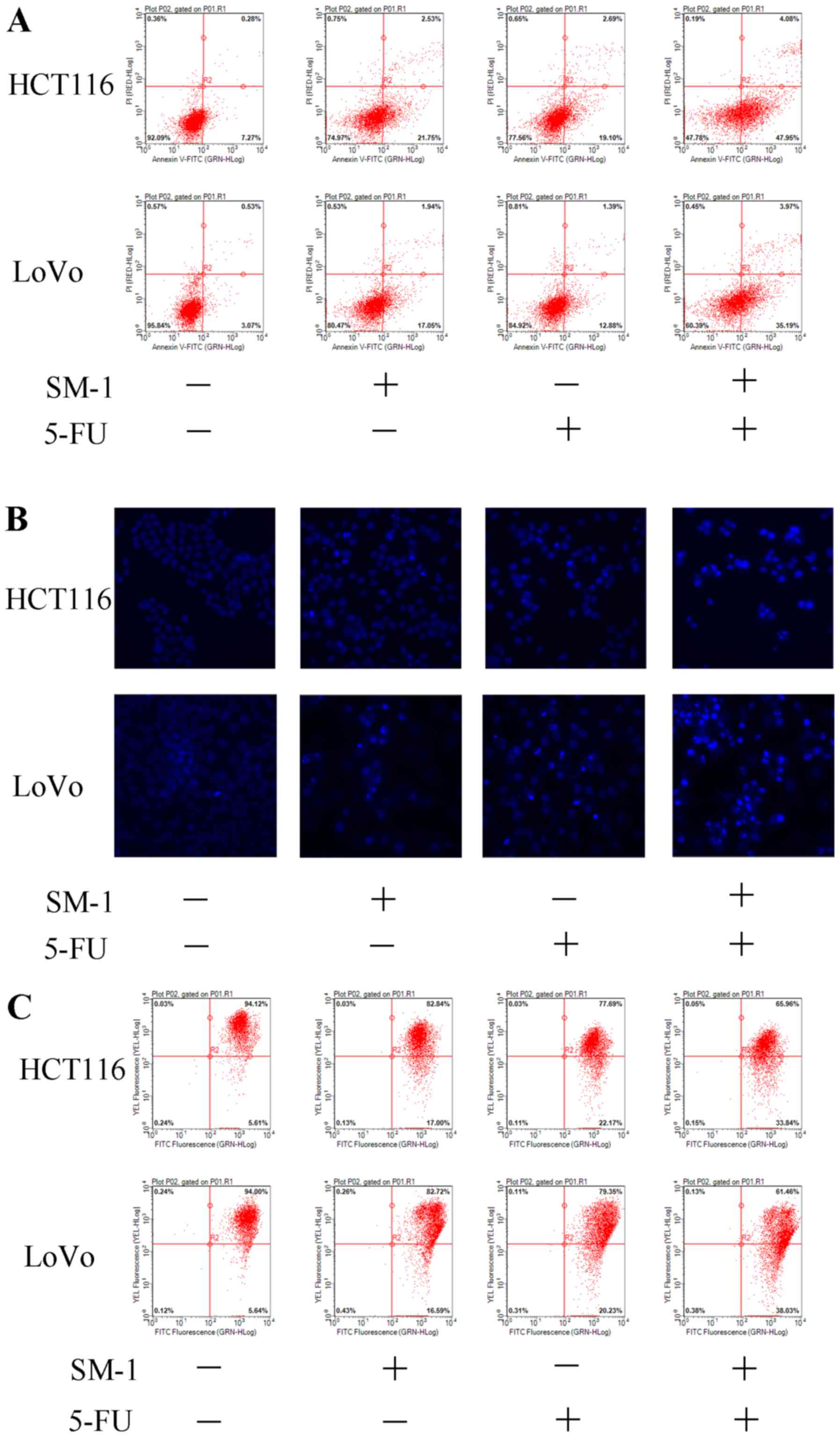 | Figure 2.SM-1 and 5-FU combination treatment
induces apoptosis in HCT116 and LoVo cells. (A) HCT116 and LoVo
cells were incubated with SM-1 (1 µmol/l), 5-FU (8 µmol/l) or a
combination of SM-1 and 5-FU (1 and 8 µmol/l, respectively) for 72
h. Phosphatidylserine exposure was measured by annexin V/PI
co-staining. The combination group demonstrated a marked increase
in apoptosis. (B) HCT116 and LoVo cells were incubated with SM-1 (1
µmol/), 5-FU (8 µmol/l) or a combination of SM-1 and 5-FU (1 and 8
µmol/l, respectively) for 72 h, and cells were observed using
fluorescence microscopy (magnification, ×400). (C) HCT116 and LoVo
cells were treated with SM-1 (1 µmol/l), 5-FU (8 µmol/l) or a
combination of SM-1 and 5-FU (1 and 8 µmol/l, respectively) for 72
h, prior to incubation with 5,5′, 6,6′-tetrachloro-1,1′,
3,3′-tetraethylbenzimidazolylcarbocyanine iodide for 20 min.
Mitochondrial membrane depolarization was measured using flow
cytometry. 5-FU, 5-fluorouracil; PI, propidium iodide; FITC,
fluorescein isothiocyanate. |
Expression of apoptosis-associated
genes in HCT-116 and LoVo cells
qPCR was used to detect expression of Bax, Bcl-2,
Survivin, X-linked inhibitor of apoptosis protein (XIAP) and PARP
mRNA following cotreatment with SM-1 and 5-FU (Fig. 3). In HCT-116 and LoVo cells,
significantly increased expression levels of Bax and PARP were
observed following cotreatment with 5-FU and SM-1 compared with
5-FU or SM-1 alone (Fig. 3). Compared
with 5-FU alone, cotreatment with SM-1 significantly decreased the
mRNA expression levels of Bcl-2, Survivin and XIAP (Fig. 3).
 | Figure 3.Effects of SM-1 and 5-FU cotreatment
on XIAP, Survivin, Bax, Bcl-2 and PARP mRNA levels were measured
using quantitative polymerase chain reaction. Bax and PARP levels
were significantly increased when treated with SM-1 and 5-FU in
combination compared with the untreated control, or SM-1 or 5-FU
alone. XIAP, Survivin and Bcl-2 levels were significantly decreased
when treated with SM-1 and 5-FU in combination compared with the
untreated control, or SM-1 or 5-FU alone. *P<0.05, **P<0.01,
***P<0.001 vs. control group; #P<0.05,
##P<0.01, ###P<0.001 vs. SM-1 and 5-FU
combination group. 5-FU, 5-fluorouracil; XIAP, X-linked inhibitor
of apoptosis protein; Bcl-2, B-cell lymphoma 2; Bax,
Bcl-2-associated X protein; PARP, poly (ADP-ribose) polymerase. |
Detection of apoptosis-associated
proteins by western blotting
As aforementioned, SM-1 induces apoptosis by
targeting procaspase-3 and allowing it to autoactivate. Therefore,
the effects of SM-1 and 5-FU in combination on the level of
caspase-3 were investigated. HCT116 and LoVo cells were pretreated
for 72 h with SM-1 (1 µmol/l), 5-FU (8 µmol/l) or a combination of
SM-1 and 5-FU (1 and 8 µmol/l, respectively), and expression of
caspase-3 was measured by western blotting. Minimal activation of
caspase-3 was observed when cells were treated with SM-1 and 5-FU
alone. However, when SM-1 and 5-FU were used in combination,
markedly increased levels of cleaved caspase-3 were observed
(Fig. 4).
 | Figure 4.Western blot analysis of the apoptotic
molecules caspase-3, Bcl-2, Bax, Survivin and PARP in HCT116 and
LoVo cells incubated with SM-1 and/or 5-FU. β-actin was used as a
loading control. For HCT116 and LoVo cells, caspase-3 levels were
markedly increased following treatment with SM-1 and 5-FU in
combination. In addition, cotreatment with SM-1 and 5-FU markedly
increased the levels of Bax and PARP, and markedly decreased the
levels of Bcl-2 and Survivin. Bcl-2, B-cell lymphoma 2; Bax,
Bcl-2-associated X protein; PARP, poly (ADP-ribose); 5-FU,
5-fluorouracil. |
The effects of SM-1 and 5-FU in combination on other
proapoptotic and antiapoptotic proteins, including Bcl-2, Bax,
Survivin and PARP were also investigated. The levels of Survivin
and Bcl-2 were decreased markedly in HCT116 and LoVo cells
following cotreatment with SM-1 and 5-FU, whereas low or no
expression of these proteins was observed with SM-1 or 5-FU
treatment alone at the same concentrations evaluated (Fig. 4). Similarly, cotreatment of HCT116 and
LoVo cells with SM-1 and 5-FU resulted in marked increases in Bax
and PARP expression compared with incubation with SM-1 or 5-FU
alone. These results indicated that a combination of SM-1 and 5-FU
induces apoptosis of colorectal cancer cells via Bcl-2, Survivin
and PARP.
SM-1 combined with 5-FU inhibits tumor
proliferation in vivo
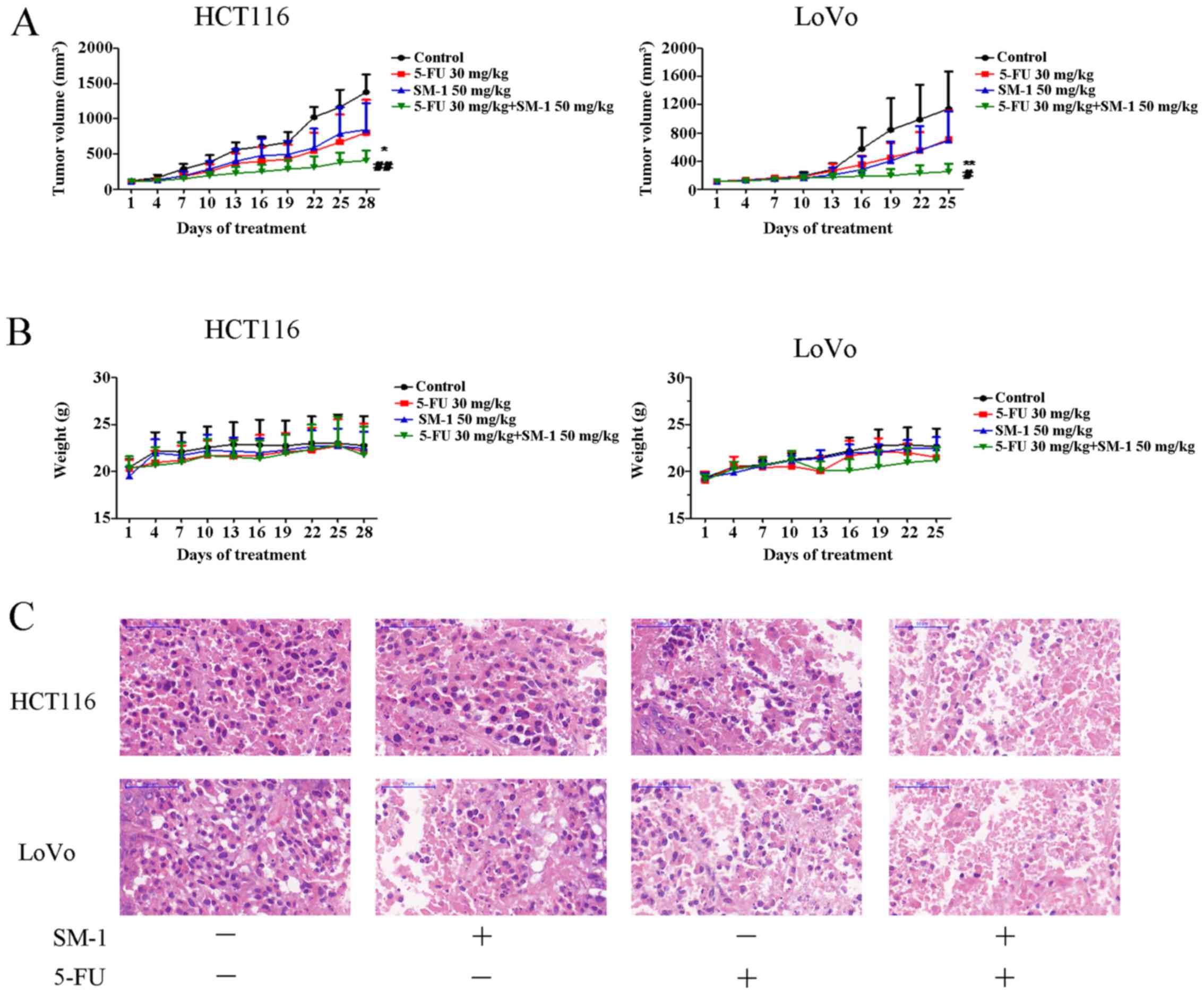
To evaluate the antitumor potential of SM-1 combined
with 5-FU in vivo, the ability of SM-1 and/or 5-FU to inhibit tumor
proliferation in HCT116 and LoVo xenograft models was examined. For
the HCT116 xenograft model, all treatments were able to markedly
decrease tumor cell proliferation compared with the control. The
tumor suppression rates for SM-1, 5-FU and the combination of SM-1
and 5-FU were 41.69, 38.81 and 70.42%, respectively. Furthermore,
mice cotreated with SM-1 and 5-FU exhibited the most increased
inhibition of tumor cell proliferation, compared with SM-1 alone
(P<0.01), 5-FU alone (P<0.05) and control (P<0.001)
groups. In the LoVo xenograft model, 25 days following the start of
treatment, the combination treatment group demonstrated a
statistically significant decrease in tumor cell proliferation
compared with SM-1 alone (P<0.05), 5-FU alone (P<0.01) and
control (P<0.001) treatment groups. The inhibition rates of
tumor cell proliferation were 38.09, 39.64 and 78.07%, respectively
(Fig. 5A). However, no significant
difference in tumor volume between the SM-1, 5-FU group and control
groups was identified. Neither significant weight loss nor
mortality was observed in any of the groups during the course of
the experiment (Fig. 5B).
The tumor tissues were further analyzed using
H&E staining. As presented in Fig.
5C, the tissue from the control group exhibited compact tumor
cells and a limited number of cells exhibiting small hyperchromic
fragmented nuclei. In the 5-FU or SM-1 experimental groups,
numerous gaps between tumor cells were observed. In the 5-FU and
SM-1 experimental group, the tumor tissue exhibited increased
damage compared with that in the 5-FU or SM-1 group; in addition,
the cellular arrangement was disordered and karyopyknosis was
observed. As a result, the combination group was identified to
ameliorate the severity of tumor.
Discussion
As a first-line chemotherapeutic drug, 5-FU is
widely used in clinical treatment of colon cancer (2,3). However,
tumor cells have demonstrated resistance to 5-FU (4). Combined chemotherapy has been considered
as an alternative treatment strategy, providing the potential for
enhanced efficacy (21,22). Despite these improvements, drug
resistance remains and novel combined treatment strategies are
urgently required. Previous studies have indicated that abnormal
apoptosis may be involved in drug resistance to 5-FU (2), including mutation of Bcl-2 or p53
proteins (23,24). Therefore, the combination of 5-FU and
drugs that induce apoptosis are frequently used in the treatment of
colorectal cancer (25). SM-1 has
exhibited antitumor and proapoptotic effects (16), therefore, in the present study, the
antitumor effects of a combination of 5-FU and SM-1 were
investigated.
An MTT assay demonstrated that the IC50
values in HCT-116 and LoVo cells were significantly decreased
following cotreatment with 5-FU and SM-1, compared with treatment
using 5-FU alone. Therefore, a combination of 5-FU and SM-1
treatment may decrease the dose of 5-FU required to achieve maximal
antitumor efficacy, without additional side effects.
The failure of 5-FU therapy in colorectal cancer was
partly because of dysfunctional apoptosis (3). Therefore, it was investigated in the
present study whether apoptosis was involved in the synergistic
combination of 5-FU and SM-1. Hoechst 33342 staining identified
that increased amounts of condensed chromatin and cell debris were
observed in 5-FU- and SM-1-cotreated HCT-116 and LoVo cells
compared with HCT-116 and LoVo cells treated with 5-FU and SM-1
alone, indicating that apoptosis was enhanced in HCT-116 and LoVo
cells. Following combined treatment with SM-1 and 5-FU, the
apoptotic rates in HCT-116 and LoVo cells were 47.95 and 35.19%,
respectively, which were significantly increased compared with
those following single treatment, as determined using Annexin V/PI
treatment and flow cytometry. It was also determined whether SM-1
and 5-FU were able to trigger mitochondrial disorders in a
synergistic manner. The results demonstrated that SM-1 and 5-FU
markedly induced a loss of MMP, suggesting that mitochondrial
depolarization may be triggered. These results suggested that 5-FU
and SM-1 in combination was able to effectively induce the
apoptosis of HCT-116 and LoVo cells.
Caspase-3 is the key point in the apoptotic
signaling pathway, being the intersection of the external and
internal apoptotic signaling pathways (26,27). The
small molecule SM-1 directly activates procaspase-3 into caspase-3;
therefore, the effect of SM-1 in combination with 5-FU on caspase-3
was investigated. Results revealed that SM-1 and 5-FU each led to
minor activation of procaspase-3 in both cell lines. However,
cotreatment with SM-1 and 5-FU markedly upregulated the level of
caspase-3. Expression levels of upstream and downstream critical
apoptotic indicators, including Bax, Bcl-2, Survivin and PARP, were
investigated. Notably, cotreatment markedly upregulated Bax and
cleaved PARP protein levels, and also decreased Bcl-2 and Survivin
protein levels in HCT116 and LoVo cells. These results suggested
that the caspase-dependent apoptosis pathway was able to be
activated and enhanced.
The in vitro results were confirmed in
vivo using colorectal cancer xenograft models. Compared with
the control group, 5-FU or SM-1 treatment alone has specific tumor
inhibition effects. However, 5-FU and SM-1 treatment in combination
exhibited increased antitumor activity compared with treatment
using either 5-FU or SM-1 alone.
Combination treatment of SM-1 with 5-FU was able to
enhance the antitumor activity of 5-FU in vitro and in
vivo. These enhanced effects were due to activation of the
caspase-dependent apoptosis signaling pathway. Therefore,
combination treatment with SM-1 and 5-FU is a potential therapy for
colorectal cancer.
References
|
1
|
Siegel R, DeSantis C and Jemal A:
Colorectal cancer statistics, 2014. CA Cancer J Clin. 64:104–117.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wolpin BM and Mayer RJ: Systemic treatment
of colorectal cancer. Gastroenterolog. 134:1296–1310. 2008.
View Article : Google Scholar
|
|
3
|
Longley DB, Harkin DP and Johnston PG:
5-Fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sobrero A, Guglielmi A, Grossi F, Puglisi
F and Aschele C: Mechanism of action of fluoropyrimidines:
Relevance to the new developments in colorectal cancer
chemotherapy. Semin Oncol. 27 5 Suppl 10:S72–S77. 2000.
|
|
5
|
Pardini B, Kumar R, Naccarati A, Novotny
J, Prasad RB, Forsti A, Hemminki K, Vodicka P and Bermejo J
Lorenzo: 5-Fluorouracil based chemotherapy for colorectal cancer
and MTHFR/MTRR genotypes. Br J Clin Pharmacol. 72:162–163. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Macdonald JS: Toxicity of 5-fluorouracil.
Oncology (Williston Park). 13 7 Suppl 3:S33–S34. 1999.
|
|
7
|
Lee SY and Oh SC: Advances of targeted
therapy in treatment of unresectable metastatic colorectal cancer.
Biomed Res Int. 2016:75902452016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Patel BB, Sengupta R, Qazi S, Vachhani H,
Yu Y, Rishi AK and Majumdar AP: Curcumin enhances the effects of
5-fluorouracil and oxaliplatin in mediating growth inhibition of
colon cancer cells by modulating EGFR and IGF-1R. Int J Cancer.
122:267–273. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee MS, Helms TL, Feng N, Gay J, Chang QE,
Tian F, Wu JY, Toniatti C, Heffernan TP, Powis G, et al: Efficacy
of the combination of MEK and CDK4/6 inhibitors in vitro and in
vivo in KRAS mutant colorectal cancer models. Oncotarget.
7:39595–39608. 2016.PubMed/NCBI
|
|
10
|
Pohl M and Schmiegel W: Colorectal
cancer-personalized, stage-adjusted tumour therapy. Dtsch Med
Wochenschr. 138:1790–1795. 2013.(In German). PubMed/NCBI
|
|
11
|
Kumar S: Caspase function in programmed
cell death. Cell Death Differ. 14:32–43. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Green DR: Apoptotic pathways: Paper wraps
stone blunts scissors. Cell. 102:1–4. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Peterson QP, Hsu DC, Goode DR, Novotny CJ,
Totten RK and Hergenrother PJ: Procaspase-3 activation as an
anti-cancer strategy: Structure-activity relationship of
procaspase-activating compound 1 (PAC-1) and its cellular
co-localization with caspase-3. J Med Chem. 52:5721–5731. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Putt KS, Chen GW, Pearson JM, Sandhorst
JS, Hoagland MS, Kwon JT, Hwang SK, Jin H, Churchwell MI, Cho MH,
et al: Small-molecule activation of procaspase-3 to caspase-3 as a
personalized anticancer strategy. Nat. Chem. Biol. 2:543–550.
2006.
|
|
15
|
Chen Y, Sun M, Ding J and Zhu Q: SM-1, a
novel PAC-1 derivative, activates procaspase-3 and causes cancer
cell apoptosis. Cancer Chemother Pharmacol. 78:643–654. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yuan HZ, Cao YT, Li LN, Wang SS, Yang DX,
Zhong XB, Tang SB and Yuan SJ: SM-1 induces apoptosis of BGC-823
cells by activating procaspase-3 and exerts antitumor effect.
Military Medical Sciences. 40:326–330. 2016.(In Chinese).
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Demchenko AP: The change of cellular
membranes on apoptosis: Fluorescence detection. Exp Oncol.
34:263–268. 2012.PubMed/NCBI
|
|
19
|
Sgonc R and Gruber J: Apoptosis detection:
An overview. Exp Gerontol. 33:525–533. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bedner E, Li X, Gorczyca W, Melamed MR and
Darzynkiewicz Z: Analysis of apoptosis by laser scanning cytometry.
Cytometry. 35:181–195. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gustavsson B, Carlsson G, Machover D,
Petrelli N, Roth A, Schmoll HJ, Tveit KM and Gibson F: A review of
the evolution of systemic chemotherapy in the anagement of
colorectal cancer. Clin Colorectal Cancer. 14:1–101. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Meyerhardt JA and Mayer RJ: Systemic
therapy for colorectal cancer. N Engl J Med. 352:476–487. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fakih MG: Metastatic colorectal cancer:
Current state and future directions. J Clin Oncol. 33:1809–1824.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang SY, Sales KM, Fuller B, Seifalian AM
and Winslet MC: Apoptosis and colorectal cancer: Implications for
therapy. Trends Mol Med. 15:225–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Azrak RG, Cao S, Slocum HK, Tóth K,
Durrani FA, Yin MB, Pendyala L, Zhang W, McLeod HL and Rustum YM:
Terapeutic synergy between irinotecan and 5-fluorouracil against
human tumor xenografs. Clin Cancer Res. 10:1121–1129. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Riedl SJ and Shi Y: Molecular mechanisms
of caspase regulation during apoptosis. Nat Rev Mol Cell Biol.
5:897–907. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hengartner MO: The biochemistry of
apoptosis. Nature. 407:770–776. 2000. View
Article : Google Scholar : PubMed/NCBI
|