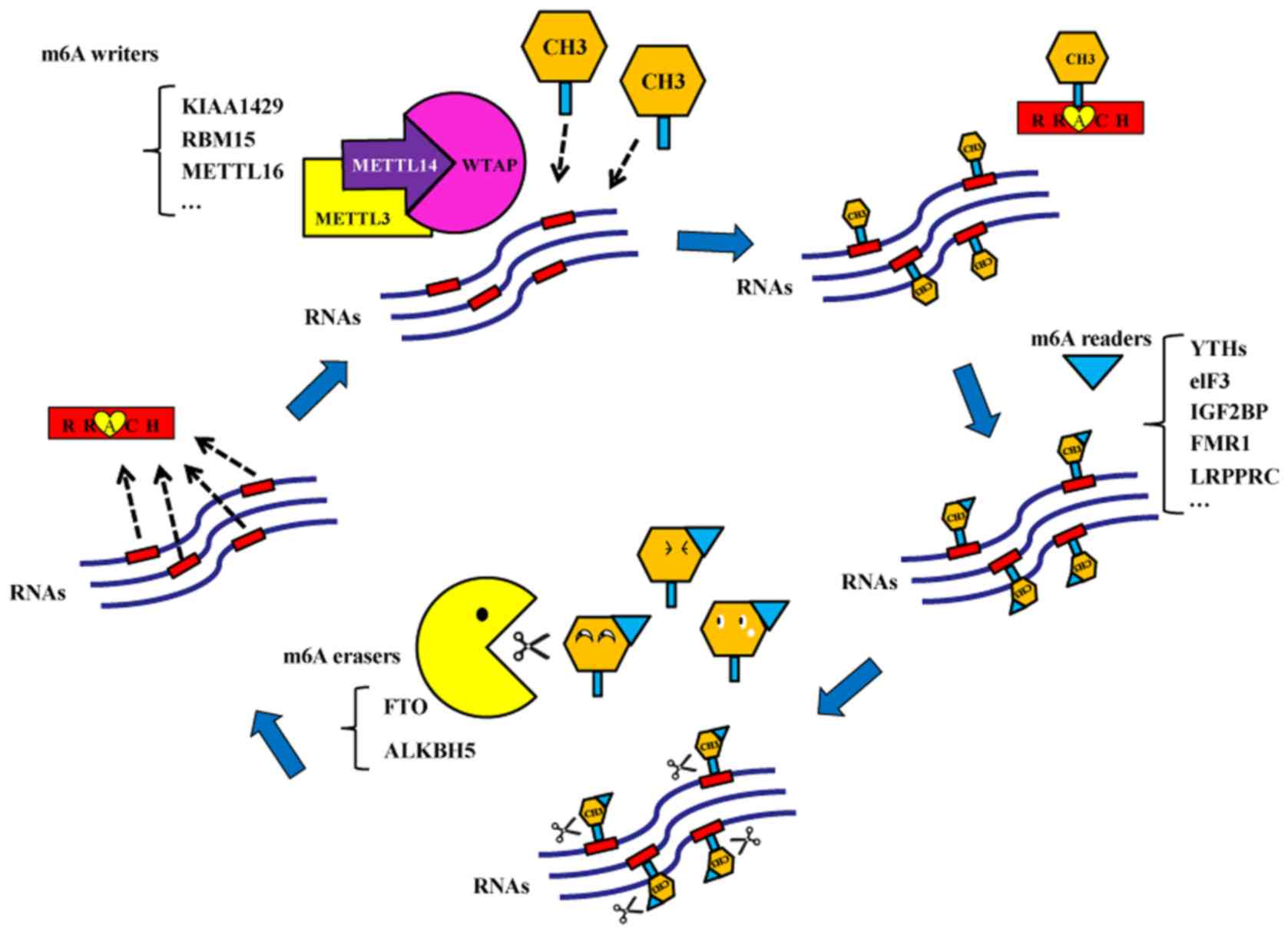
Over 100 different types of post-transcriptional
chemical modifications of RNA have been identified in living
organisms (1). A total of three
types of RNA modifications present in eukaryotic messenger RNA
(mRNA) have become prevalent in recent years: N6-methyladenosine
(m6A), 5-methylcytosine (m5C) and N1-methyladenosine (m1A)
(2). m6A was first identified by
Desrosiers et al (3) in 1974
and is the most commonly observed internal modification between
long noncoding RNA (lncRNA) and mRNA in eukaryotes. Dominissini
et al (4) used
high-throughput sequencing to demonstrate that m6A modifications
are frequently present at stop codons, 3′-untranslated regions
(3′-UTRs) and internal long exons, whereas m6A modifications are
typically present at RRm6ACH motifs (5).
m6A methylation is a methylation modification of
adenine (A) at position 6N and is catalyzed by the
methyltransferases (6). Bokar et
al (7) first discovered that a
methyltransferase complex catalyzed the formation of an m6A
modification in 1994. m6A methylation modifications are typically
present in mRNAs, transport RNAs, ribosomal RNAs and non-coding
RNAs (8). It was demonstrated that
m6A methylation may be present on microRNAs (miRNAs) (9). There are three types of enzymes that
participate in the dynamic and reversible m6A methylation and
demethylation modifications. The first type is the
methyltransferases, which serve a crucial role in RNA
transformation into m6A-modified RNA (7). Their coding genes are called m6A
‘writers’. Methyltransferase-like 3 (METTL3) was the first protein
reported to exhibit methyltransferase activity in 1997 (10). Subsequently, additional m6A writers
were discovered in mammals, including methyltransferase-like 14
(METTL14), Wilms' tumor 1-associating protein (WTAP), KIAA1429, RNA
binding motif protein 15/15B and methyltransferase-like 16
(11–14). The second type of enzymes is the
demethylases, which may alter the m6A-modified RNA back to RNA
(15). Demethylase coding genes are
called m6A ‘erasers’. Fat mass and obesity-associated protein (FTO)
and alkB homolog 5 (ALKBH5) may function as m6A erasers (15,16). The
last type of enzymes is the m6A ‘readers’, which read the
m6A-mediated physiological effects and influence RNA behaviors
(17). There are several m6A
readers, including YT521-B homology (YTH) domain family-YTHDF
(YTHDF1, YTHDF2 and YTHDF3) and YTHDC subtypes (YTHDC1 and YTHDC2)
(4), eukaryotic translation
initiation factor 3 (eIF3), heterogeneous nuclear ribonucleoprotein
A2B1 and C, insulin-like growth factor 2 mRNA-binding proteins
(IGF2BP), fragile X mental retardation 1 (FMR1) and leucine rich
pentatricopeptide repeat containing (LRPPRC; Fig. 1).
Recently, increasing evidence has demonstrated that
aberrantly expressed m6A modifications are associated with human
tumors (18–20). In the present review, the roles of
m6A regulators in various types of cancer are summarized.
Recently, METTL3-mediated m6A modifications were
discovered to promote tumor progression via interacting with
various miRNAs in numerous types of cancer (29–35). Du
et al (29) demonstrated that
miR-33a suppressed the proliferation of non-small-cell lung cancer
(NSCLC) cells via targeting METTL3 mRNA. Jin et al (30) revealed that METTL3 may initiate m6A
modification and promote the translation of the yes-associated
protein (YAP) mRNA to increase the resistance of NSCLC to
anticancer therapeutics and metastasis via a metastasis associated
lung adenocarcinoma transcript 1-miR-1914-3p-YAP axis. Cai et
al (31) revealed that Hepatitis
B virus X-interacting protein (HBXIP) modulated METTL3 by reducing
the expression of miRNA let-7 g, thus promoting breast cancer
progression. Wei et al (32)
demonstrated that miR-600 inhibited the migration and proliferation
of lung cancer cells via downregulation of METTL3 expression. Han
et al (33) revealed that
METTL3 interacted with DiGeorge Syndrome Critical Region 8 (DGCR8)
and upregulated pre-miR-221/222, which restricted PTEN expression,
thereby resulting in the proliferation of bladder cell carcinoma.
Cheng et al (34)
demonstrated that METTL3 enhanced the progression of bladder cell
carcinoma via AF4/FMR2 family member 4/NF-κB/MYC signaling. Peng
et al (35) revealed that the
upregulation of METTL3 promoted CRC metastasis via
miR-1246/sprouty-related, EVH1 domain-containing protein
2/mitogen-activated protein kinase signaling.
METTL3 has not only been revealed to promote the
proliferation and metastasis of tumor cells, but also to inhibit
the progression of tumors. Li et al (36) demonstrated that reduced expression of
METTL3 was associated with higher histological grades and larger
tumor sizes in nude mice. Furthermore, patients with renal cell
carcinoma (RCC) and upregulated expression of METTL3 exhibited
longer survival times.
Collectively, the results from these previous
studies suggested that METTL3-mediated m6A modifications may
promote tumor progression in different types of cancer; however the
underlying mechanisms, such as the specific signaling pathways
involved, may differ in different types of tumors. METTL3 may also
affect tumors independently of its catalytic activity, for example
via enhancing the translation of oncogenes. Currently, RCC is the
sole tumor type where METTL3 is demonstrated to inhibit the
progression of cancer. Additional studies are required to unravel
the specific mechanisms by which METTL3-mediated inhibition affects
RCC progression.
METTL14, which is a homolog of METTL3, is another
methyltransferase that catalyzes m6A modifications on RNA (11,12,37).
METTL14 interacts with METTL3 and modifies the m6A content, and
together, they form the core m6A methylation complex (38). METTL14 has been demonstrated to
occupy a degraded, non-functional SAM-binding domain, based on the
crystal structure analysis of the METTL3-METTL14 complex (39). On the other hand, METTL14 is
important for the positioning of the methyl group and the binding
of the RNA substrate to promote the catalytic activity of METTL3
(40,41). Thus, METTL14 is an indispensable
factor for METTL3 activity.
METTL14 may inhibit the initiation and progression
of tumors. A previous study revealed that the mRNA levels of
METTL14 in patients with metastatic HCC were significantly lower
compared with that in patients with non-metastatic HCC (42). Further experiments demonstrated that
METTL14 interacted with DGCR8 and upregulated the expression of
miRNA-126. Overexpression of METTL14 inhibited the metastasis of
HCC in an established liver metastasis mouse model. Based on these
results, it was hypothesized that METTL14 regulated the expression
of miRNA-126 via m6A modifications, thereby regulating its
downstream targets to inhibit the metastasis of HCC. Therefore,
METTL14 expression may be used as a prognostic factor for HCC
(42). Previous studies revealed
that low methylation levels of m6A, caused by mutations in the
METTL14 gene, were observed in endometrial cancer cells (43,44).
Using the CRISPR technology, METTL14 was removed from hec-1-a
endometrial cancer cells and a lack of METTL14 was demonstrated to
increase the tumor cell proliferation, colony formation and
metastasis. It was observed that the levels of m6A methylation in
endometrial cancer cells were reduced compared with adjacent normal
tissues. Thus, it was deduced that the mutations of METTL14
decreased m6A methylation levels, which served a key role in the
development of endometrial cancer. Subsequently, it was
demonstrated in patient samples and endometrial cancer cell lines
that downregulation of m6A methylation altered the activity of the
Akt/protein kinase B signaling pathway, thereby promoting the
proliferation and metastasis of cancer cells. Liu et al
(45) also discovered that the
downregulation of METTL14 decreased PH domain and leucine rich
repeat protein phosphatase 2 expression, which is a negative
regulator of the AKT signaling pathway. Downregulation of METTL14
also increased the expression of mTOR Complex 2, a positive
regulator of the AKT signaling pathway, which resulted in the
proliferation of endometrial cancer cells. Gong et al
(46) revealed that METTL14
downregulated the translation of P2X purinoceptor 6 (P2RX6), thus
reducing the RCC cell invasion and metastasis via
ATP-P2RX6-Ca2+-p-ERK1/2-matrix
metalloproteinase-9-mediated signaling. METTL14-mediated m6A
modifications may also suppress tumor progression in CRC and
bladder cancer (47,48).
The METTL14-driven m6A modifications consistently
result in the inhibition of tumor progression, including HCC,
endometrial cancer, RCC, CRC and bladder cancer. Whether METTL14
promotes tumor progression in an m6A modification-dependent manner
remains to be elucidated.
Downregulation of WTAP may result in the formation
of METTL14 and METTL3 degradation complexes and considerably
decrease m6A levels (11). In
addition, Schwartz et al (12) reported the WTAP-independent and
-dependent m6A modification sites upon WTAP depletion.
WTAP-independent sites are located at the cap structure of the
transcripts, whereas WTAP-dependent sites are present at internal
positions (12), highlighting the
complexity of the co-transcriptional regulation. Ping et al
(51) revealed that WTAP assisted in
the binding of METTL3 and METTL14 with the target RNA, whereas the
absence of WTAP resulted in the failure of METTL3 and METTL14
binding with the RNA in the nuclear speckle; however the protein
levels of METTL3 and METTL14 remained unaltered. Schöller et
al (52) discovered that WTAP
bound to a region of METTL3 in the first 150 amino acid region.
Even if WTAP was truncated from the C-terminal, it would still bind
with METTL3, as long as the remaining structure contained the first
150 amino acids from the N-terminal.
Recently, WTAP was demonstrated to regulate tumor
progression in an m6A modification-dependent manner. Chen et
al (53) revealed that WTAP
expression was upregulated in HCC. WTAP-mediated m6A modifications
contributed to the progression of HCC via human antigen R/protein
C-ets-1/p21/p27 signaling.
In comparison with METTL3 and METTL14, there is a
decreased number of publications reporting the relationship between
WTAP and cancer. The primary reason for this may be that WTAP does
not exhibit an independent catalytic activity, while it mainly
forms methyltransferase complexes with METTL3 and METTL14 to
catalyze m6A modifications on RNA.
KIAA1429. KIAA1429 was first identified in 2003 on
account of the discovery of its homologs, which exhibited an
interaction with WTAP in a sex-specific manner (54). Subsequent studies demonstrated that
METTL14, METTL3 and KIAA1429 are required for methylation in
mammals (12). Recently, two studies
revealed that METTL14, METTL3, WTAP and KIAA1429 form a
methyltransferase complex (55,56). The
downregulation of KIAA1429 resulted in a higher decrease of the m6A
index in the mRNA compared with the downregulation of either
METTL14 or METTL3, suggesting that KIAA1429 is required for the
complete catalytic activity of the methyltransferase complex.
As in the case of WTAP, KIAA1429 also requires the
formation of a methyltransferase complex with other m6A writers to
perform its function. KIAA1429 may affect tumorigenesis in both an
m6A-dependent and -independent manner.
In recent years, it has been demonstrated that FTO
is closely associated with the onset and progression of various
malignant tumors, such as breast cancer (65), lung cancer (66,67),
gastric cancer (68,69) and AML (70–72).
FTO is an important m6A demethylation enzyme. At
present, it has been demonstrated that FTO promotes tumorigenesis.
However, whether FTO inhibits tumor progression requires further
investigation.
ALKBH5 is an additional m6A demethylase. ALKBH5 is a
2-oxoglutarate- and ferrous iron-dependent nucleic acid oxygenase
that catalyzes the demethylation of m6A on RNA (76). ALKHB5 downregulation is required to
increase m6A levels, whereas its overexpression decreases the m6A
modification on mRNA in human cell lines. ALKBH5 is upregulated
under hypoxic conditions and serves a role in spermatogenesis
(16,77).
Although ALKBH5 and FTO are both demethylases, they
display opposing effects on tumor progression. ALKBH5 inhibits
tumor progression, whereas FTO promotes tumorigenesis. The
elucidation of the mechanisms underlying the differences in their
effects requires further investigation.
The YTH domain- containing proteins include five
members: YTHDF1, YTHDF2, YTHDF3, YTHDC1 and YTHDC2. Although YTHDF
proteins share high similarity, their functions may differ. In the
cytoplasm, YTHDF1 assists in the translation of m6A-modified mRNAs,
whereas YTHDF2 expedites the recycling of m6A-modified transcripts
(83). YTHDF3 facilitates the
protein synthesis in synergy with YTHDF1 and promotes the recycling
of methylated mRNA, mediated by YTHDF2 (84). All three YTHDF proteins function in a
coordinated manner to influence biological processes, associated
with m6A RNA methylation. YTHDC1 regulates mRNA splicing (85), whereas YTHDC2 binds to certain
noncoding RNAs to perform its function (86). The binding sites of each protein are
known; all the YTH domain-containing proteins, except YTHDC2, bind
to m6A, which was identified using the
individual-nucleotide-resolution UV crosslinking and
immunoprecipitation method (87).
A recent study revealed that YTHDF1 promoted the
proliferation of CRC cells and protected the tumor cells from
antitumor drugs (91). In addition,
c-Myc, which is an oncogenic transcription factor, is an upstream
gene of YTHDF1 and is associated with its expression. Knocking out
YTHDF1 inhibited the proliferation of tumor cells, whereas knocking
out c-Myc has been indicated to reduce the expression of YTHDF1,
thus restricting the proliferation of CRC cells. Bai et al
(92) obtained similar results when
YTHDF1 was overexpressed in CRC. Silencing YTHDF1 significantly
reduced Wnt/β-catenin signaling in CRC cells, thus highlighting a
YTHDF1 as a potential therapeutic target for the treatment of
CRC.
YTHDF2 has been known to be involved in the
development of AML for a considerable amount of time (93). In 2014, Wang et al (94) first reported that m6A was selectively
recognized by the YTHDF2 protein to regulate mRNA degradation. It
was demonstrated that reversible m6A deposition dynamically tuned
the stability and localization of the target RNAs via YTHDF2.
YTHDF2 may specifically interact with m6A-modified mRNAs via its
C-terminal YTH domain and recruit the mRNAs to cytoplasmic foci to
control the mRNA degradation via its N-terminal region.
YTHDF2 also acts as a tumor suppressor protein in
addition to acting as an oncogene. Hypoxia resulted in a decrease
of YTHDF2 expression in HCC (98).
It was demonstrated that the overexpression of YTHDF2 inhibited the
proliferation of HCC cells and activated the mitogen-activated
protein kinase kinase- and ERK-mediated signaling. YTHDF2 directly
targeted the m6A modification site in the 3′-UTR of the EGFR mRNA,
leading to the degradation of the EGFR mRNA in HCC cells. In
addition, hypoxia-induced phosphorylation of ERK was also inhibited
by YTHDF2. Therefore, YTHDF2 may inhibit the ERK/MAPK signaling via
reducing the stability of the EGFR mRNA in HCC cells to reduce the
proliferation of HCC (98).
Dysregulation of translation initiation results in
abnormal gene expression, leading to altered cell growth and
potentially cancer (99).
Translation initiation is regulated by many eIFs and one of these,
eIF3, is a critical factor controlling translation, with a
molecular weight of 550–700 kDa in mammalian cells. eIF3 consists
of ~13 subunits ranging in mass from 35–170 kDa, which associate
with the 40S ribosome in an early stage of translation initiation
and facilitate the interaction of mRNA with methionyl-tRNAi
(99). Several subunits, known as
eIF3a-eIF3m, have previously been revealed to serve vital roles in
modulating the translation of specific mRNAs encoding proteins
important for cell growth and oncogenesis (100). The abnormal expression of eIF3 may
result in alterations in the translation efficiency of certain
mRNAs, including those involved in angiogenesis, cellular growth
and malignancy (101,102).
Previous studies demonstrated that eIF3 expression
was upregulated in various types of cancer. Bachmann et al
(104) discovered that the
expression of eIF3 was increased in breast cancer compared with
adjacent normal tissues. Subsequently, Dellas et al
(105) revealed that eIF3
expression was upregulated in cervical cancer and it was higher
during the earlier stages of cancer.
m6A modifications have been investigated for >40
years. However, this dynamic and reversible modification is still
garnering increasing attention, particularly in relation to cancer
research. m6A writers perform the modification, whereas m6A erasers
reverse it. m6A readers are responsible for reading the
modification to regulate the mRNA fate. Thus, m6A modifications
serve as another level of regulation of RNAs in addition to
histones and DNA modifications (106).
Aberrant m6A modifications are involved in the onset
and progression of various types of cancer, including HCC, breast
cancer, RCC, endometrial cancer, AML and lung cancer (45,46,65–72,74).
Numerous genes are regulated by m6A modifications and participate
in tumorigenesis, such as SOX2, HBXIP, MZF1, NANOG and SOCS2
(24,31,67,77,95). m6A
modifications also participate in the modulation of lncRNAs at the
post-transcriptional level (81,82).
Furthermore, m6A may alter the local structure of mRNAs and lncRNAs
to regulate the gene expression and RNA maturation (107,108).
These findings may facilitate the discovery of novel therapeutic
strategies.
Although considerable progress has been made in
understanding the relevance of m6A modifications on RNA, several
domains remain still unknown. Firstly, certain m6A enzymes have
been investigated extensively in relation to tumorigenesis, whereas
studies on the involvement of other m6A enzymes is limited, such as
KIAA1429, RBM15, FMR1 and LRPPRC. Additional studies are required
to elucidate the complex pathological processes of tumors. Second,
whether m6A modifications interact with other RNA modifications,
such as m5C and m1A, and whether these interactions affect
tumorigenesis remains to be explored. Finally, additional clinical
trials are required to determine the potential diagnostic and
therapeutic functions of m6A modifications in human tumors.
Not applicable.
This study was funded by National Natural Science
Foundation of China (grant. no. 81502208), Department of Education
of Liaoning Province (grant. no. L2014310), Natural Science
Foundation of Liaoning Province (grant. no. 2019-MS-360), Shenyang
Science and Technology Bureau Plan Projects (grant. no.
F16-206-9-09) and 345 Talent Project of Shengjing Hospital of China
Medical University, to K Wang.
Not applicable.
KW, JY and JC conceived of the review. KW, XF and XW
wrote the manuscript. All authors have read and approved the final
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Boccaletto P, Machnicka MA, Purta E,
Piatkowski P, Baginski B, WireckI TK, de Crécy-Lagard V, Ross R,
Limbach PA, Kotter A, et al: MODOMICS: A database of RNA
modification pathways. 2017 update. Nucleic Acids Res. 46(D1):
D303–D307. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Liu ZX, Li LM, Sun HL and Liu SM: Link
between m6A modification and cancers. Front Bioeng Biotechnol.
6:892018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Desrosiers R, Friderici K and Rottman F:
Identification of methylated nucleosides in messenger RNA from
Novikoff hepatoma cells. Proc Natl Acad Sci USA. 71:3971–3975.
1974. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dominissini D, Moshitch-Moshkovitz S,
Schwartz S, Salmon- Divon M, Ungar L, Osenberg L, Cesarkas K,
Jacob-Hirsch J, Amariglio N, Kupiec M, et al: Topology of the human
and mouse m6A RNA methylomes revealed by m6A-seq. Nature.
485:201–206. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kane SE and Beemon K: Precise localization
of m6A in Rous sarcoma virus RNA reveals clustering of methylation
sites: Implications for RNA processing. Mol Cell Biol. 5:2298–2306.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li YL, Yu J and Song SH: Recent progresses
in RNA N6-methyladenosine research. Yi Chuan. 35:1340–1351.
2013.(In Chinese). View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bokar JA, Rath-Shambaugh ME, Ludwiczak R,
Narayan P and Rottman F: Caracterization and partial purification
of mRNA N6-adenosine methyltransferase from Hela cell nuclei.
Internal mRNA methylation requires a multisubunit complex. J Biol
Chem. 269:17697–17704. 1994.PubMed/NCBI
|
|
8
|
Wu X, Sang L and Gong Y: N6-methyladenine
RNA modification and cancers. Am J Cancer Res. 8:1957–1966.
2018.PubMed/NCBI
|
|
9
|
Alarcón CR, Lee H, Goodarzi H, Halberg N
and Tavazoie SF: N6-methyladenosine marks primary microRNAs for
processing. Nature. 519:482–485. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bokar JA, Shambaugh ME, Polayes D, Matera
AG and Rottman FM: Purification and cDNA cloning of the
AdoMet-binding subunit of the human mRNA
(N6-adenosine)-methyltransferase. RNA. 3:1233–1247. 1997.PubMed/NCBI
|
|
11
|
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang
L, Jia G, Yu M, Lu Z, Deng X, et al: A METTL3-METTL14 complex
mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem
Biol. 10:93–95. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schwartz S, Mumbach MR, Jovanovic M, Wang
T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N,
Cacchiarelli D, et al: Perturbation of m6A writers reveals two
distinct classes of mRNA methylation at internal and 5′ sites. Cell
Rep. 8:284–296. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Visvanathan A and Somasundaram K: mRNA
traffic control reviewed: N6-methyladenosine (M6A) takes
the driver's seat. Bioessays. 40:2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pendleton KE, Chen B, Liu K, Hunter OV,
Xie Y, Tu BP and Conrad NK: The U6 snRNA m6A
methyltransferase METTL16 regulates SAM synthetase intron
retention. Cell. 169:824–835.e14. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang
Y, Yi C, Lindahl T, Pan T, Yang YG and He C: N6-methyladenosine in
nuclear RNA is a major substrate of the obesity-associated FTO. Nat
Chem Biol. 7:885–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zheng G, Dahl JA, Niu Y, Fedorcsak P,
Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al: ALKBH5
is a mammalian RNA demethylase that impacts RNA metabolism and
mouse fertility. Mol Cell. 49:18–29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li A, Chen YS, Ping XL, Yang X, Xiao W,
Yang Y, Sun HY, Zhu Q, Baidya P, Wang X, et al: Cytoplasmic
m6A reader YTHDF3 promotes mRNA translation. Cell Res.
27:444–447. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN,
Chen ZH, Zeng ZL, Wang F, Zheng J, et al: METTL3 facilitates tumor
progression via an m6A-IGF2BP2-dependent mechanism in
colorectal carcinoma. Mol Cancer. 18:1122019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang Q, Chen C, Ding QQ, Zhao Y, Wang ZD,
Chen JJ, Jiang ZR, Zhang Y, Xu GF, Zhang JJ, et al: METTL3-mediated
m6A modification of HDGF mRNA promotes gastric cancer
progression and has prognostic significance. Gut. Oct 3–2019.(Epub
ahead of print).
|
|
20
|
Yue B, Song C, Yang L, Cui R, Cheng X,
Zhang Z and Zhao G: METTL3-mediated N6-methyladenosine modification
is critical for epithelial-mesenchymal transition and metastasis of
gastric cancer. Mol Cancer. 18:1422019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Leach RA and Tuck MT: Expression of the
mRNA (N6-adenosine)-methyltransferase S-adenosyl-L-methionine
binding subunit mRNA in cultured cells. Int J Biochem Cell Biol.
33:984–999. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen T, Hao YJ, Zhang Y, Li MM, Wang M,
Han W, Wu Y, Lv Y, Hao J, Wang L, et al: m(6)A RNA methylation is
regulated by microRNAs and promotes reprogramming to pluripotency.
Cell Stem Cell. 16:289–301. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lin S, Choe J, Du P, Triboulet R and
Gregory RI: The m(6)A Methyltransferase METTL3 promotes translation
in human cancer cells. Mol Cell. 62:335–345. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Visvanathan A, Patil V, Arora A, Hegde AS,
Arivazhagan A, Santosh V and Somasundaram K: Essential role of
METTL3- mediated m6A modification in glioma stem-like
cells maintenance and radioresistance. Oncogene. 37:522–533. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Barbieri I, Tzelepis K, Pandolfini L, Shi
J, Millán-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister
AJ, Han N, et al: Promoter-bound METTL3 maintains myeloid leukaemia
by m6A-dependent translation control. Nature.
522:126–131. 2017. View Article : Google Scholar
|
|
26
|
Taketo K, Konno M, Asai A, Koseki J,
Toratani M, Satoh T, Doki Y, Mori M, Ishii H and Ogawa K: The
epitranscriptome m6A writer METTL3 promotes chemo- and
radioresistance in pancreatic cancer cells. Int J Oncol.
52:621–629. 2018.PubMed/NCBI
|
|
27
|
Choe J, Lin S, Zhang W, Liu Q, Wang L,
Ramirez-Moya J, Du P, Kim W, Tang S, Sliz P, et al: mRNA
circularization by METTL3-eIF3h enhances translation and promotes
oncogenesis. Nature. 561:556–560. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu T, Yang S, Sui J, Xu SY, Cheng YP,
Shen B, Zhang Y, Zhang XM, Yin LH, Pu YP and Liang GY: Dysregulated
N6-methyladenosine methylation writer METTL3 contributes to the
proliferation and migration of gastric cancer. J Cell Physiol.
235:548–562. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Du M, Zhang Y, Mao Y, Mou J, Zhao J, Xue
Q, Wang D, Huang J, Gao S and Gao Y: MiR-33a suppresses
proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem
Biophys Res Commun. 482:582–589. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jin D, Guo J, Wu Y, Du J, Yang L, Wang X,
Di W, Hu B, An J, Kong L, et al: m6A mRNA methylation
initiated by METTL3 directly promotes YAP translation and increases
YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to
induce NSCLC drug resistance and metastasis. J Hematol Oncol.
12:1352019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang
Z, Liu Y, Zhang X, Zhang W and Ye L: HBXIP-elevated
methyltransferase METTL3 promotes the progression of breast cancer
via inhibiting tumor suppressor let-7 g. Cancer Lett. 415:11–19.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wei W, Huo B and Shi X: miR-600 inhibits
lung cancer via downregulating the expression of METTL3. Cancer
Manag Res. 11:1177–1187. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu
HC, Yuan WB, Lu JC, Zhou ZJ, Lu Q, et al: METTL3 promote tumor
proliferation of bladder cancer by accelerating pri-miR221/222
maturation in m6A-dependent manner. Mol Cancer. 18:1102019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H,
Wu M, Liang Y, Zhu F, Zhang Y, Zhang X, et al: The m6A
methyltransferase METTL3 promotes bladder cancer progression via
AFF4/NF-κB/MYC signaling network. Oncogene. 38:3667–3680. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Peng W, Li J, Chen R, Gu Q, Yang P, Qian
W, Ji D, Wang Q, Zhang Z, Tang J and Sun Y: Upregulated METTL3
promotes metastasis of colorectal cancer via miR-1246/SPRED2/MAPK
signaling pathway. J Exp Clin Cancer Res. 38:3932019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li X, Tang J, Huang W, Wang F, Li P, Qin
C, Qin Z, Zou Q, WeI J, Hua L, et al: The M6A methyltransferase
METTL3: Acting as a tumor suppressor in renal cell carcinoma.
Oncotarget. 8:96103–96116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang Y, Li Y, Toth JI, Petroski MD, Zhang
Z and Zhao JC: N6-methyladenosine modification destabilizes
developmental regulators in embryonic stem cells. Nat Cell Biol.
16:191–198. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tong J, Flavell RA and Li HB: RNA
m6A modification and its function in diseases. Front
Med. 12:481–489. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Śledź P and Jinek M: Structural insights
into the molecular mechanism of the m(6)A writer complex. Elife.
5:e184342016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang P, Doxtader KA and Nam Y: Structural
basis for cooperative function of Mettl3 and Mettl14
methyltransferases. Mol Cell. 63:306–317. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang X, Feng J, Xue Y, Guan Z, Zhang D,
Liu Z, Gong Z, Wang Q, Huang J, Tang C, et al: Structural basis of
N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature.
534:575–578. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH,
Wang F, Wang TT, Xu QG, Zhou WP and Sun SH: METTL14 suppresses the
metastatic potential of hepatocellular carcinoma by modulating
N6 methyladenosine-dependent primary MicroRNA
processing. Hepatology. 65:529–543. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Manning BD and Toker A: AKT/PKB signaling:
Navigating the network. Cell. 169:381–405. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sarbassov DD, Guertin DA, Ali SM and
Sabatini DM: Phosphorylation and regulation of Akt/PKB by the
rictor-mTOR complex. Science. 307:1098–1101. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu J, Eckert MA, Harada BT, Liu SM, Lu Z,
Yu K, Tienda SM, Chryplewicz A, Zhu AC, Yang Y, et al:
m6A mRNA methylation regulates AKT activity to promote
the proliferation and tumorigenicity of endometrial cancer. Nat
Cell Biol. 20:1074–1083. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gong D, Zhang J, Chen Y, Xu Y, Ma J, Hu G,
Huang Y, Zheng J, ZhaI W and Xue W: The m6A-suppressed
P2RX6 activation promotes renal cancer cells migration and invasion
through ATP-induced Ca2+ Influx Modulating ERK1/2
phosphorylation and MMP9 signaling pathway. J Exp Clin Cancer Res.
38:2332019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen X, Xu M, Xu X, Zeng K, Liu X, Sun L,
Pan B, He B, Pan Y, Sun H, et al: METTL14 suppresses crc
progression via regulating N6-methyladenosine-dependent primary
miR-375 processing. Mol Ther. 28:599–612. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gu C, Wang Z, Zhou N, Li G, Kou Y, Luo Y,
Wang Y, Yang J and Tian F: Mettl14 inhibits bladder TIC
self-renewal and bladder tumorigenesis through
N6-methyladenosine of Notch1. Mol Cancer. 18:1682019.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Little NA, Hastie ND and Davies RC:
Identification of WTAP, a novel Wilms' tumour 1-associating
protein. Hum Mol Genet. 9:2231–2239. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhong S, Li H, Bodi Z, Button J, Vespa L,
Herzog M and Fray RG: MTA is an Arabidopsis messenger RNA adenosine
methylase and interacts with a homolog of a sex-specific splicing
factor. Plant Cell. 20:1278–1288. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ping XL, Sun BF, Wang L, Xiao W, Yang X,
Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al: Mammalian WTAP is
a regulatory subunit of the RNA N6-methyladenosine
methyltransferase. Cell Res. 24:177–189. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Schöller E, Weichmann F, Treiber T, Ringle
S, Treiber N, Flatley A, Feederle R, Bruckmann A and Meister G:
Interactions, localization, and phosphorylation of the
m6A generating METTL3-METTL14-WTAP complex. RNA.
24:499–512. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chen Y, Peng C, Chen J, Chen D, Yang B, He
B, Hu W, Zhang Y, Liu H, Dai L, et al: WTAP facilitates progression
of hepatocellular carcinoma via m6A-HuR-dependent epigenetic
silencing of ETS1. Mol Cancer. 18:1272019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ortega A, Niksic M, Bachi A, Wilm M,
Sánchez L, Hastie N and Valcárcel J: Biochemical function of
female-lethal(2)D/Wilms' tumor suppressor-1-associated proteins in
alternative pre-mRNA splicing. J Biol Chem. 278:3040–3047. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Guo J, Tang HW, Li J, Perrimon N and Yan
D: Xio is a component of the Drosophila sex determination pathway
and RNA N6-methyladenosine methyltransferase complex.
Proc Natl Acad Sci USA. 115:3674–3679. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Robinson M, Shah P, Cui YH and He YY: The
role of dynamic m6A RNA methylation in photobiology. Photochem
Photobiol. 95:95–104. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cheng X, Li M, Rao X, Zhang W, Li XP, Wang
L and Huang G: KIAA1429 regulates the migration and invasion of
hepatocellular carcinoma by altering m6A modification of ID2 mRNA.
Onco Targets Ther. 12:3421–3428. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Qian JY, Gao J, Sun X, Cao MD, Shi L, Xia
TS, Zhou WB, Wang S, Ding Q and Wei JF: KIAA1429 acts as an
oncogenic factor in breast cancer by regulating CDK1 in an
N6-methyladenosine-independent Manner. Oncogene. 38:6123–6141.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Frayling TM, Timpson NJ, Weedon MN,
Zeggini E, Freathy RM, Lindgren CM, Perry JRB, Elliott KS, Lango H,
Rayner NW, et al: A common variant in the FTO gene is associated
with body mass index and predisposes to childhood and adult
obesity. Science. 316:889–894. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Jia G, Yang CG, Yang S, Jian X, Yi C, Zhou
Z and He C: Oxidative demethylation of 3-methylthymine and
3-methyluracil in single-stranded DNA and RNA by mouse and human
FTO. FEBS Lett. 582:3313–3319. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mauer J, Luo X, Blanjoie A, Jiao X,
Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur JJ, Chen Q,
et al: Reversible methylation of m6Am in the
5′ cap controls mRNA stability. Nature. 541:371–375. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Linder B, Grozhik AV, Olarerin-George AO,
Meydan C, Mason CE and Jaffrey SR: Single-nucleotide-resolution
mapping of m6A and m6Am throughout the transcriptome. Nat Methods.
12:767–772. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC,
Shi H, Cui X, Su R, Klungland A, et al: Differential
m6A, m6Am, and m1A demethylation
mediated by FTO in the cell nucleus and cytoplasm. Mol Cell.
71:973–985 e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wu WC, Feng J, Jiang D, Zhou X, Jiang Q,
Cai M, Wang X, Shan T and Wang Y: AMPK regulates lipid accumulation
in skeletal muscle cells through FTO-dependent demethylation of
N6-methyladenosine. Sci Rep. 7:416062017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun
L, Wang Y, Li X, Xiong XF, Wei B, et al: RNA N6-methyladenosine
demethylase FTO promotes breast tumor progression through
inhibiting BNIP3. Mol Cancer. 18:462019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Li J, Han Y, Zhang H, Qian Z, Jia W, Gao
Y, Zheng H and Li B: The m6A demethylase FTO promotes the growth of
lung cancer cells by regulating the m6A level of USP7 mRNA. Biochem
Biophys Res Commun. 512:479–485. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Liu J, Ren D, Du Z, Wang H, Zhang H and
Jin Y: m6A demethylase FTO facilitates tumor progression
in lung squamous cell carcinoma by regulating MZF1 expression.
Biochem Biophys Res Commun. 502:456–464. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Xu D, Shao W, Jiang Y, Wang X, Liu Y and
Liu X: FTO expression is associated with the occurrence of gastric
cancer and prognosis. Oncol Rep. 38:2285–2292. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Li Y, Zheng D, Wang F, Xu Y, Yu H and
Zhang H: Expression of demethylase gene, FTO and ALKBH1, is
associated with prognosis of gastric cancer. Dig Dis Sci.
64:1503–1513. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Li Z, Weng H, Su R, Weng X, Zuo Z, Li C,
Huang H, Nachtergaele S, Dong L, Hu C, et al: FTO plays an
oncogenic role in acute myeloid leukemia as a
N6-methyladenosine RNA demethylase. Cancer Cell.
31:127–141. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Su R, Dong L, Li C, Nachtergaele S,
Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, et al: R-2HG
exhibits anti-tumor activity by targeting
FTO/m6A/MYC/CEBPA signaling. Cell. 172:90–105.e23. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu
H, Ni T, Zhang ZS, Zhang T, Li C, et al: Small-molecule targeting
of oncogenic Fto demethylase in acute myeloid leukemia. Cancer
Cell. 35:677–691.e10. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhou S, Bai ZL, Xia D, Zhao ZJ, Zhao R,
Wang YY and Zhe H: FTO regulates the chemo-radiotherapy resistance
of cervical squamous cell carcinoma (CSCC) by targeting β-catenin
through mRNA demethylation. Mol Carcinog. 57:590–597. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Li J, Zhu L, Shi Y, Liu J, Lin L and Chen
X: m6A demethylase FTO promotes hepatocellular carcinoma
tumorigenesis via mediating PKM2 demethylation. Am J Transl Res.
11:6084–6092. 2019.PubMed/NCBI
|
|
75
|
Yang S, Wei J, Cui YH, Park G, Shah P,
Deng Y, Aplin AE, Lu Z, Hwang S, He C and He YY: m6A
mRNA demethylase FTO regulates melanoma tumorigenicity and response
to anti-PD-1 blockade. Nat Commun. 10:27822019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Aik W, Scotti JS, Choi H, Gong L,
Demetriades M, Schofield CJ and McDonough MA: Structure of human
RNA N6-methyladenine demethylase ALKBH5 provides insights into its
mechanisms of nucleic acid recognition and demethylation. Nucleic
Acids Res. 42:4741–4754. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhang C, Samanta D, Lu H, Bullen JW, Zhang
H, Chen I, He X and Semenza GL: Hypoxia induces the breast cancer
stem cell phenotype by HIF-dependent and ALKBH5-mediated
m6A-demethylation of NANOG mRNA. Proc Natl Acad Sci USA.
113:E2047–E2056. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Nettersheim D, Berger D, Jostes S,
Kristiansen G, Lochnit G and Schorle H: N6-Methyladenosine detected
in RNA of testicular germ cell tumors is controlled by METTL3,
ALKBH5, YTHDC1/F1/F2, and HNRNPC as writers, erasers, and readers.
Andrology. 7:498–506. 2019.PubMed/NCBI
|
|
79
|
Zhang C, Zhi WI, Lu H, Samanta D, Chen I,
Gabrielson E and Semenza GL: Hypoxia-inducible factors regulate
pluripotency factor expression by ZNF217- and ALKBH5-mediated
modulation of RNA methylation in breast cancer cells. Oncotarget.
7:64527–64542. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S,
Lu Z, Chen Y, Sulman EP, Xie K, Bögler O, et al: m6A
demethylase ALKBH5 maintains tumorigenicity of glioblastoma
stem-like cells by sustaining FOXM1 expression and cell
proliferation program. Cancer Cell. 31:591–606 e6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
He Y, Hu H, Wang Y, Yuan H, Lu Z, Wu P,
Liu D, Tian L, Yin J, Jiang K and Miao Y: ALKBH5 inhibits
pancreatic cancer motility by decreasing long non-coding RNA
KCNK15-AS1 methylation. Cell Physiol Biochem. 48:838–846. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhang J, Guo S, Piao HY, Wang Y, Wu Y,
Meng XY, Yang D, Zheng ZC and Zhao Y: ALKBH5 promotes invasion and
metastasis of gastric cancer by decreasing methylation of the
lncRNA NEAT1. J Physiol Biochem. 75:379–389. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Wang X, Zhao BS, Roundtree IA, Lu Z, Han
D, Ma H, Weng X, Chen K, Shi H and He C: N(6)-methyladenosine
modulates messenger RNA translation efficiency. Cell.
161:1388–1399. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu
PJ, Liu C and He C: YTHDF3 facilitates translation and decay of
N6-methyladenosine-modified RNA. Cell Res. 27:315–328.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Xu C, Wang X, Liu K, Roundtree IA, Tempel
W, Li Y, Lu Z, He C and Min J: Structural basis for selective
binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol.
10:927–929. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Tanabe A, Tanikawa K, Tsunetomi M, Takai
K, Ikeda H, Konno J, Torigoe T, Maeda H, Kutomi G, Okita K, et al:
RNA helicase YTHDC2 promotes cancer metastasis via the enhancement
of the efficiency by which HIF-1α mRNA is translated. Cancer Lett.
376:34–42. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Patil DP, Chen CK, Pickering BF, Chow A,
Jackson C, Guttman M and Jaffrey SR: m(6)A RNA methylation promotes
XIST-mediated transcriptional repression. Nature. 537:369–373.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zhao X, Chen Y, Mao Q, Jiang X, Jiang W,
Chen J, Xu W, Zhong L and Sun X: Overexpression of YTHDF1 is
associated with poor prognosis in patients with hepatocellular
carcinoma. Cancer Biomark. 21:859–868. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Han D, Liu J, Chen C, Dong L, Liu Y, Chang
R, Huang X, Liu Y, Wang J, Dougherty U, et al: Anti-tumour immunity
controlled through mRNA m6A methylation and YTHDF1 in
dendritic cells. Nature. 566:270–274. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kim DJ and Iwasaki A: YTHDF1 control of
dendritic cell cross-priming as a possible target of cancer
immunotherapy. Biochemistry. 58:1945–1946. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Nishizawa Y, Konno M, Asai A, Koseki J,
Kawamoto K, Miyoshi N, Takahashi H, Nishida N, Haraguchi N, Sakai
D, et al: Oncogene c-Myc promotes epitranscriptome m6A
reader YTHDF1 expression in colorectal cancer. Oncotarget.
9:7476–7486. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Bai Y, Yang C, Wu R, Huang L, Song S, Li
W, Yan P, Lin C, Li D and Zhang Y: YTHDF1 regulates tumorigenicity
and cancer stem cell-like activity in human colorectal carcinoma.
Front Oncol. 9:3322019. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Nguyen TT, Ma LN, Slovak ML, Bangs CD,
Cherry AM and Arber DA: Identification of novel Runx1 (AML1)
translocation partner genes SH3D19, YTHDf2, and ZNF687 in acute
myeloid leukemia. Genes Chromosomes Cancer. 45:918–932. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han
D, Fu Y, Parisien M, Dai Q, Jia G, et al:
N6-methyladenosine-dependent regulation of messenger RNA stability.
Nature. 505:117–120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Chen M, Wei L, Law CT, Tsang FH, Shen J,
Cheng CL, Tsang LH, Ho DW, Chiu DK, Lee JM, et al: RNA
N6-methyladenosine methyltransferase-like 3 promotes liver cancer
progression through YTHDF2-dependent posttranscriptional silencing
of SOCS2. Hepatology. 67:2254–2270. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Li J, Meng S, Xu M, Wang S, He L, Xu X,
Wang X and Xie L: Downregulation of N6-methyladenosine
binding YTHDF2 protein mediated by miR-493-3p suppresses prostate
cancer by elevating N6-methyladenosine levels.
Oncotarget. 9:3752–3764. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Paris J, Morgan M, Campos J, Spencer GJ,
Shmakova A, Ivanova I, Mapperley C, Lawson H, Wotherspoon DA,
Sepulveda C, et al: Targeting the RNA m6A reader YTHDF2
selectively compromises cancer stem cells in acute myeloid
leukemia. Cell Stem Cell. 25:137–148.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Zhong L, Liao D, Zhang M, Zeng C, Li X,
Zhang R, Ma H and Kang T: YTHDF2 suppresses cell proliferation and
growth via destabilizing the EGFR mRNA in hepatocellular carcinoma.
Cancer Lett. 442:252–261. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Asano K, Kinzy TG, Merrick WC and Hershey
JW: Conservation and diversity of eukaryotic translation initiation
factor eIF3. J Biol Chem. 272:1101–1109. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Dong Z and Zhang JT: Initiation factor
eIF3 and regulation of mRNA translation, cell growth, and cancer.
Crit Rev Oncol Hematol. 59:169–180. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
De Benedetti A and Harris AL: eIF4E
expression in tumors: Its possible role in progression of
malignancies. Int J Biochem Cell Biol. 31:59–72. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
De Benedetti A and Graff JR: eIF-4E
expression and its role in malignancies and metastases. Oncogene.
23:3189–3199. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Meyer KD, Patil DP, Zhou J, Zinoviev A,
Skabkin MA, Elemento O, Pestova TV, Qian SB and Jaffrey SR: 5′UTR
m(6)A promotes cap-independent translation. Cell. 163:999–1010.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Bachmann F, Banziger R and Burger MM:
Cloning of a novel protein overexpressed in human mammary
carcinoma. Cancer Res. 57:988–994. 1997.PubMed/NCBI
|
|
105
|
Dellas A, Torhorst J, Bachmann F, Bänziger
R, Schultheiss E and Burger MM: Expression of p150 in cervical
neoplasia and its potential value in predicting survival. Cancer.
83:1376–1383. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Wang S, Sun C, Li J, Zhang E, Ma Z, Xu W,
Li H, Qiu M, Xu Y, Xia W, et al: Roles of RNA methylation by means
of N6-methyladenosine (m6A) in human cancers. Cancer
Lett. 408:112–120. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Yang D, Qiao J, Wang G, Lan Y, Li G, Guo
X, Xi J, Ye D, Zhu S, Chen W, et al: N6-methyladenosine
modification of lincRNA 1281 is critically required for mESC
differentiation potential. Nucleic Acids Res. 46:3906–3920. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Liu N, Dai Q, Zheng G, He C, Parisien M
and Pan T: N(6)-methyladenosine-dependent RNA structural switches
regulate RNA-protein interactions. Nature. 518:560–564. 2015.
View Article : Google Scholar : PubMed/NCBI
|