Introduction
Gastrointestinal (GI) cancers include several
malignancies of the gastrointestinal tract and organs such as the
stomach, colon, liver, intrahepatic bile duct, gallbladder, and
pancreas. GI cancers have epithelial cell or stromal origin, and GI
cancers account for approx. 30% of all tumor cases in 2018.
Together GI cancers have been responsible for over 3.5 million
deaths which corresponds to 37% of the deaths from all human
malignancies (1).
While clinical assessment of GI cancer has been
performed with a physical examination, blood test, imaging, and
endoscopy, recent advancements in genomics have led to the
development of genetic analysis for diagnosis (2,3). In
the modern era of precision medicine, genetic testing has been
incorporated into routine clinical practice to assist
decision-making regarding appropriate genetically matched
treatments for patients with GI cancers (4). To acquire genetic information in a
timely and cost-effective manner, a variety of gene panel tests,
which is based on next-generation sequencing (NGS), have been
progressed and some have been approved by the U.S. Food and Drug
Administration (FDA) as companion diagnostics for multiple
molecular-targeted therapies (5).
In contrast, the clinical value of genetic testing for GI cancer
receptiveness is not well established. This review describes the
current genomic landscape in GI cancers, and testing modalities
that have prognostic, predictive, and therapeutic value. We provide
an outline of the clinical use of multi-gene testing in GI cancers,
and discuss the practical utility and potential of a liquid biopsy
and whole genome sequencing.
Methods
Non-systematic review was conducted in a basis of an
electronic search through the medical literature using PubMed and
Google Scholar. The keywords ‘genetic testing’, ‘Multi-gene panel
testing’, ‘Whole genome sequencing’, ‘Next-generation sequencing’,
‘gastrointestinal cancer’, ‘colorectal cancer’, ‘pancreatic
cancer’, ‘gastric cancer’, ‘hepatobiliary cancer’, ‘esophageal
cancer’, were searched. Guidelines and review articles from
gastroenterology, oncology, endoscopy and genetics were included.
When more than one guideline concerning the same subject was
available, the latest one was picked up. Only full articles in the
English language published in the last ten years were considered to
be suitable for review. The exclusion criteria consisted duplicated
articles, studies absent of diagnostic outcomes. Case reports,
editorials, book chapters, correspondences, letters, and non-human
research were not included. First, the titles were screened and
appropriate studies were selected. Of these studies, the full text
was acquired. A total of 258 articles were identified (Fig. 1).
Multi-gene panel testing
Comprehensive genomic profiling by NGS enables the
detection of multiple genomic features in GI cancers. Multi-gene
testing is conducted based on NGS platforms, and before sequencing,
genomic regions of attention are chosen from the DNA sample
(6). The sustainability and quality
assurance of the molecular tumor board, named Expert Panel,
examines the appropriate treatment methods such as drug treatment
and participation in clinical trials, and the results were
informed. Multi-gene panels are largely utilized in clinical
settings for the identification of somatic and germline mutations
in GI cancers, which lead to molecular classification, and
prediction of therapeutic effect. It can also detect the gene which
is involved in drug resistance. Additionally, microsatellite
instability (MSI) and tumor mutational burden (TMB) are approved by
the FDA as companion diagnostics for GI cancers.
Recently, the survival benefit of matched therapy
using panel testing has been established. An encouraging impact on
tumor response rates, patient outcome, and on detecting novel tools
of molecularly-targeted therapy has been suggested by some clinical
trials by utilizing multi-gene testing which may lead to
personalized cancer treatments (7–9). The
Know Your Tumor program testing matched therapies after multi-omics
profiling provided suggestions for elucidated clinical trials and
personalized therapy for patients with pancreatic cancer (PC). The
outcomes of this trial revealed that the patients with BRCA
mutations after Poly (ADP-Ribose) Polymerase (PARP) inhibitor
treatment or those with mismatch repair deficiency after immune
checkpoint blockade (ICB) treatment demonstrated 1-year survival
benefit compared with patients who received unmatched therapies or
those without an actionable molecular change (10). This study also showed that mutations
in the DNA damage response (DDR) pathway were the most popular
actionable alteration. These data would indicate a guarantee for
this precision approach. Although the frequency of druggable
genetic alterations in GI cancers is lower than that in breast
cancer and non-small cell lung cancer, a variety of noble candidate
genes have been identified over the past few years.
In this section, we summarize the current status of
genetic tests and molecular-targeted therapies for GI cancer that
are expected in the future (Table
I).
 | Table I.Key genetic alterations of
gastrointestinal cancer and the corresponding targeted
therapies. |
Table I.
Key genetic alterations of
gastrointestinal cancer and the corresponding targeted
therapies.
| Genomic
profiling | Targeted agent |
|---|
| Gastric cancer |
|
|
HER2 | Trastuzumab,
Trastuzumab- |
|
| Deruxtecan |
|
FGFR | Bemarituzumab |
|
VEGFR | N/A |
|
MSI | Pembrolizumab |
| Colorectal
cancer |
|
|
EGFR | Cetuximab,
Panitumumab |
|
BRAFV600E | Encorafenib |
|
PIK3CA | N/A |
|
MEK | Binimetinib |
|
HER2 | Trastuzumab,
Trastuzumab- |
|
| Deruxtecan |
| KRAS
G12C | Sotorasib,
Adagrasib |
|
NTRK | Larotrectinib,
Entrectinib |
|
MSI | Pembrolizumab,
ipilimumab |
| Pancreatic
cancer |
|
|
KRAS | N/A |
|
TP53 | N/A |
|
CDKN2A | N/A |
|
SMAD4 | N/A |
|
EGFR | Erlotinib |
|
NTRK | Larotrectinib,
Entrectinib |
|
ALK | N/A |
|
BRAF | Encorafenib |
|
PIK3CA | N/A |
|
BRCA/HRD | Olaparib |
|
MSI | Pembrolizumab |
| Hepatocellular
carcinoma |
|
|
TERT | N/A |
|
TP53 | N/A |
|
CTNNB1 | N/A |
|
VEGFR | Lenvatinib,
Cabozantinib |
|
ARID1A | N/A |
|
CCND1 | N/A |
|
MET | N/A |
|
PTEN | N/A |
| Biliary tract
cancer |
|
|
FGFR | Pemigatinib,
Infigratinib |
|
IDH1/2 | Ivosidenib |
|
BRAF | N/A |
|
TP53 | N/A |
|
HER2 | Trastuzumab,
Pertuzumab |
|
PIK3CA | N/A |
|
NTRK | Larotrectinib,
Entrectinib |
Colorectal cancer
For a decade, patients with KRAS/NRAS wild type are
acceptable for therapy targeting the epidermal growth factor
receptor (EGFR) signaling (11).
BRAFV600E mutation is identified in approximately 8–10% of
colorectal cancer (CRC) and generates a RAS-independent
constitutional activation of the mitogen-activated protein kinases
(MAPK) pathway, which leads to cell growth and survival and is a
prognostic biomarker for patients with CRC (12). Even if some BRAF mutations
are identified beyond the V600 hotspot in CRC, they do not present
similar clinical, biological, and therapeutic results as the V600E
mutation (13). These BRAF
non-V600E mutated tumors tend to be well differentiated with
left-sided tumor site and were correlated with improved prognosis
and resistance to BRAF inhibitors, whereas some have a sensitivity
to EGFR (14,15). Notably, the MSI phenotype, which can
predict the efficiency of immune checkpoint blockade (ICB)
therapies, was identified in approximately 20% of BRAFV600E CRC,
regardless of the BRAF mutational status (12). BRAF inhibition has been said to
cause a rapid feedback EGFR activation, which assists MAPK
constitutive signaling. Continued proliferation and resistance of
these tumors to BRAF inhibitor monotherapy may occur by
EGFR-mediated stimulation of downstream signaling (16). In light of these, the combination
strategy with the BRAF inhibitor, anti-EGFR agents,
phosphatidylinositol-3-kinase (PI3K) inhibitors, or MEK inhibitors
was investigated (17–20). These studies assisted the scheme of
the BEACON CRC phase III study, which elucidated encorafenib,
binimetinib, and cetuximab or encorafenib and cetuximab, or other
treatment options, such as cetuximab and irinotecan or cetuximab
and FOLFIRI (folinic acid, fluorouracil, and irinotecan).
Metastatic CRC (mCRC) patients harboring a BRAF exon 15
p.V600E point mutation, with disease progression after one or two
prior treatment approaches, were randomized. Conclusively, the
median overall survival (OS) was prolonged over 3 months in the
triplet and the doublet experimental regimens, compared to the
control. Notably, median progression-free survival (PFS) was
superior in the triple-combination group and in the association of
the doublet group compared with the other group. These data
indicated the clinical benefit of the molecular-targeting
combination therapy in previously treated patients with mCRC
harboring a BRAF exon 15 p.V600E point mutation (21). Although the two experimental
regimens were not compared in the study the combination of
encorafenib plus cetuximab, without the MEK inhibitor, is currently
positioned as the standard for second- and third-line BRAF
V600E-mutated mCRC (22).
KRASG12C (glycine 12 to aspartic acid) is one of the
most popular KRAS mutations in CRC. A novel production of
KRAS inhibitors may result in a revolutionary change in the
treatment for patients with CRC (23). In a recent, convincing potencies of
a direct KRASG12C inhibitor were described. AMG 510 is a
new small molecule that exclusively and irretrievably impaired
KRASG12C activity, by locking it in an inactive
guanosine diphosphate-bound state. The initial study using AMG 510
in patients with advanced or metastatic KRASG12C
mutant solid tumors (CodeBreak-100; NCT03600883) demonstrated that
ORR and DCR were 12.0 and 80.0%, respectively, in
KRASG12C-mutated mCRC patients (24). Although the majority of CRCs
initiate through the chromosomal instability pathway, 10–15% of
CRCs occur based on the MSI pathway. MSI/dMMR CRCs are
characterized by a high TMB with highly immunogenic neoantigens
arising from frameshift mutations that induce high infiltration
through activated cytotoxic T CD8+ lymphocytes (25,26).
for the therapy of CRC patients with MSI/dMMR, who progressed after
first or second chemotherapy (27).
HER2 gene amplification was found in approximately 1 to 8%
of CRC patients (28). Although the
prognostic implication of HER2 amplifications is
controversial, a negative predictive value of HER2
amplifications for anti-EGFRs efficacy tends to be familiar
(29). Phase II studies, named
Heracles-A, and MyPathway, evaluated the combination therapy of
trastuzumab plus lapatinib, and trastuzumab with pertuzumab.
Convincing response rates of 30 and 32% and median PFS of 4.7 and,
2.9 months, respectively were demonstrated (30,31).
Trastuzumab-Deruxtecan (T-DXd) is a new antibody drug conjugate
consisting of a humanized anti-human epidermal growth factor
receptor2 (HER2) antibody, a cleavable, peptide-based linker, and a
potent topoisomerase I inhibitor. T-DXd showed a preliminary effect
on HER2-positive mCRC refractory to standard therapy, which may
lead to the increased advancement of precision treatment of
HER2-positive CRC (32).
Recently, NTRK gene fusions emerged as a greatly
attractive target for the treatment of patients with cancer. A
remarkable clinical significance is demonstrated by TRK inhibitors
(larotrectinib, entrectinib). The ALK and ROS1 genes,
which encode for the homonym tyrosine kinase receptors, mediate
various cellular biological activity via diverse signal
transduction (33). ALK,
ROS1, and NTRK fusions occur in 0.2 to 2.4% of CRCs
(34). Hence, it needs to choose
the population to be examined.
Pancreatic cancer
A recent study utilized whole genome sequencing
(WGS) to map the genome of 100 pancreatic ductal adenocarcinoma
(PDAC) specimens (35).
Acknowledged common drivers of PDAC (KRAS, TP53, CDKN2A, and
SMAD4) were emphasized in this study, and also various other
mutations at greatly lower frequencies were shown. Activating
KRAS mutations are found in more than 90% of PDAC. Besides,
ALK rearrangements, NRG1 rearrangements, NTRK
fusions, ROS, BRAF, PIK3CA, and a variety of
cancer-associated genes identified as potential drivers have been
detected (e.g., STK11, RB1, GNAS, CHEK2, and ERBB2),
which may lead to potential targets. Inactivation of tumor
suppressor genes, including SMAD4, CDKN2A, and, TP53
is detected in advancing pancreatic intraepithelial neoplasia
progression and arises in up to 50% (36). The frequency of persistently mutated
genes then diminishes to less than 10%, which accumulates into
central molecular pathways, such as KRAS, wingless and int, TGF-β
signaling, DDR, NOTCH, RNA processing, cell cycle regulation
(37). Associations of numerous
pathways with survival have been detected by analyses of pathways
in PDAC patients. DNA repair-related pathways were shown to
contribute to a poor outcome (38).
In preclinical vivo models and the clinical setting, several of
these pathways can be actionable targets for treatment. BRAF
mutation and NTRK gene fusions in KRASWT, MMR-D/MSI-H, and
genetic alterations in homologous recombination deficiency (HRD)
are considered to be prospective actionable mutations. The American
Society of Clinical Oncology (ASCO) guidelines recommended early
examination for actionable genetic alterations for PC patients who
can be convincing candidates for subsequent therapy following
first-line therapy (39). Patients
with BRCA mutations, NTRK gene fusions, and MSI-H/MMR-D are
likely to be provided personalized therapies, such as PARP
inhibitors, TRK fusion inhibitors, and ICB therapy,
respectively (10).
In ovarian and breast cancers, discriminate defects
in Homologous Recombination DNA repair genes, such as germline
mutations in BRCA1, 2, and PALB2, somatic mutations
in BRCA1, 2, and promoter methylation of BRCA1, have
been represented (40,41). BRCA mutations also contributed to
promoting the risk for PC. BRCA genes encode for proteins
involved in the HR repair of DNA double-stranded breaks. PC
patients with deficient HR repair are predicted to be responsible
for PARP inhibition. Hence, PARP inhibitors are efficient for
selective patients with HRD owning to BRCA1 or BRCA2
mutations (42). According to
recent experiments of genomic profiling in large populations of
PDAC, the importance of HRD in predicting the efficacy on PARP
inhibitors and platinum-based therapy was accumulated (10,43).
ASCO guidelines recommended treatment with PARP inhibitor or
platinum-based chemotherapy for patients with a germline BRCA1,
2 mutations. In a recent randomized phase III study (POLO), the
efficacy of olaparib was demonstrated in germline
BRCA-mutated metastatic PDAC (44), suggesting that HRD can effectively
be targeted in pancreatic cancer. Due to Lynch syndrome or somatic
MMR gene mutations, about 1% of PDAC patients have MMR-D/MSI-H
(45,46). ASCO guidelines reported that
pembrolizumab is advocated as a second-line therapy for PDAC
patients with MMR-D/MSI-H (39).
Gastric cancer
The Phase III TOGA trial assessed the
trastuzumab-containing regimen compared with standard first-line
chemotherapy. Trastuzumab yielded a statistical improvement in
terms of median OS, median PFS, and overall response rate (ORR)
(47). According to these results,
trastuzumab combined therapy come to be the standard treatment for
advanced HER2-positive gastric cancer (GC). Although the continuous
administration of trastuzumab after progression failed to improve
PFS in patients with HER2-positive GC, a phase II trial that
assessed the efficacy and safety of T-DXd vs. a physician's choice
of chemotherapy in patients with HER2-positive GC treated with two
prior lines including trastuzumab (The DESTINY-Gastric01 trial)
showed that OS, as a key secondary endpoint with T-DXd, was
significantly improved (median OS 12.5 vs. 8.4 months, HR 0.59,
P=0.01). According to these results of the DESTINY-Gastric01 trial,
T-DXd was approved for the treatment of patients with HER2-positive
unresectable GC in Japan (48,49).
Alterations in fibroblast growth factor receptor
(FGFR) genes are found in gastric and gastro-esophageal junction
cancers and frequency ranges between 3–7%. The most familiar
alteration is amplifications, which are followed by rearrangements
and mutations (50). The
amplification level was revealed to be negatively associated with
patients' prognosis (51). A
first-in-class humanized fucosylated IgG1 monoclonal antibody
directed against FGFR2b, bemarituzumab has demonstrated convincing
results in a phase I study in solid tumors and FGFR2b-positive GC
patients (52). The addition of
bemarituzumab to chemotherapy was evaluated in first-line therapy
in locally advanced, unresectable, metastatic HER2-negative and
FGFR2b-positive GC patients. Notably, median PFS was prolonged to
9.5 months in the bemarituzumab group, compared to 7.4 months in
the placebo group. Median OS was not reached in the bemarituzumab
group vs. 12.9 months in the placebo arm (HR, 0.58, 95% confidence
interval 0.35–0.95; P=0.03) and ORR was prolonged from 40 to 53% in
the bemarituzumab group (53).
Evaluation of Phase III trials in the near future is expected
(NCT03694522).
Hepatocellular carcinoma
Although the etiology of hepatocellular carcinoma
(HCC) is unsatisfactorily evaluated, recent developments in genomic
studies have provided a profound understanding of HCC advancement
and may result in new approaches for prevention and treatment.
TP53, CTNNB1, ARID1A, ARID2, AXIN1, RB1, and NFE2L2
are the most common mutations in HCC. In a recent, catalytic
telomerase reverse transcriptase (TERT) has been distinguished as a
frequent driver mutation which is identified in 40–65% of HCC
patients (54,55). VEGFA, MYC, CCND1, and
MET are other oncogenes frequently amplified (56,57),
PTEN is often suppressed (58) and p16 is commonly deleted in
HCCs (59). Present guidelines
recommend chemotherapy, with sorafenib being the only first-line
therapy for unresectable HCC because of its approval in 2007. A
recent REFLECT trial demonstrated that lenvatinib revealed OS
non-inferiority to sorafenib (60).
The IMbrave 150 trial displayed a combination of atezolizumab plus
bevacizumab showed better PFS and OS than that associated with
sorafenib (61). Therefore,
atezolizumab plus bevacizumab has been positioned as a first-line
HCC therapy. According to the RESORCE trial, which showed the
superiority of prognosis in patients with HCC whose disease
progressed during sorafenib treatment, Regorafenib has been
approved as a second-line therapy (62). Additionally, in a phase III trial
(CELESTIAL), cabozantinib was presented to have met clinical
endpoints, compared with control, as a second-line treatment
(63). However, there are no
molecular-targeted drugs that match these major genetic
abnormalities, and personalized medicine is rarely conducted.
Biliary tract cancers
Biliary tract cancers have poor prognoses even when
cytotoxic chemotherapy is applied. Based on the phase 3 ABC-02 and
BT-22 trials, combined cisplatin with gemcitabine is the recent
standard treatment in unresectable, metastatic biliary tract
cancers (55,64). In the second-line setting, FOLFOX
(folinic acid, 5-fluorouracil, and oxaliplatin) showed a prolonged
OS compared with best supportive care in the phase 3 ABC-06 trial
(65). The molecular analysis of
biliary tract cancers has significantly improved understanding of
the underlying pathological mechanism which may lead to novel
targeted therapeutic approaches. FGFR2 fusions and
IDH1/2 mutations are the most ordinary and clinically
important genetic aberrations in intrahepatic cholangiocarcinoma,
whereas TP53 mutations, KRAS mutations, and
HER2 amplifications are the most meaningful genetic
aberrations in extrahepatic cholangiocarcinoma (66).
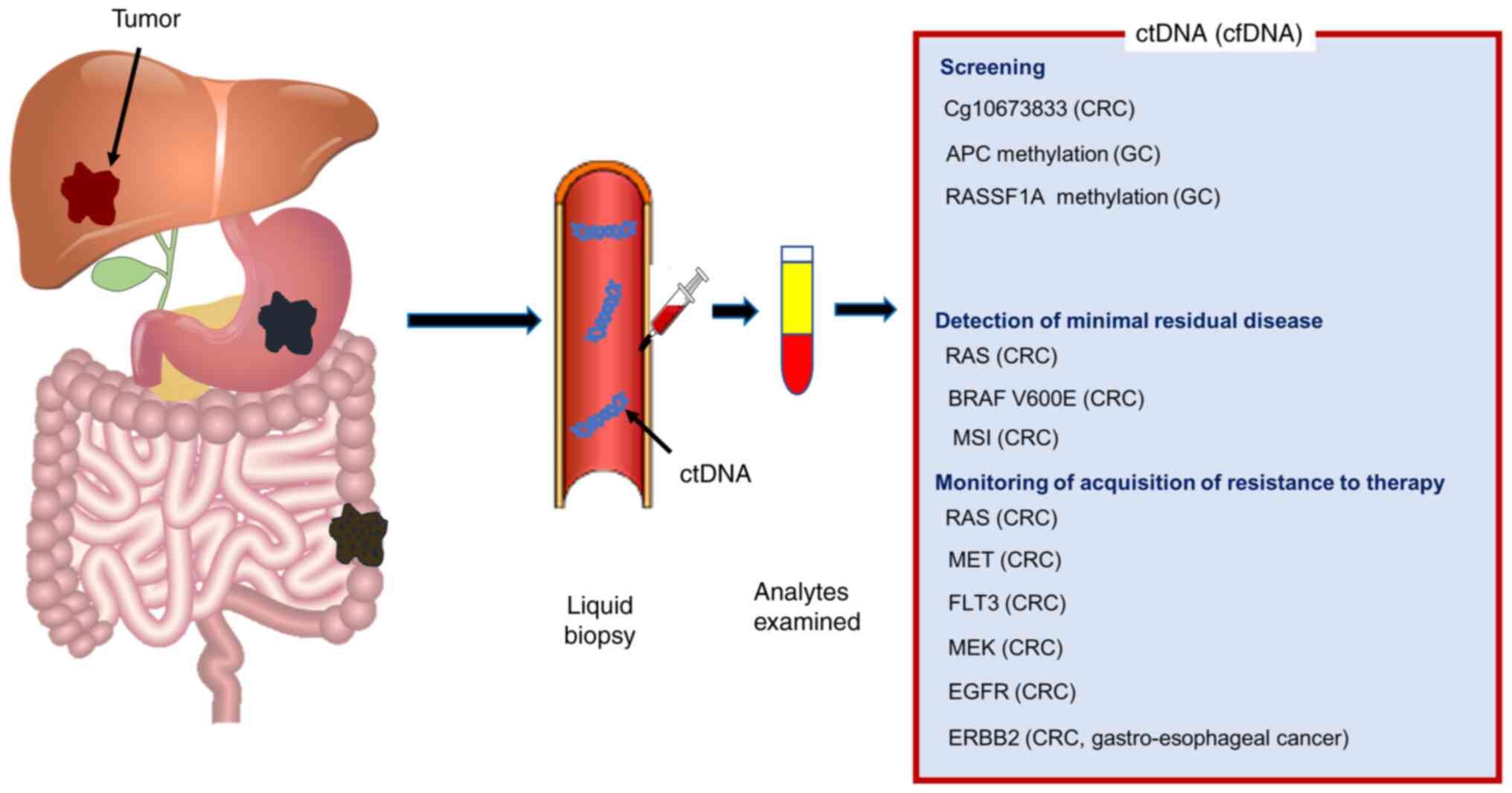
Liquid biopsy
GI cancers account for a significant proportion of
mortality worldwide (1). For these
tumors, staging at diagnosis persists as the most principal
prognostic factor. With the advancement of tumor biology, it has
become important to search for basic knowledge such as pathology as
well as for biomarkers that characterize tumors for a treatment
approach. Although genome-based precision medicine is convincing,
tissue-based genomic sequencing for first-line therapy
decision-making in GI cancer remains obstacles owning to the long
turnaround time between the receipt of tissue samples and reporting
results. Liquid biopsy has the potential to detect circulating
tumor DNA (ctDNR) from all tumors that shed into the circulation
and can be used to assess intratumor genetic heterogeneity and
overcome the limitations of tissue analysis. Circulating tumor
cells, ctDNR, exosome, and microRNAs exist in the blood or other
body fluids and exhibit the tumor condition in real-time. More
recently, methods based on NGS have enabled ctDNA profiling as a
replacement for tumor tissue sequencing (67,68).
So far, assays applied for ctDNA can be categorized into two
classes: those targeted for a single or small number of variants
including CAPP-Seq, Safe-seq, Signatera, or ArcherDX, which have a
limit of identification no more than 0.01% variant allele
frequency, and those aimed at a broader coverage. These
comprehensive panel-based sequencing assays which integrate genomic
alterations as well as methylation status, are used for genotyping
or early diagnosis and achieve a detection limit of approximately
0.2% in the Guardant Health Reveal test (69). Furthermore, cancer genomic testing
using ctDNA has been commercialized and approved with an insurance.
Meanwhile, although cell-free DNA (cfDNA)-based liquid biopsy test
has been approved by the U.S. FDA to detect EGFR mutations in the
ctDNA of patients with NSCLC who are candidates for targeted
therapy with erlotinib and osimertinib (70), further studies are still required to
confirm the clinical usefulness of ctDNA as prescribed by ASCO
(71).
Several studies evaluated the possibility of ctDNA
as a screening device for tumor progression. A recent study
presented that in a high-risk population of 1493 enrolled patients
in a prospective cohort study, a single ctDNA methylation marker,
cg10673833, revealed distinguished diagnostic accuracy, with the
sensitivity of 89.7%, and specificity of 86.8% for the finding of
CRC and precancerous lesions (72).
The promoter methylation of APC and RASSF1A in cfDNA
was illustrated as frequent epigenetic results in patients with
operable GC at an early stage (73). In HCC, when using NGS technology
with a panel of regularly altered genes, in a prospective cohort of
30 patients, the ctDNA detection rate reached 63% with stage A
based on the Barcelona Clinic Liver Cancer score (74). 81% of concordance rate was obtained
between tissue and liquid biopsy. Distinguishment of HCC specimens
from control cirrhotic and not cirrhotic tissue samples was
reported with a specificity of 95% by a combination of five
aberrant methylation biomarkers (75). Mutations of exons 9, 11, 13, and 17
of KIT, and exons 12, 14, and 18 of PDGFRA are important
drivers of oncogenesis and exist in around 85–90% of
gastrointestinal stromal tumors. Hence, the main part of the
studies assessing the usefulness of ctDNA in gastrointestinal
stromal tumors was focused on KIT and PDGFRA
alterations (76).
In GI cancers, evaluation of minimal residual
disease (MRD) through the study of ctDNA is not still defined, but
has already been assessed in diverse analyses. In the TRACC study
(NCT04050345) designed on stage II–III CRC, 6 out of 14 (43%)
MRD-positive patients recurred whereas only 8 out of 93 (9%)
MRD-negative patients did.
The TRACC study (NCT04050345) demonstrated that 6
out of 14 (43%) MRD-positive patients with stage II–III CRC
recurred whereas only 8 out of 93 (9%) MRD-negative patients did.
The most meaningful prognostic factor related with recurrence-free
survival was shown to be ctDNA status. CRC patients at high risk of
recurrence and who will really receive benefit from adjuvant
therapy may be identified (77).
ctDNA measurements provide the capability to guide surveillance
while detecting latent candidates for escalated or de-escalated
adjuvant therapy approaches in resected, stage I–III CRC. A report
at the conference in ESMO 2021 evaluated somatic tissue mutations
using MSK-IMPACT, and ctDNA utilizing Guardant360, FoundationOne,
or MSK-ACCESS. ctDNA identification predicted the risk of
recurrence in resected MSI-high patients and evaluated the effect
of ICI (atezolizumab) on these MRD positive patients (NCT03832569).
Meanwhile, the retrospective CORRECT trial, analysis of ctDNA,
could predict the clinical utility of regorafenib and evaluate the
survival in mCRC patients (78).
Recently, the CIRCULATE-Japan trial, which involved a prospective
nationwide patient-screening registry named GALAXY using the
Signatera ctDNA assay, reported preliminary findings (79). The sample size of this observational
study is 5,000 and 301 patients had clinical stages I, II, and III
CRC with preoperative ctDNA identified in 50 (77%), 267 (95%), and
288 (96%) patients, respectively. Interestingly, ctDNA-positive
status at 4 weeks showed a negative correlation with survival
despite the association with RAS, BRAF V600E, and MSI status
were not demonstrated. Notably, 99% of patients with ctDNA-negative
clinical stage I–III survive for the postoperative 6-months.
Liquid biopsy is nearly ready to be approved not
only for diagnosis but also for monitoring the acquisition of
resistance to therapy in real-time due to its minimal invasiveness
and easy collection. For example, in clinical and preclinical
studies, RAS mutant clones have been elucidated as drivers
of acquired resistance to anti-EGFR treatment (80). The appearance of acquired RAS
mutations and alterations in other genes, such as ERBB2, MET,
FLT3, MEK, and EGFR was suggested by an extensive
observation of ctDNA using a ctDNA assay based on NGS during
anti-EGFR treatment (81). In
metastatic HER2-positive gastro-esophageal cancer, a longitudinal
surveillance of serial plasma samples utilizing a ctDNA assay
demonstrated that to be correlated to the resistance to trastuzumab
in patients treated with trastuzumab in addition to chemotherapy
(82). The CRICKET phase II study,
the first prospective trial evaluating the efficacy of rechallenge
approach with cetuximab and irinotecan, displayed the benefit in
RAS/BRAF wild-type mCRC patients with acquired resistance to
cetuximab. RAS mutation was not identified in patients who
partially responded to treatment (83,84).
Numerous clinical studies are currently assessing the role of
liquid biopsy in anti-EGFR rechallenge (CHRONOS, NCT03227926;
RASINTRO, NCT03259009) (83). We
summarize the clinical relevance of liquid biopsy with ctDNA in GI
cancer in Fig. 2.
In the near future, liquid biopsies with ctDNA will
be essential for GI cancer treatment. A recent study showed that
ctDNA analysis significantly reduced the screening period and
improved the study enrollment rate compared with sequencing of
tumor specimens in GI cancer. Collectively, ctDNA was found in
91.4% of patients (85). Besides,
liquid biopsy permits the collection of repetitive samples during
the course of the patient's therapy and the collection of clones
that show resistance to ongoing treatment. ctDNA as circulating
biomarkers can assess the response to ongoing treatments, thus
rapidly guiding the medical choice for further chemotherapy regimen
and the requirement to switch treatment strategy. Thus, the
application of ctDNA-based analysis may provide great benefits in
supporting clinical decision-making and improving patient
prognosis, which may lead to personalized medicine.
Hereditary gastrointestinal cancer
In addition to the main purpose of predicting the
drug effects, genetic testing may result in findings regarding
germline variants (secondary findings). Multi-gene panel testing
has been increasingly required and become broadly available in the
research of hereditary cancer syndromes (86). Analysis of secondary findings has
been discussed, and the results will be disclosed if the patient
wishes to reveal them after discussing whether they should be
disclosed in the expert panel. For patients and their families
identified at risk by genetic testing, strategies for rigorous
screening and risk-decreasing approaches for cancer prevention are
important in their outcomes. They can accept genetic counseling
consisting of medical geneticists and genetic counselors. Nowadays,
guidelines state that patients with suspected hereditary CRC and PC
should receive genetic counseling and be offered comprehensive
genetic testing (87). The National
Comprehensive Cancer Network guideline (88) plans a series of clinical outlines
the way to approve multi-gene panel testing: i) when personal
medical and/or family cancer history meets criteria for more than
one hereditary cancer syndrome ii) when family cancer history does
not meet established testing guidelines, but consideration of
inherited cancer risk persists iii) in individuals concerned about
cancer predisposition for whom family cancer history is limited or
unknown. Hereditary breast and ovarian cancer have been
investigated extensively by utilizing a multi-gene panel. It
consists of genes as BRCA1, BRCA2, PALB2, ATM, MLH1, MSH2, MSH6,
TP53, CHEK2, STK11 and PTEN (89). A sensitivity to GI cancer was
distributed among these genes, which led to personalized treatment
and follow-up (88,89).
In GI cancers, the major organs involved with
inherited cancer are the colon, pancreas, and stomach. Table II provides an overview of
hereditary GI cancers, along with their genetic cause, cancer
risks, and drug sensitivity.
| Table II.Characteristic feature of hereditary
gastrointestinal cancers. |
Table II.
Characteristic feature of hereditary
gastrointestinal cancers.
| Disease | Causative
genes | Inheritance
trait | Gastrointestinal
tumors (lifetime cancer risk) | Other
malignancies | Drug
sensitivity |
|---|
| Lynch syndrome | MLH1,
MSH2, | AD | CRC (22–74%) | Endometrium,
ovary, | ICI, |
|
| MSH6,
PMS2, |
| GC (11–19%) | uterus, brain |
|
|
| EPCAM |
| Small bowel cancer
(1–4%) |
|
|
|
|
|
| PC (3–4%) |
|
|
|
|
|
| HCC and bile
tract |
|
|
|
|
|
| cancer (2–7%) |
|
|
| FAP | APC | AD | CRC (100 %) | Thyroid,
adrenal | NSAID |
|
|
|
| Duodenum and
ampullary | gland, brain |
|
|
|
|
| (4–12%) |
|
|
|
|
|
| GC (<1%) |
|
|
|
|
|
| PC (2%) |
|
|
| JPS | SMAD4, | AD | CRC (39%) | None | N/A |
|
| BMPR1A |
| GC, PC, Small
bowel |
|
|
|
|
|
| cancer (21%) |
|
|
| PJS | STK11 | AD | CRC (39%) | Breast, lung,
ovary, | N/A |
|
|
|
| GC (29%) | uterus, testis,
cervix |
|
|
|
|
| PC (11–36%) |
|
|
|
|
|
| Small bowel cancer
(13%) | Sarcoma, breast,
adrenal | N/A |
| LFS | TP53 | AD | CRC (12.5%) | gland brain,
lung |
|
|
|
|
| GC (4.8%) |
|
|
|
MUTYH-associated | MUTYH | AR | CRC (40–100%) | Thyroid, | N/A |
| polyposis |
|
| Duodenum (4%) |
|
|
| Cowden
syndrome | PTEN | AD | CRC (9–16%) | Breast,
thyroid, | N/A |
|
|
|
|
| endometrium,
brain, |
|
|
|
|
|
| kidney |
|
| HDGC | CDH1 | AD | GC (70–80%) | Breast | N/A |
| HBOS | BRCA1,
BRCA2, | AD | PC (1–7%) | Breast, ovary, | PPAP |
|
| PALB2 |
|
| prostate, skin | inhibitor |
| FAMMM | CDKM2A | AD | PC (17%) | Skin
(melanoma), | N/A |
|
|
|
|
| lung, larynx,
breast |
|
Colorectal cancer
Lynch syndrome (LS) is one of the most familiar
hereditary cancer syndromes that is caused by germline pathogenic
variants in DNA MMR, including EPCAM, MLH1, MSH2, MSH6, and
PMS2 (90). Families with LS
have a high risk of developing colorectal, small intestine,
ureteral, urological, endometrial, ovarian, and hepatobiliary
cancer, and are prone to progress cancer at a youthful age. The
risk of advancing cancer in LS varies according to the causal gene
(91). MSI testing is recommended
by the American guidelines for all CRC patients with newly
diagnosed CRC to find LS patients (92). When a pathogenic germline variant in
MMR genes is detected by following genetic testing, LS is
diagnosed. Microsatellite regions are involved in various genes
contained in cancer initiation, and the accumulation of aberrations
in these regions caused MSI-H. GI cancers with MSI-H are remarkably
sensitive to ICI, suggesting that ICI should be efficient in LS
(93).
Pancreatic cancer
Hereditary cancers caused by germline pathogenic
variants are present in approximately 5–10% of PC. Individuals with
at least one first-degree relative (FDR) with PC are at higher risk
(OR 1.76). Patients with no less than one FDR with PC have elevated
risk. The more FDR additionally increases this risk. If a family
involves two concerned FDRs, the colleagues of this family are
identified as FPC kindreds. Risk elevates promptly depending on the
number of affected family members; 4.6-fold with one, 6.4-fold with
two, and 32-fold with three affected FDRs (94). Well-defined genetic cancer
sensitivity syndromes correlated with PC clarify a minority of this
familial accumulation, as shown in Table II. In recent studies using
gene-panel testing, some PCs harbor actionable BRCA1/2
pathogenic or likely pathogenic variants (0–3% for BRCA1 and
1–6% for BRCA2) were presented (95–97).
While screening every person for PC is expensive due to the
comparatively low occurrence of this disease and the deficiency of
precise, and noninvasive screening methods, screening may have
significance for patients who reveal elevated risk (98,99).
Genetic testing seems to be worthwhile for patients with an
increased risk of carrying a pancreas-related cancer susceptibility
gene.
Gastric cancer
Hereditary Diffuse Gastric Cancer (HDGC is
characterized by a high prevalence of diffuse-type GC in the family
lineage. HDGC is an autosomal dominant inheritance caused by a
germline CDH1 mutation encoding the adhesion molecules
E-cadherin. In Western countries, approximately 40% of HDGC
families are shown to have germline mutations in CHD1
(100). Genetic testing is
recommended for HDGC candidates because it involves multiplex
ligation-dependent probe amplification is recommended for HDGC
candidates. Truncating mutations in CDH1 and CTNNA1
are thought to be responsible for this syndrome. Recently, exome
sequencing identified germline mutations of some related genes,
such as INSR, FBXO24, MAP3K6, PALB2, RAD51C, MET, and
DOT1L as other latent candidate genes for HDGC (101).
The increasing use of multigene panel testing has
redefined gene-cancer associations, and consecutively, cancer risk
assesses that penetrance values range from low to high. Cancer
screening approvals and preventive strategies adapted by germline
mutation will enable us to improve clinical prognosis for patients
at greatest risk of cancer and their kindreds.
Comprehensive genetic analysis
As mentioned above, standards of cancer molecular
diagnostics, including multi-gene panels have been launched and
developed in clinical settings. In contrast, these tests cover only
a certain number of associated genomic alterations in coding
regions of the genome. Because cancer genomes evolve in a while, it
is recommended to utilize comprehensive NGS techniques over
restricted-gene tests. Recent advances in NGS as large-scale
sequencing technology allow one to investigate the entire genome
(WGS), the exons within all known genes (whole exome sequencing,
WES), or total RNA (whole transcriptome sequencing) (102). WGS is theoretically
straightforward. DNA is randomly fragmented by physical shearing,
and 30–50× sequence depth (90–150 Gb) of the individual human whole
genome is ordinarily sequenced for both cancer and normal genomes,
which result in comprise 99% of the total human genome (103). WGS strategies can identify
unexplored mutations, such as untranslated regions, introns,
promoters, non-coding functional RNA, and mitochondrial genomes, as
well as coding mutations and somatic copy number alterations. WGS
also provides a range of diagnostic significance, including new
detection in rare cancer mutations (104). WGS analysis will enable us to
clarify the functions of these unknown genomic regions and further
understand the whole landscape of cancer genomes (105). The comprehensive genetic testing
for GI cancer now being examined in clinical studies is reviewed in
Table III.
| Table III.Ongoing clinical trials of
gastrointestinal cancer classified on comprehensive entire genetic
testing. |
Table III.
Ongoing clinical trials of
gastrointestinal cancer classified on comprehensive entire genetic
testing.
| Sequencing/NCT
number | Type of trial | Clinical
purpose | Results | Detection
method | Comments |
|---|
| WGS |
|
|
|
|
|
|
NCT02759657 (COMPASS) | Cohort | Diagnostic | Active, not
recruiting | Tissue | Comprehensive
molecular characterization of PDAC for better treatment
selection |
|
NCT03254121 (HEPCASUS) | Cohort | Diagnostic | Completed | Tissue | Genome studies of
HCC developed in hepatitis C patients with sustained virological
response |
|
NCT03718897 |
Cohort/prospective | Prognostic | Recruiting | Tissue | Identification of
prognostic gene Mutations in biliary tract cancer Using WGS |
|
NCT04597710 |
Cohort/prospective |
Diagnostic/predictive | Recruiting | Tissue | Utility of WGS to
aid clinical decision making in patients referred for liver
resection |
|
NCT05242237 |
Cohort/prospective | Prognostic | Recruiting | Blood | The prognostic
value of CTC isolated by a novel microfluidic platform in liver
cancer patients |
| WES |
|
|
|
|
|
|
NCT04694391 |
Case-Control/prospective | Diagnostic | Recruiting | Tissue/blood | Genomic study of
relapse EC after radiotherapy |
|
NCT02127359 |
Cohort/prospective | Diagnostic | Completed | Tissue | The study is to
perform WES on cancer cells and normal tissues to develop better
ways to treat and prevent cancers |
|
NCT03486574 |
Family-based/prospective | Diagnostic | Enrolled by
invitation | Blood | Research for
associated genes for developing GC in family member with
first-degree relatives |
|
NCT03982173 (MATILDA) | Single Group | Therapeutic | Active, not
recruiting | Tissue | A phase II
WES-based basket trial for combination therapy with durvalumab and
tremelimumab in patients with metastatic solid tumors |
|
NCT03108885 |
Case-Only/prospective | Predictive | Enrolled by
invitation | Tissue/blood | Measuring cfDNA
during the course of treatment for EC as a marker of response and
recurrence |
|
NCT04955808 |
Case-Only/prospective | Diagnostic | Recruiting | Tissue/blood | The utility of
biospecimen collection in identifying genetic changes in patients
with solid tumors or multiple myeloma undergoing surgery |
|
NCT02851004 | Single Group | Therapeutic | Terminated | Tissue/blood | The efficacy and
safety of BBI608 in combination with pembrolizumab in mCRC |
|
NCT05048524 | Single Group | Diagnostic | Recruiting | Tissue/blood | The feasibility of
SLOG regimen in patients with localized PC. |
|
NCT03832621 (MAYA study) | Single Group, open
label | Diagnostic | Active, not
recruiting | Tissue/blood | The efficacy and
safety of nivolumab, ipilimumab and temozolomide combination in
patients with MSS, MGMT-silenced mCRC. |
|
NCT03023436 | Single Group, open
label |
Diagnostic/therapeutic | Recruiting | Tissue/blood | The survival
benefit and safety of cytoreductive surgery combined with
Hyperthermic Intraperitoneal Chemotherapy and chemotherapy in
gastric cancer with peritoneal metastasis. |
| WTS |
|
|
|
|
|
|
NCT03886571 |
Cohort/prospective | Diagnostic | Recruiting | Tissue/blood | An observational,
biospecimen collection protocol to develop a bank of pancreatic
cancer tissue and normal tissue. |
|
NCT03573791 |
Case-control/prospective | Diagnostic | Recruiting | Tissue | The purpose of this
trail is to identify the biomarkers to predict resistance to
neoadjuvant therapy. |
|
NCT03840460 |
Cohort/prospective | Diagnostic | Recruiting | Tissue/blood | A study in PDAC to
enable further disease characterization and the development of
predictive and prognostic biomarkers |
|
NCT04249739 | Non-Randomized | Therapeutic | Recruiting | Tissue | Pembrolizumab +
capecitabin/oxaliplatin or pembrolizumab + trastuzumab +
capecitabine/cisplatin in GC |
|
NCT02015169 | Single Group | Therapeutic | Completed | Tissue | Phase II study of
neoadjuvant XELOX + Lapatinib in HER2- positive GC patients with
liver metastasis |
|
NCT03841799 (COLON-IM) |
Cohort/prospective | Diagnostic | Recruiting | Tissue | Assessment of
colorectal tissue microenvironment (neutrophils infiltrate) of
patients with benign or malignant colorectal lesion |
|
NCT03260712 | Single Group | Predictive | Active, not
recruiting | Tissue | Evaluation of
pathological predictive factors for response and toxicity which are
responsible for chemotherapy and pembrolizumab. |
|
NCT04554771 (BASALT) | Randomized | Treatment | Recruiting | Blood | Blood-borne
assessment of stromal activation in EC to guide tocilizumab
therapy |
A recent genomic-based study of glioblastoma
patients examined the usefulness of WGS/RNA-seq vs. targeted panels
(106). WGS/RNA-seq detected more
conceivably criminal clinical findings than targeted panels in 90%
of cases, with an average of 16-fold more unique conceivably
criminal variants identified for each patient. In PC, WES of
germline DNA from whole blood of Japanese familial pancreatic
cancer patients revealed novel germline susceptibility genes,
FAT1 and FAT4, which encode the large transmembrane
proteins protocadherins (107).
Thus, WES for PC patients would offer significant information about
high-risk pathogenic germline variants in hereditary cancer
syndromes. A study focusing on rare genetic variants using WGS
through analysis of heterozygous premature truncating variants
showed that 20 significant genes, including PALD1, LRP1B,
COL4A2, CYLC2, ZFYVE9, BRD3, AHDC1 were identified, which would
play an important role in risk prediction of high-risk patients in
families identified at risk (108).
In CRC, a novel tumor suppressor, ARID2 was
detected based on WES analysis of younger patients (109). Substantial augmentation for
mutations in 4 out of 23 coding and 12 out of 15 noncoding driver
genes was shown in the mCRC cohort compared with primary CRC by
using WES. Mutations in PIK3CA were significantly reduced in
mCRC among detected putative drivers (110). Six of the newly found coding
driver genes, ZFP36L2, BCL, BCL9L, ELF3, LMTK3, and
TGIF1 are not detected in the CRC-specific MSK-IMPACT panel.
Similarly, WGS analysis of metastatic vs. matched primary
colorectal lesions, 65% of somatic mutations originate from a
common progenitor, with 15% being tumor- and 19%
metastasis-specific (111). Both
primary- and metastasis-specific mutations maintain high levels of
BRCAness. Recurrently mutated non-coding elements such as ncRNAs
RP11-594N15.3, AC010091, SNHG14, 3′ UTRs of FOXP2, DACH2, TRPM3,
XKR4, ANO5, CBL, CBLB and efferocytosis-/PD-L1were identified.
Numerous metastasis-specific mutations were detected, including
non-silent mutations of FAT1, FGF1, BRCA2, TP53, and
KDR, splice site mutations of JAK2 and 3′-UTR
mutations in KDR, PDGFRA, and AKT2 genes, suggesting
the existence of a high degree of mutational discordance between
metastatic and primary tumor (111). An original dataset containing
whole genomes analysis from 60 single-cell collecting samples
before therapy and after metastatic relapse resection demonstrated
that three non-synonymous and one stop codon mutations specific to
the recurrent lineage in four different genes, PKHD1, PCDHB15,
CSF1R, and CC2D1B, were detected in CRC patients. Moreover, a
distinctive mutagenic prototype distinguishing the cancer cells
from the recurrent lesion illustrated by a substantial contribution
of COSMIC signatures SBS35 and SBS17b was identified (112).
In esophageal cancer (EC), somatic mutations and
copy number alterations in multiple chromosome segments, encoding
MYC on 8q24.21, PIK3CA and SOX2 on 3q26,
CCND1, SHANK2, CTTN on 11q13.3, and KRAS on 12p12
were detected using WES. Amplifications of EGFRvIII and
EGFRvIVa mutants were identified, representing a novel
finding in African-American EC that may lead to clinical practice
(113). WGS can lead to the
detection of novel treatment targets and the discovery of new
genomic biomarkers, which may eventually develop the treatment
modalities for patients with GI tumors.
Discussion and future perspective
In this review, we focused on the latest advances in
genetic testing for the diagnosis and management of GI cancer. With
the introduction of Sanger sequencing and polymerase chain
reaction, laboratory genetic testing became an important instrument
for the genomic profiling of cancers in clinical settings. However,
although a variety of genes that are mutated in GI malignancies are
known, none of the mutations has had clear actionability, and DNA
analyses of GI cancers were not a part of clinical oncology until
relatively recently. The concept of massively parallel sequencing
led to the development of multi-gene panels, that cover the entire
spectrum of all acknowledged targeted genes and assist in selecting
a useful therapy (5). Studies of
germline variants that contribute to cancer predisposition now help
detect individuals who have a high-risk for some heritable cancers.
Multi-gene panel testing has the capacity to provide significant
advances in daily oncology practice. However, there are still
several obstacles to be addressed before multigene panel testing
can be effectively applied to patients with GI cancer.
One of the major issues regarding the use of
multi-gene panel for precision medicine is the lack of appropriate
treatment. The proportion of GI cancers that have clearly
actionable genetic alterations is comparatively low, and there is
no gene-tailored therapy for the majority of patients with GI
cancer. From a translational viewpoint, the persistent success of
comprehensive genetic testing will depend principally on the
testing's clinical utility and ability to identify the treatable
targets. To increase the number of patients whose tumors can be
successfully treated, it would be indispensable to use strategies
such as large-scale analyses in preclinical settings to increase
our understanding of the biological processes driving cancer and to
identify biomarkers for cancer diagnostics and new drugs.
Comprehensive genome profiling might represent one of these
strategies, and the continued progress in such profiling may lead
to genetic testing as a first option in the treatment of GI
cancers.
Liquid biopsies are an ideal sample source that
reflects individual characteristics and the heterogeneity of GI
cancers. The widespread clinical applications of ctDNA-based assays
for therapy decision-making and monitoring of tumors are based on
promising preliminary findings, but many challenges remain. The
ctDNA levels in plasma are prone to be inconsistent and low,
causing a variety of detection thresholds. Additionally, a negative
ctDNA finding may be attributed to low copy number identification
instead of the absence of ctDNA. Hence, the restricted sensitivity
of a ctDNA examination is an essential issue in patients who have
early-stage cancer as well as a lower level of plasma ctDNA. The
low level of ctDNA in the plasma may require the usage of
ultrahigh-depth sequencing and sophisticated statistical models for
the purpose of decreasing background error rates for very low
variant allele frequencies (114).
False-positive ctDNA results can also be caused by DNA fragments
from the clonal hematopoiesis of indeterminate potential or
non-neoplastic hematopoietic stem cells can be reduced by
conducting an advanced bioinformatics analysis or by comparing the
results of ctDNA sequencing with the findings obtained from
leukocytes and/or matched tumor tissues (115). A high-intensity cfDNA sequencing
analysis based on the combination of cfDNA and white blood cell DNA
analysis provides both the de novo detection of
tumor-derived changes and the clarification of MSI, the TMB,
mutational profiles, and the sources of somatic mutations
identified in cfDNA (115). A
quantitative ctDNA evaluation and methylation uncovering may
increase the specificity of ctDNA identification and consequently
allow to distinguish benign from cancerous GI disease, even at
early tumor stages. Further explorations by a large number of
clinical trials are necessary for the standardization of the
detection process as well as the clinical application of liquid
biopsies.
Among the multiple technical platforms that are now
available, the WGS strategy is now the most effective way to
construct a comprehensive image of the genomic variation in a
tumor. The widespread use of WGS technologies in clinical settings
seems no longer a distant dream, but the application of WGS
strategy possesses tremendous challenges in light of the sequencing
costs, computational processing, long-term storage, and meaningful
biological interpretation. Moreover, WGS needs particular ethical
and regulatory frameworks to handle accidental and secondary
genomic detections in the germline. However, in light of the
estimation that the costs of sequencing will result in the historic
descending tendency, a more gradual approval of WGS approaches for
a more improved stratification and subtyping of rare tumors may be
attainable in the short period. Algorithms that can dependably
support the latent significance of new genetic issues and then
associate these issues to theoretical or assumed clinical activity
with limited manual interference are required. The advancement of
those algorithms will be essential for decreasing analysis and
explanation costs and reducing the turnaround time for clinical
strategy The most modern WGS platforms such as Illumina NovaSeq
6000 system can handle a great volume of specimens in comparatively
short turnaround times, which makes WGS more practical (116).
Recent advances in cancer research have revealed
intratumor heterogeneity at the cell levels, epigenetic profiles,
and interferences with the tumor microenvironment. Hence, the
incorporation of multiple layers of information for individual
cancer cells is crucial for a comprehensive knowledge of the
mechanisms of cancer initiation (117). The addition of ‘omics’ to a
molecular word suggests a comprehensive, or worldwide evaluation of
a set of molecules. A multi-omics study is a data-driven biological
analysis in which the data sets are diverse individual omic
analyses, such as genomics, epigenomics, transcriptomics,
proteomics, metagenomics, and microbiomics that are used to
investigate physiological or pathological phenomena and
characterize biomolecular systems at different levels. The recent
advances in high-throughput technologies for genomics and
transcriptomics have resulted in a paradigm shift toward
multi-omics investigations, large-scale research collaborations,
and the design of computational algorithms (118). Multi-omics studies for GI cancer
currently being evaluated in clinical trials are summarized in
Table IV. Multi-omics
investigations have been applied in a variety of clinical studies
for a better detection of clinical subtypes or drug resistance, the
prediction of efficient combined therapies, and the exploration of
novel biomarkers. For instance, integrated proteogenomic data
together with genomic and transcriptomic data of CRCs, which were
illustrated by The Cancer Genome Atlas, demonstrated that a
chromosome 20q amplicon was correlated with the great inclusive
alterations at both messenger RNA and protein levels. In addition,
the incorporation of proteomics data provides the detection of
important 20q candidates, including HNF4A (hepatocyte
nuclear factor 4, alpha), TOMM34 (translocase of outer
mitochondrial membrane 34), and SRC (SRC proto-oncogene,
nonreceptor tyrosine kinase), suggesting that incorporated
proteogenomic analyses will enable novel developments in cancer
diagnosis and treatment (119). A
study that performed a multi-omics characterization of molecular
features of GC, using WGS, WES, and RNA-seq for 35 GC patients
before and after their neoadjuvant chemotherapy, showed that
C10orf71 was associated with treatment resistance, whereas
MYC and MDM2 amplification mutations were associated
with treatment sensitivity (120).
| Table IV.Selective ongoing clinical trials of
multi-omics study for gastrointestinal cancer. |
Table IV.
Selective ongoing clinical trials of
multi-omics study for gastrointestinal cancer.
| Multi-omics
study | Type of trial | Clinical
purpose | Phase | Results | Detection
method | Comments |
|---|
| NCT02342158
(PERMED-01 trial) | Single Group | Diagnostic | N/A | Active, not
recruiting | Tissue/blood | Identification
molecular alterations to guide individualized treatment in advanced
solid tumor |
| NCT03546127
(MULTIPLI-0) |
Cohort/prospective | Diagnostic | N/A | Completed | Tissue/blood | A feasibility study
in France to assess sample circuit and to perform analyzes within a
limited time in CRC. |
| NCT03951792 |
Case-control/prospective | Diagnostic | N/A | Enrolled by
invitation | Tissue/stool | Time longitudinal
study of the microbiome in CRC |
| NCT04318834
(COMPASS-B-MUHC) | Single Group, open
label | Diagnostic |
| Recruiting | Tissue | Identification of
actionable molecular alterations of biliary tract cancer through
WTS. |
| NCT04622423 |
Cohort/prospective | Diagnostic | N/A | Recruiting | Tissue | Advanced therapies
for liver metastasis in CRC and PC. |
| NCT04871321 |
Cohort/prospective | Diagnostic | N/A | Recruiting | Tissue/blood | Biomarker discovery
in patients within patients with advanced biliary tract cancer who
received nab-paclitaxel plus gemcitabine-cisplatin |
| NCT05234450 |
Case-only/prospective | Diagnostic | N/A | Recruiting | Tissue | Identification of
different tumor subgroups in pancreatic neuroendocrine tumors and
carcinomas regardless of their grade and stage. |
| NCT03429816
(OPPOSITE) | Single Group, open
label | Therapeutic | N/A | Active, not
recruiting | Tissue | Correlation of
molecular subtypes with histological response after neoadjuvant
therapy in patients with EC and GC. |
Conclusion
Multi-gene testing should be widely applied in
clinical settings, not only for greater insights into tumor biology
but also to drive cancer treatment. New clinical studies should
apply multigene testing toward the goal of finding novel targeted
therapies. The rapid analysis of genetic alterations with real-time
monitoring of therapy responses by ctDNA can optimize new
therapeutic strategies. The comprehensive characterization of GI
cancers by genetic testing will contribute to a better
molecular-level understanding of cancer, and it will contribute to
more appropriate and effective genomic-driven therapies for
patients who might not benefit from standardized therapy or
experimental interferences in the context of clinical studies. Due
to several challenges to be resolved such as the costs, restricted
sensitivity, and time consumption to carry out, genetic testing
should be used when standard therapeutic approaches have been
completed at present.
Acknowledgements
Not applicable.
Funding
This study was funded by KAKENHI (Grant-in-Aid for Scientific
Research) (grant no. 18H02883).
Availability of data and materials
Not applicable.
Authors' contributions
TM and MY performed the literature research. TM
wrote the manuscript and performed the revision and approval of the
final version. MY designed research, coordinated and corrected the
writing of the paper. Both authors read and approved the final
manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Authors' information
Dr Tasuku Matsuoka (ORCID: 0000-0001-5019-8519); Dr
Masakazu Yashiro (ORCID: 0000-0001-5743-7228).
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
GI
|
gastrointestinal
|
|
NGS
|
next-generation sequencing
|
|
FDA
|
U.S. Food and Drug Administration
|
|
MSI
|
microsatellite instability
|
|
TMB
|
tumor mutational burden
|
|
OS
|
overall survival
|
|
PARP
|
poly (ADP-ribose) polymerase
|
|
ICB
|
immune checkpoint blockade
|
|
PC
|
pancreatic cancer
|
|
DDR
|
DNA damage response
|
|
EGFR
|
epidermal growth factor receptor
|
|
MAPK
|
mitogen-activated protein kinases
|
|
CRC
|
colorectal cancer
|
|
mCRC
|
metastatic CRC
|
|
HR
|
hazard ratio
|
|
T-DXd
|
Trastuzumab-Deruxtecan
|
|
HER2
|
human epidermal growth factor
receptor 2
|
|
WGS
|
whole genome sequencing
|
|
PDAC
|
pancreatic ductal adenocarcinoma
|
|
HRD
|
homologous recombination
deficiency
|
|
ASCO
|
American Society of Clinical
Oncology
|
|
PFS
|
progression-free survival
|
|
GC
|
gastric cancer
|
|
FGFR
|
fibroblast growth factor receptor
|
|
HCC
|
hepatocellular carcinoma
|
|
TERT
|
telomerase reverse transcriptase
|
|
ctDNR
|
circulating tumor DNA
|
|
cfDNA
|
cell-free DNA
|
|
MRD
|
minimal residual disease
|
|
LS
|
Lynch syndrome
|
|
FDR
|
first-degree-relative
|
|
HDGC
|
hereditary diffuse gastric cancer
|
|
DGC
|
diffuse type gastric cancer
|
|
WES
|
whole exome sequencing
|
|
EC
|
esophageal cancer
|
References
|
1
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ceasovschih A, Voloc G, Sorodoc V, Vâță D,
Lupașcu CD, Preda C, Lionte C, Stoica A, Sîrbu O, Grigorescu ED, et
al: From chronic pruritus to neuroendocrine tumor: A case report.
Exp Ther Med. 23:1892022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Watanabe M, Baba H, Ishioka C, Nishimura Y
and Muto M: Recent advances in diagnosis and treatment for
malignancies of the gastrointestinal tract. Digestion. 85:95–98.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Matsuoka T and Yashiro M: Precision
medicine for gastrointestinal cancer: Recent progress and future
perspective. World J Gastrointest Oncol. 12:1–20. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nagahashi M, Shimada Y, Ichikawa H,
Kameyama H, Takabe K, Okuda S and Wakai T: Next generation
sequencing-based gene panel tests for the management of solid
tumors. Cancer Sci. 110:6–15. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bahassi el M and Stambrook PJ:
Next-generation sequencing technologies: Breaking the sound barrier
of human genetics. Mutagenesis. 29:303–310. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sicklick JK, Kato S, Okamura R,
Schwaederle M, Hahn ME, Williams CB, De P, Krie A, Piccioni DE,
Miller VA, et al: Molecular profiling of cancer patients enables
personalized combination therapy: The I-PREDICT study. Nat Med.
25:744–750. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Massard C, Michiels S, Ferté C, Le Deley
MC, Lacroix L, Hollebecque A, Verlingue L, Ileana E, Rosellini S,
Ammari S, et al: High-throughput genomics and clinical outcome in
hard-to-treat advanced cancers: Results of the MOSCATO 01 trial.
Cancer Discov. 7:586–595. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cobain EF, Wu YM, Vats P, Chugh R, Worden
F, Smith DC, Schuetze SM, Zalupski MM, Sahai V, Alva A, et al:
Assessment of clinical benefit of integrative genomic profiling in
advanced solid tumors. JAMA Oncol. 7:525–533. 2021.PubMed/NCBI
|
|
10
|
Pishvaian MJ, Blais EM, Brody JR, Lyons E,
DeArbeloa P, Hendifar A, Mikhail S, Chung V, Sahai V, Sohal DPS, et
al: Overall survival in patients with pancreatic cancer receiving
matched therapies following molecular profiling: A retrospective
analysis of the know your tumor registry trial. Lancet Oncol.
21:508–518. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Van Cutsem E, Cervantes A, Adam R, Sobrero
A, Van Krieken JH, Aderka D, Aranda Aguilar E, Bardelli A, Benson
A, Bodoky G, et al: ESMO consensus guidelines for the management of
patients with metastatic colorectal cancer. Ann Oncol.
27:1386–1422. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Venderbosch S, Nagtegaal ID, Maughan TS,
Smith CG, Cheadle JP, Fisher D, Kaplan R, Quirke P, Seymour MT,
Richman SD, et al: Mismatch repair status and BRAF mutation status
in metastatic colorectal cancer patients: A pooled analysis of the
CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res.
20:5322–5330. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jones JC, Renfro LA, Al-Shamsi HO, Schrock
AB, Rankin A, Zhang BY, Kasi PM, Voss JS, Leal AD, Sun J, et al:
Non-V600 BRAF mutations define a clinically distinct
molecular subtype of metastatic colorectal cancer. J Clin Oncol.
35:2624–2630. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yaeger R, Kotani D, Mondaca S, Parikh AR,
Bando H, Van Seventer EE, Taniguchi H, Zhao H, Thant CN, de
Stanchina E, et al: Response to anti-EGFR therapy in patients with
BRAF non-V600-mutant metastatic colorectal cancer. Clin Cancer Res.
25:7089–7097. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Johnson B, Loree JM, Jacome AA, Mendis S,
Syed M, Morris Ii VK, Parseghian CM, Dasari A, Pant S, Raymond VM,
et al: Atypical, non-V600 BRAF mutations as a potential mechanism
of resistance to EGFR inhibition in metastatic colorectal cancer.
JCO Precis Oncol. 3:PO.19.00102. 2019.
|
|
16
|
Corcoran RB, Ebi H, Turke AB, Coffee EM,
Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D,
Hung KE, et al: EGFR-mediated re-activation of MAPK signaling
contributes to insensitivity of BRAF mutant colorectal cancers to
RAF inhibition with vemurafenib. Cancer Discov. 2:227–235. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yaeger R, Cercek A, O'Reilly EM, Reidy DL,
Kemeny N, Wolinsky T, Capanu M, Gollub MJ, Rosen N, Berger MF, et
al: Pilot trial of combined BRAF and EGFR inhibition in BRAF-mutant
metastatic colorectal cancer patients. Clin Cancer Res.
21:1313–1320. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van Geel RMJM, Tabernero J, Elez E,
Bendell JC, Spreafico A, Schuler M, Yoshino T, Delord JP, Yamada Y,
Lolkema MP, et al: A phase Ib dose-escalation study of encorafenib
and cetuximab with or without alpelisib in metastatic BRAF-mutant
colorectal cancer. Cancer Discov. 7:610–619. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kopetz S, Guthrie KA, Morris VK, Lenz HJ,
Magliocco AM, Maru D, Yan Y, Lanman R, Manyam G, Hong DS, et al:
Randomized trial of irinotecan and cetuximab with or without
vemurafenib in BRAF-mutant metastatic colorectal cancer (SWOG
S1406). J Clin Oncol. 39:285–294. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Corcoran RB, André T, Atreya CE, Schellens
JHM, Yoshino T, Bendell JC, Hollebecque A, McRee AJ, Siena S,
Middleton G, et al: Combined BRAF, EGFR, and MEK inhibition in
patients with BRAFV600E-mutant colorectal cancer. Cancer
Discov. 8:428–443. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Roviello G, D'Angelo A, Petrioli R,
Roviello F, Cianchi F, Nobili S, Mini E and Lavacchi D:
Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated
colorectal cancer. Transl Oncol. 13:1007952020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Grothey A, Fakih M and Tabernero J:
Management of BRAF-mutant metastatic colorectal cancer: A review of
treatment options and evidence-based guidelines. Ann Oncol.
32:959–967. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nagasaka M, Li Y, Sukari A, Ou SHI,
Al-Hallak MN and Azmi AS: KRAS G12C Game of Thrones, which direct
KRAS inhibitor will claim the iron throne? Cancer Treat Rev.
84:1019742020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang C and Fakih M: Targeting KRAS in
colorectal cancer. Curr Oncol Rep. 23:282021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stadler ZK, Battaglin F, Middha S,
Hechtman JF, Tran C, Cercek A, Yaeger R, Segal NH, Varghese AM,
Reidy-Lagunes DL, et al: Reliable detection of mismatch repair
deficiency in colorectal cancers using mutational load in
next-generation sequencing panels. J Clin Oncol. 34:2141–2147.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Maby P, Tougeron D, Hamieh M, Mlecnik B,
Kora H, Bindea G, Angell HK, Fredriksen T, Elie N, Fauquembergue E,
et al: Correlation between density of CD8+ T-cell infiltrate in
microsatellite unstable colorectal cancers and frameshift
mutations: A rationale for personalized immunotherapy. Cancer Res.
75:3446–3455. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Andre T, Amonkar M, Norquist JM, Shiu KK,
Kim TW, Jensen BV, Jensen LH, Punt CJA, Smith D, Garcia-Carbonero
R, et al: Health-related quality of life in patients with
microsatellite instability-high or mismatch repair deficient
metastatic colorectal cancer treated with first-line pembrolizumab
versus chemotherapy (KEYNOTE-177): An open-label, randomised, phase
3 trial. Lancet Oncol. 22:665–677. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Richman SD, Southward K, Chambers P, Cross
D, Barrett J, Hemmings G, Taylor M, Wood H, Hutchins G, Foster JM,
et al: HER2 overexpression and amplification as a potential
therapeutic target in colorectal cancer: Analysis of 3256 patients
enrolled in the QUASAR, FOCUS and PICCOLO colorectal cancer trials.
J Pathol. 238:562–570. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang G, He Y, Sun Y, Wang W, Qian X, Yu X
and Pan Y: Prevalence, prognosis and predictive status of HER2
amplification in anti-EGFR-resistant metastatic colorectal cancer.
Clin Transl Oncol. 22:813–822. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sartore-Bianchi A, Trusolino L, Martino C,
Bencardino K, Lonardi S, Bergamo F, Zagonel V, Leone F, Depetris I,
Martinelli E, et al: Dual-targeted therapy with trastuzumab and
lapatinib in treatment-refractory, KRAS codon 12/13 wild-type,
HER2-positive metastatic colorectal cancer (HERACLES): A
proof-of-concept, multicentre, open-label, phase 2 trial. Lancet
Oncol. 17:738–746. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Meric-Bernstam F, Hurwitz H, Raghav KPS,
McWilliams RR, Fakih M, VanderWalde A, Swanton C, Kurzrock R,
Burris H, Sweeney C, et al: Pertuzumab plus trastuzumab for
HER2-amplified metastatic colorectal cancer (MyPathway): An updated
report from a multicentre, open-label, phase 2a, multiple basket
study. Lancet Oncol. 20:518–530. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Siena S, Di Bartolomeo M, Raghav K,
Masuishi T, Loupakis F, Kawakami H, Yamaguchi K, Nishina T, Fakih
M, Elez E, et al: Trastuzumab deruxtecan (DS-8201) in patients with
HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): A
multicentre, open-label, phase 2 trial. Lancet Oncol. 22:779–789.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hallberg B and Palmer RH: The role of the
ALK receptor in cancer biology. Ann Oncol. 27 (Suppl 3):iii4–iii15.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pietrantonio F, Di Nicolantonio F, Schrock
AB, Lee J, Tejpar S, Sartore-Bianchi A, Hechtman JF, Christiansen
J, Novara L, Tebbutt N, et al: ALK, ROS1, and NTRK rearrangements
in metastatic colorectal cancer. J Natl Cancer Inst. 109:2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ruela AL, de Figueiredo EC, de Araújo MB,
Carvalho FC and Pereira GR: Molecularly imprinted microparticles in
lipid-based formulations for sustained release of donepezil. Eur J
Pharm Sci. 93:114–122. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hosoda W, Chianchiano P, Griffin JF,
Pittman ME, Brosens LA, Noë M, Yu J, Shindo K, Suenaga M, Rezaee N,
et al: Genetic analyses of isolated high-grade pancreatic
intraepithelial neoplasia (HG-PanIN) reveal paucity of alterations
in TP53 and SMAD4. J Pathol. 242:16–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Waddell N, Pajic M, Patch AM, Chang DK,
Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al:
Whole genomes redefine the mutational landscape of pancreatic
cancer. Nature. 518:495–501. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Witkiewicz AK, McMillan EA, Balaji U, Baek
G, Lin WC, Mansour J, Mollaee M, Wagner KU, Koduru P, Yopp A, et
al: Whole-exome sequencing of pancreatic cancer defines genetic
diversity and therapeutic targets. Nat Commun. 6:67442015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sohal DPS, Kennedy EB, Cinar P, Conroy T,
Copur MS, Crane CH, Garrido-Laguna I, Lau MW, Johnson T,
Krishnamurthi S, et al: Metastatic pancreatic cancer: ASCO
guideline update. J Clin Oncol. 38:3217–3230. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Robson M, Im SA, Senkus E, Xu B, Domchek
SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A, et al:
Olaparib for metastatic breast cancer in patients with a germline
BRCA mutation. N Engl J Med. 377:523–533. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Del Campo JM, Matulonis UA, Malander S,
Provencher D, Mahner S, Follana P, Waters J, Berek JS, Woie K, Oza
AM, et al: Niraparib maintenance therapy in patients with recurrent
ovarian cancer after a partial response to the last platinum-based
chemotherapy in the ENGOT-OV16/NOVA trial. J Clin Oncol.
37:2968–2973. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Curtin NJ and Szabo C: Poly(ADP-ribose)
polymerase inhibition: Past, present and future. Nat Rev Drug
Discov. 19:711–736. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Park W, Chen J, Chou JF, Varghese AM, Yu
KH, Wong W, Capanu M, Balachandran V, McIntyre CA, El Dika I, et
al: Genomic methods identify homologous recombination deficiency in
pancreas adenocarcinoma and optimize treatment selection. Clin
Cancer Res. 26:3239–3247. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Golan T, Hammel P, Reni M, Van Cutsem E,
Macarulla T, Hall MJ, Park JO, Hochhauser D, Arnold D, Oh DY, et
al: Maintenance olaparib for germline BRCA-mutated metastatic
pancreatic cancer. N Engl J Med. 381:317–327. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hu ZI, Shia J, Stadler ZK, Varghese AM,
Capanu M, Salo-Mullen E, Lowery MA, Diaz LA Jr, Mandelker D, Yu KH,
et al: Evaluating mismatch repair deficiency in pancreatic
adenocarcinoma: Challenges and recommendations. Clin Cancer Res.
24:1326–1336. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Latham A, Srinivasan P, Kemel Y, Shia J,
Bandlamudi C, Mandelker D, Middha S, Hechtman J, Zehir A,
Dubard-Gault M, et al: Microsatellite instability is associated
with the presence of lynch syndrome pan-cancer. J Clin Oncol.
37:286–295. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bang YJ, Van Cutsem E, Feyereislova A,
Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T,
et al: Trastuzumab in combination with chemotherapy versus
chemotherapy alone for treatment of HER2-positive advanced gastric
or gastro-oesophageal junction cancer (ToGA): A phase 3,
open-label, randomised controlled trial. Lancet. 376:687–697. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Takegawa N, Tsurutani J, Kawakami H,
Yonesaka K, Kato R, Haratani K, Hayashi H, Takeda M, Nonagase Y,
Maenishi O and Nakagawa K: [fam-] trastuzumab deruxtecan, antitumor
activity is dependent on HER2 expression level rather than on HER2
amplification. Int J Cancer. 145:3414–3424. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Shitara K, Bang YJ, Iwasa S, Sugimoto N,
Ryu MH, Sakai D, Chung HC, Kawakami H, Yabusaki H, Lee J, et al:
Trastuzumab deruxtecan in previously treated HER2-positive gastric
cancer. N Engl J Med. 382:2419–2430. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Helsten T, Elkin S, Arthur E, Tomson BN,
Carter J and Kurzrock R: The FGFR landscape in cancer: Analysis of
4,853 tumors by next-generation sequencing. Clin Cancer Res.
22:259–267. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hur JY, Chao J, Kim K, Kim ST, Kim KM,
Klempner SJ and Lee J: High-level FGFR2 amplification is associated
with poor prognosis and lower response to chemotherapy in gastric
cancers. Pathol Res Pract. 216:1528782020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Catenacci DVT, Rasco D, Lee J, Rha SY, Lee
KW, Bang YJ, Bendell J, Enzinger P, Marina N, Xiang H, et al: Phase
I escalation and expansion study of bemarituzumab (FPA144) in
patients with advanced solid tumors and FGFR2b-selected
gastroesophageal adenocarcinoma. J Clin Oncol. 38:2418–2426. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Catenacci DV, Tesfaye A, Tejani M, Cheung
E, Eisenberg P, Scott AJ, Eng C, Hnatyszyn J, Marina N, Powers J
and Wainberg Z: Bemarituzumab with modified FOLFOX6 for advanced
FGFR2-positive gastroesophageal cancer: FIGHT Phase III study
design. Future Oncol. 15:2073–2082. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nault JC, Mallet M, Pilati C, Calderaro J,
Bioulac-Sage P, Laurent C, Laurent A, Cherqui D, Balabaud C and
Zucman-Rossi J: High frequency of telomerase reverse-transcriptase
promoter somatic mutations in hepatocellular carcinoma and
preneoplastic lesions. Nat Commun. 4:22182013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Schulze K, Imbeaud S, Letouzé E,
Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C,
Shinde J, Soysouvanh F, et al: Exome sequencing of hepatocellular
carcinomas identifies new mutational signatures and potential
therapeutic targets. Nat Genet. 47:505–511. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Jhunjhunwala S, Jiang Z, Stawiski EW, Gnad
F, Liu J, Mayba O, Du P, Diao J, Johnson S, Wong KF, et al: Diverse
modes of genomic alteration in hepatocellular carcinoma. Genome
Biol. 15:4362014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cancer Genome Atlas Research Network.
Electronic address, . simplewheeler@bcm.edu; Cancer
Genome Atlas Research Network: Comprehensive and integrative
genomic characterization of hepatocellular carcinoma. Cell.
169:1327–1341.e23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Xu Z, Hu J, Cao H, Pilo MG, Cigliano A,
Shao Z, Xu M, Ribback S, Dombrowski F, Calvisi DF and Chen X: Loss
of Pten synergizes with c-Met to promote hepatocellular carcinoma
development via mTORC2 pathway. Exp Mol Med. 50:e4172018.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Jin M, Piao Z, Kim NG, Park C, Shin EC,
Park JH, Jung HJ, Kim CG and Kim H: p16 is a major inactivation
target in hepatocellular carcinoma. Cancer. 89:60–68. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kudo M, Finn RS, Qin S, Han KH, Ikeda K,
Piscaglia F, Baron A, Park JW, Han G, Jassem J, et al: Lenvatinib
versus sorafenib in first-line treatment of patients with
unresectable hepatocellular carcinoma: A randomised phase 3
non-inferiority trial. Lancet. 391:1163–1173. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Finn RS, Qin S, Ikeda M, Galle PR, Ducreux
M, Kim TY, Kudo M, Breder V, Merle P, Kaseb AO, et al: Atezolizumab
plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J
Med. 382:1894–1905. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Bruix J, Qin S, Merle P, Granito A, Huang
YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, et al:
Regorafenib for patients with hepatocellular carcinoma who
progressed on sorafenib treatment (RESORCE): A randomised,
double-blind, placebo-controlled, phase 3 trial. Lancet. 389:56–66.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Abou-Alfa GK, Meyer T, Cheng AL,
El-Khoueiry AB, Rimassa L, Ryoo BY, Cicin I, Merle P, Chen Y, Park
JW, et al: Cabozantinib in patients with advanced and progressing
hepatocellular carcinoma. N Engl J Med. 379:54–63. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Valle J, Wasan H, Palmer DH, Cunningham D,
Anthoney A, Maraveyas A, Madhusudan S, Iveson T, Hughes S, Pereira
SP, et al: Cisplatin plus gemcitabine versus gemcitabine for
biliary tract cancer. N Engl J Med. 362:1273–1281. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Lamarca A, Palmer DH, Wasan HS, Ross PJ,
Ma YT, Arora A, Falk S, Gillmore R, Wadsley J, Patel K, et al:
Second-line FOLFOX chemotherapy versus active symptom control for
advanced biliary tract cancer (ABC-06): A phase 3, open-label,
randomised, controlled trial. Lancet Oncol. 22:690–701. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Javle M, Bekaii-Saab T, Jain A, Wang Y,
Kelley RK, Wang K, Kang HC, Catenacci D, Ali S, Krishnan S, et al:
Biliary cancer: Utility of next-generation sequencing for clinical
management. Cancer. 122:3838–3847. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Nakamura Y and Yoshino T: Clinical utility
of analyzing circulating tumor DNA in patients with metastatic
colorectal cancer. Oncologist. 23:1310–1318. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Nakamura Y and Shitara K: Development of
circulating tumour DNA analysis for gastrointestinal cancers. ESMO
Open. 5 (Suppl 1):e0006002020. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Parikh AR, Van Seventer EE, Siravegna G,
Hartwig AV, Jaimovich A, He Y, Kanter K, Fish MG, Fosbenner KD,
Miao B, et al: Minimal residual disease detection using a
plasma-only circulating tumor DNA assay in patients with colorectal
cancer. Clin Cancer Res. 27:5586–5594. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kwapisz D: The first liquid biopsy test
approved. Is it a new era of mutation testing for non-small cell
lung cancer? Ann Transl Med. 5:462017.PubMed/NCBI
|
|
71
|
Merker JD, Oxnard GR, Compton C, Diehn M,
Hurley P, Lazar AJ, Lindeman N, Lockwood CM, Rai AJ, Schilsky RL,
et al: Circulating tumor DNA analysis in patients with cancer:
American society of clinical oncology and college of american
pathologists joint review. J Clin Oncol. 36:1631–1641. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Luo H, Zhao Q, Wei W, Zheng L, Yi S, Li G,
Wang W, Sheng H, Pu H, Mo H, et al: Circulating tumor DNA
methylation profiles enable early diagnosis, prognosis prediction,
and screening for colorectal cancer. Sci Transl Med.
12:eaax75332020. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Balgkouranidou I, Matthaios D,
Karayiannakis A, Bolanaki H, Michailidis P, Xenidis N, Amarantidis
K, Chelis L, Trypsianis G, Chatzaki E, et al: Prognostic role of
APC and RASSF1A promoter methylation status in cell free
circulating DNA of operable gastric cancer patients. Mutat Res.
778:46–51. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Ng CKY, Di Costanzo GG, Tosti N, Paradiso
V, Coto-Llerena M, Roscigno G, Perrina V, Quintavalle C, Boldanova
T, Wieland S, et al: Genetic profiling using plasma-derived
cell-free DNA in therapy-naïve hepatocellular carcinoma patients: A
pilot study. Ann Oncol. 29:1286–1291. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Kisiel JB, Dukek BA, V S R Kanipakam R,
Ghoz HM, Yab TC, Berger CK, Taylor WR, Foote PH, Giama NH,
Onyirioha K, et al: Hepatocellular carcinoma detection by plasma
methylated DNA: Discovery, phase I pilot, and phase II clinical
validation. Hepatology. 69:1180–1192. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Maleddu A, Pantaleo MA, Nannini M and
Biasco G: The role of mutational analysis of KIT and PDGFRA in
gastrointestinal stromal tumors in a clinical setting. J Transl
Med. 9:752011. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Larribère L and Martens UM: Advantages and
challenges of using ctDNA NGS to assess the presence of minimal
residual disease (MRD) in solid tumors. Cancers (Basel).
13:56982021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Tabernero J, Lenz HJ, Siena S, Sobrero A,
Falcone A, Ychou M, Humblet Y, Bouché O, Mineur L, Barone C, et al:
Analysis of circulating DNA and protein biomarkers to predict the
clinical activity of regorafenib and assess prognosis in patients
with metastatic colorectal cancer: A retrospective, exploratory
analysis of the CORRECT trial. Lancet Oncol. 16:937–948. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Taniguchi H, Nakamura Y, Kotani D, Yukami
H, Mishima S, Sawada K, Shirasu H, Ebi H, Yamanaka T, Aleshin A, et
al: CIRCULATE-Japan: Circulating tumor DNA-guided adaptive platform
trials to refine adjuvant therapy for colorectal cancer. Cancer
Sci. 112:2915–2920. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Misale S, Yaeger R, Hobor S, Scala E,
Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M,
Siravegna G, et al: Emergence of KRAS mutations and acquired
resistance to anti-EGFR therapy in colorectal cancer. Nature.
486:532–536. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Siravegna G, Mussolin B, Buscarino M,
Corti G, Cassingena A, Crisafulli G, Ponzetti A, Cremolini C, Amatu
A, Lauricella C, et al: Clonal evolution and resistance to EGFR
blockade in the blood of colorectal cancer patients. Nat Med.
21:795–801. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wang DS, Liu ZX, Lu YX, Bao H, Wu X, Zeng
ZL, Liu Z, Zhao Q, He CY, Lu JH, et al: Liquid biopsies to track
trastuzumab resistance in metastatic HER2-positive gastric cancer.
Gut. 68:1152–1161. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Martinelli E, Ciardiello D, Martini G,
Troiani T, Cardone C, Vitiello PP, Normanno N, Rachiglio AM,
Maiello E, Latiano T, et al: Implementing anti-epidermal growth
factor receptor (EGFR) therapy in metastatic colorectal cancer:
Challenges and future perspectives. Ann Oncol. 31:30–40. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Cremolini C, Rossini D, Dell'Aquila E,
Lonardi S, Conca E, Del Re M, Busico A, Pietrantonio F, Danesi R,
Aprile G, et al: Rechallenge for patients with RAS and BRAF
wild-type metastatic colorectal cancer with acquired resistance to
first-line cetuximab and irinotecan: A phase 2 single-arm clinical
trial. JAMA Oncol. 5:343–350. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Nakamura Y, Taniguchi H, Ikeda M, Bando H,
Kato K, Morizane C, Esaki T, Komatsu Y, Kawamoto Y, Takahashi N, et
al: Clinical utility of circulating tumor DNA sequencing in
advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA
studies. Nat Med. 26:1859–1864. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Catana A, Apostu AP and Antemie RG: Multi
gene panel testing for hereditary breast cancer-is it ready to be
used? Med Pharm Rep. 92:220–225. 2019.PubMed/NCBI
|
|
87
|
Stjepanovic N, Moreira L, Carneiro F,
Balaguer F, Cervantes A, Balmaña J and Martinelli E; ESMO
Guidelines Committee. Electronic address, : simpleclinicalguidelines@esmo.org:
Hereditary gastrointestinal cancers: ESMO clinical practice
guidelines for diagnosis, treatment and follow-up†. Ann Oncol.
30:1558–1571. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Daly MB, Pal T, Berry MP, Buys SS, Dickson
P, Domchek SM, Elkhanany A, Friedman S, Goggins M, Hutton ML, et
al: Genetic/familial high-risk assessment: Breast, ovarian, and
pancreatic, version 2.2021, NCCN clinical practice guidelines in
oncology. J Natl Compr Canc Netw. 19:77–102. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Taylor A, Brady AF, Frayling IM, Hanson H,
Tischkowitz M, Turnbull C and Side L; UK Cancer Genetics Group
(UK-CGG), : Consensus for genes to be included on cancer panel
tests offered by UK genetics services: Guidelines of the UK Cancer
Genetics Group. J Med Genet. 55:372–377. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Cerretelli G, Ager A, Arends MJ and
Frayling IM: Molecular pathology of Lynch syndrome. J Pathol.
250:518–531. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Win AK, Lindor NM, Young JP, Macrae FA,
Young GP, Williamson E, Parry S, Goldblatt J, Lipton L, Winship I,
et al: Risks of primary extracolonic cancers following colorectal
cancer in lynch syndrome. J Natl Cancer Inst. 104:1363–1372. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Syngal S, Brand RE, Church JM, Giardiello
FM, Hampel HL and Burt RW; American College of Gastroenterology, :
ACG clinical guideline: Genetic testing and management of
hereditary gastrointestinal cancer syndromes. Am J Gastroenterol.
110:223–263. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Le DT, Kim TW, Van Cutsem E, Geva R, Jäger
D, Hara H, Burge M, O'Neil B, Kavan P, Yoshino T, et al: Phase II
open-label study of pembrolizumab in treatment-refractory,
microsatellite instability-high/mismatch repair-deficient
metastatic colorectal cancer: KEYNOTE-164. J Clin Oncol. 38:11–19.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Overbeek KA, Cahen DL, Canto MI and Bruno
MJ: Surveillance for neoplasia in the pancreas. Best Pract Res Clin
Gastroenterol. 30:971–986. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Shindo K, Yu J, Suenaga M, Fesharakizadeh
S, Cho C, Macgregor-Das A, Siddiqui A, Witmer PD, Tamura K, Song
TJ, et al: Deleterious germline mutations in patients with
apparently sporadic pancreatic adenocarcinoma. J Clin Oncol.
35:3382–3390. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Chaffee KG, Oberg AL, McWilliams RR,
Majithia N, Allen BA, Kidd J, Singh N, Hartman AR, Wenstrup RJ and
Petersen GM: Prevalence of germ-line mutations in cancer genes
among pancreatic cancer patients with a positive family history.
Genet Med. 20:119–127. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Hu C, Hart SN, Polley EC, Gnanaolivu R,
Shimelis H, Lee KY, Lilyquist J, Na J, Moore R, Antwi SO, et al:
Association between inherited germline mutations in cancer
predisposition genes and risk of pancreatic cancer. JAMA.
319:2401–2409. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Bartsch DK, Gress TM and Langer P:
Familial pancreatic cancer-current knowledge. Nat Rev Gastroenterol
Hepatol. 9:445–453. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Hampel H, Bennett RL, Buchanan A, Pearlman
R and Wiesner GL: Guideline Development Group, American College of
Medical Genetics and Genomics Professional Practice and Guidelines
Committee and National Society of Genetic Counselors Practice
Guidelines Committee: A practice guideline from the American
college of medical genetics and genomics and the national society
of genetic counselors: Referral indications for cancer
predisposition assessment. Genet Med. 17:70–87. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Pilonis ND, Tischkowitz M, Fitzgerald RC
and di Pietro M: Hereditary diffuse gastric cancer: Approaches to
screening, surveillance, and treatment. Annu Rev Med. 72:263–280.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Donner I, Kiviluoto T, Ristimäki A,
Aaltonen LA and Vahteristo P: Exome sequencing reveals three novel
candidate predisposition genes for diffuse gastric cancer. Fam
Cancer. 14:241–246. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Berger MF and Mardis ER: The emerging
clinical relevance of genomics in cancer medicine. Nat Rev Clin
Oncol. 15:353–365. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Nakagawa H, Wardell CP, Furuta M,
Taniguchi H and Fujimoto A: Cancer whole-genome sequencing: Present
and future. Oncogene. 34:5943–5950. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Colomer R, Mondejar R, Romero-Laorden N,
Alfranca A, Sanchez-Madrid F and Quintela-Fandino M: When should we
order a next generation sequencing test in a patient with cancer?
EClinicalMedicine. 25:1004872020. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
ICGC/TCGA Pan-Cancer Analysis of Whole
Genomes Consortium, . Pan-cancer analysis of whole genomes. Nature.
578:82–93. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Frank MO, Koyama T, Rhrissorrakrai K,
Robine N, Utro F, Emde AK, Chen BJ, Arora K, Shah M, Geiger H, et
al: Sequencing and curation strategies for identifying candidate
glioblastoma treatments. BMC Med Genomics. 12:562019. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Takai E, Nakamura H, Chiku S, Kubo E,
Ohmoto A, Totoki Y, Shibata T, Higuchi R, Yamamoto M, Furuse J, et
al: Whole-exome Sequencing reveals new potential susceptibility
genes for Japanese familial pancreatic cancer. Ann Surg.
275:e652–e658. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Tan M, Brusgaard K, Gerdes AM, Mortensen
MB, Detlefsen S, Schaffalitzky de Muckadell OB and Joergensen MT:
Whole genome sequencing identifies rare germline variants enriched
in cancer related genes in first degree relatives of familial
pancreatic cancer patients. Clin Genet. 100:551–562. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Bala P, Singh AK, Kavadipula P, Kotapalli
V, Sabarinathan R and Bashyam MD: Exome sequencing identifies ARID2
as a novel tumor suppressor in early-onset sporadic rectal cancer.
Oncogene. 40:863–874. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Mendelaar PAJ, Smid M, van Riet J, Angus
L, Labots M, Steeghs N, Hendriks MP, Cirkel GA, van Rooijen JM, Ten
Tije AJ, et al: Whole genome sequencing of metastatic colorectal
cancer reveals prior treatment effects and specific metastasis
features. Nat Commun. 12:5742021. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Ishaque N, Abba ML, Hauser C, Patil N,
Paramasivam N, Huebschmann D, Leupold JH, Balasubramanian GP,
Kleinheinz K, Toprak UH, et al: Whole genome sequencing puts
forward hypotheses on metastasis evolution and therapy in
colorectal cancer. Nat Commun. 9:47822018. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Alves JM, Prado-López S, Tomás L, Valecha
M, Estévez-Gómez N, Alvariño P, Geisel D, Modest DP, Sauer IM,
Pratschke J, et al: Clonality and timing of relapsing colorectal
cancer metastasis revealed through whole-genome single-cell
sequencing. Cancer Lett. 543:2157672022. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Erkizan HV, Sukhadia S, Natarajan TG,
Marino G, Notario V, Lichy JH and Wadleigh RG: Exome sequencing
identifies novel somatic variants in African American esophageal
squamous cell carcinoma. Sci Rep. 11:148142021. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Rose Brannon A, Jayakumaran G, Diosdado M,
Patel J, Razumova A, Hu Y, Meng F, Haque M, Sadowska J, Murphy BJ,
et al: Enhanced specificity of clinical high-sensitivity tumor
mutation profiling in cell-free DNA via paired normal sequencing
using MSK-ACCESS. Nat Commun. 12:37702021. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Razavi P, Li BT, Brown DN, Jung B, Hubbell
E, Shen R, Abida W, Juluru K, De Bruijn I, Hou C, et al:
High-intensity sequencing reveals the sources of plasma circulating
cell-free DNA variants. Nat Med. 25:1928–1937. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Modi A, Vai S, Caramelli D and Lari M: The
illumina sequencing protocol and the novaseq 6000 system. Methods
Mol Biol. 2242:15–42. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Subramanian I, Verma S, Kumar S, Jere A
and Anamika K: Multi-omics data integration, interpretation, and
its application. Bioinform Biol Insights. 14:11779322198990512020.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Basu A, Bodycombe NE, Cheah JH, Price EV,
Liu K, Schaefer GI, Ebright RY, Stewart ML, Ito D, Wang S, et al:
An interactive resource to identify cancer genetic and lineage
dependencies targeted by small molecules. Cell. 154:1151–1161.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi
Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, et al:
Proteogenomic characterization of human colon and rectal cancer.
Nature. 513:382–387. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Li Z, Gao X, Peng X, May Chen MJ, Li Z,
Wei B, Wen X, Wei B, Dong Y, Bu Z, et al: Multi-omics
characterization of molecular features of gastric cancer correlated
with response to neoadjuvant chemotherapy. Sci Adv. 6:eaay42112020.
View Article : Google Scholar : PubMed/NCBI
|