Introduction
Atypical teratoid rhabdoid tumor (ATRT) is a rare
(1–2% of pediatric brain tumors) type of potentially fatal
pediatric brain tumor (1). ATRT
accounts for nearly half of the central nervous system (CNS)
malignancies diagnosed during the first year of life (1). A total of 75% of patients with ATRT
are <3 years old. Posterior fossa tumors predominate in infants,
while supratentorial and spinal tumors are more common in children
>3 years old. Metastatic disease occurs in ~30% of ATRT cases
(1).
Rhabdoid tumors are characterized by mutations of
the integrase interactor 1 (INI1) gene on chromosome 22. The
absence of the INI1 protein, observed through immunohistochemical
staining, is used to facilitate the diagnosis of this disease
(2). Pathological features of ATRT
could highlight specific molecular subgroups that need further
confirmation through DNA methylation analysis (3). ATRT had four morphological categories:
Rhabdoid, small-round-blue, epithelial and mesenchymal. The
epithelial category is over-represented in ATRT-TYR, while the
category small-round-blue category is over-represented in ATRT-SHH.
The majority of ATRT-MYC was categorized as mesenchymal or rhabdoid
(3).
ATRTs encompass three epigenetic subgroups with
distinct genomic profiles and SWI/SNF-related matrix-associated
actin-dependent regulator of chromatin subfamily B member 1
(SMARCB1) genotypes: i) atrt-tyrosinase (TYR) tumors are more
common in the infratentorial regions; ii) ATRT-MYC tumors are more
common in the supratentorial regions; and iii) ATRT-sonic hedgehog
(SHH) tumors are found in both infra- and supratentorial areas
(4).
Currently, there is no standard chemotherapeutic
regimen for the treatment of patients with ATRT. Alkylating agents,
high-dose methotrexate or high-dose chemotherapy with stem cell
transplantation are the most recommended regimens of treatment
(5). A multimodal treatment
strategy that involved maximal safe resection, standard dose
chemotherapy, radiation therapy and high-dose chemotherapy improved
the survival rates of patients, but caused notable toxicity due to
the young age of the patients who could not tolerate this
aggressive approach (1).
The present study aimed to investigate the outcome
of pediatric ATRT at the Children's Cancer Hospital of Egypt
(Cairo, Egypt). Different prognostic factors, such as age,
metastatic phenotype (M+), primary tumor site, tumor size, extent
of resection [gross total resection (GTR) vs. subtotal resection
(STR)], field of radiotherapy, chemotherapy dose intensity (full
vs. reduced dose) and remission status after 6 weeks of induction,
were analyzed and their association with patient survival was
examined.
Materials and methods
Patient population
The present retrospective study included 47 patients
with ATRT treated at the Children's Cancer Hospital of Egypt
(Cairo, Egypt) between July 2007 and December 2017. Patients were
treated according to the Dana-Farber Cancer Institute (DFCI)
protocol 02-294 (6). All patients
diagnosed with ATRT at ≤18 years of age with available pathological
specimen and complete data in their electronic files were included.
Patients who received chemotherapy or radiotherapy outside the
hospital or their data were missing from the electronic files were
excluded.
Data were retrieved from the electronic files of
patients after ethics approval was obtained from the Institutional
Review Board of the Children's Cancer Hospital of Egypt (approval
no. 33/2019; Cairo, Egypt). The requirement for written patient
consent was waived due to the retrospective nature of the present
study. The patients or legal guardians were contacted for any
information that was unavailable in the patient files. Oral consent
was obtained from the parents or legal guardians of the patients
over the phone due to their inability to travel to the hospital.
All patient data were anonymized to protect their privacy and
confidentiality.
The following data were retrieved from the patient
files: Age, sex, initial tumor size, tumor site, the presence of
metastatic disease, response to treatment, types and grades of
different toxicities according to Common Toxicity Criteria (CTC)
version V (7), date of progression
or disease recurrence, date of the end of treatment, and date of
death. The extent of surgical resection was classified into GTR (no
evidence of residual tumor), near-total resection (NTR; ≤.1.5
cm2 residual tumor), STR (≥1.5 cm2 residual
tumor) and debulking surgery or biopsy (8). All patients were pathologically
diagnosed with grade IV ATRT with a loss of INI1 expression
according to the World Health Organization guidelines (9). Patient responses were assessed twice,
once during pre-irradiation at 6 weeks and once at the end of
treatment. The radiological response was assessed according to the
Response Assessment in Neuro-Oncology criteria (10). Complete response (CR) was defined by
the following criteria: i) Complete disappearance of all enhancing
measurable and non-measurable disease sustained for ≥4 weeks; ii)
no new lesions; iii) stable or improved non-enhancing
T2/fluid-attenuated inversion recovery (FLAIR) lesions; and iv)
stable or improved clinical condition without any medical support
as assessed by the primary physician. Partial response (PR) was
achieved if the following criteria were met: i) A total of ≥50%
decrease compared with baseline in the sum of products of
perpendicular diameters of all measurable enhancing lesions
sustained for ≥4 weeks; ii) no progression of non-measurable
disease; iii) no new lesions; iv) stable or improved non-enhancing
T2/FLAIR lesions on the same or a lower dose of corticosteroids;
and v) clinically stable or improved condition. Stable disease (SD)
was achieved if the following criteria were met: i) Patient did not
qualify for CR, PR or progressive disease (PD); and ii) stable
non-enhancing T2/FLAIR lesions were observed on the same or a lower
dose of corticosteroids compared with the baseline scan. PD was
defined using the following criteria: i) A total of ≥25% increase
in the sum of products of perpendicular diameters of enhancing
lesions compared with the smallest tumor measurement obtained
either at baseline (if no decrease) or best response on stable or
increasing doses of corticosteroids; and ii) a significant increase
in T2/FLAIR non-enhancing lesions on stable or increasing doses of
corticosteroids. Measurable disease was defined by dimensional
contrast-enhancing lesions with clearly defined margins, with two
perpendicular diameters ≥10 mm, visible on ≥2 axial slices.
Non-measurable lesions were those lesions without enhancement and
assessed by their flair uptake.
Treatment details
All patients were subjected to maximal safe
resection. The extent of resection was assessed by MRI brain and
intraoperative neurosurgical assessment to decide the resection of
the tumor without jeopardizing vital structure. Two patients had no
available data regarding the location of the surgery performed, and
the families were unable to state the hospitals where surgeries
were carried out The treatment protocol lasted 51 weeks and
included the following phases: Induction, maintenance and
continuation therapy (using either doxorubicin or actinomycin
therapy). This regimen is similar to that applied by the DFCI with
which our center had collaboration facilitating the adoption of
this protocol (6).
The induction phase consisted of 14 weeks of
chemotherapy. For the first 6 weeks, chemotherapy was administered
pre-irradiation, the following 6 weeks chemotherapy was
administered during the radiation therapy, and the last 2 weeks
chemotherapy was administered post-radiation with an interval of 3
weeks between every cycle of chemotherapy. After week 6, patients
with residual disease would undergo second-look surgery.
Age-adapted radiotherapy was implemented, whereby
patients <3 years old received focal irradiation regardless of
their metastatic status. Only M+ patients >3 years old received
craniospinal irradiation (CSI). The dose for focal radiotherapy
ranged between 4,800 and 5,900 cGy given for 6 weeks (5 days/week)
with total of 31 sessions. Younger patients were treated with a
lower dose. Regarding the craniospinal dose, 2 patients received
2,340 cGy CSI with boost radiation of the tumor bed up to 5,040
cGy, while 2 patients received a reduced dose of 3,600 cGy CSI with
boost radiation of the tumor bed ≤5,400 cGy.
The maintenance chemotherapy phase lasted 23 weeks.
The continuation therapy consisted of 3 weeks of chemotherapy and
was performed with or without doxorubicin depending on the
radiation field received. Patients receiving CSI received
non-doxorubicin continuation therapy to minimize further
cardiotoxicity. The details of the full treatment protocol are
presented in Fig. S1. The type and
grade of toxicities were collected and graded according to the CTC
version V. Patients who encountered grade 3–4 toxicity were treated
with a chemotherapy dose that was reduced by 25% in subsequent
cycles regardless of the phase of therapy.
Statistical analysis
All statistical analyses were conducted using SPSS
(version 23; IBM Corp.). The study endpoints included overall
survival (OS) and event-free survival (EFS). OS was calculated as
the time from registration until death or last contact with the
patient, while EFS was calculated as the time until recurrence,
progression, death or last contact with the patient. Survival
analyses were performed using the Kaplan-Meier estimator function
and standard errors were calculated using Greenwood's formula. The
two-sided log-rank test was used to compare survival curves of
potential prognostic factors. Median follow-up was determined using
the reverse Kaplan-Meier method. Enumeration data are presented as
n (%) and were analyzed with χ2 or Fisher's exact tests.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Patient characteristics
The present study retrospectively included 47
patients with ATRT (Table I). The
median age of patients was 2.4 years (range, 0.5–16.9 years). A
total of 30 patients (63.8%) were <3 years old, while 17
patients (36.2%) were ≥3 years old. The male-to-female ratio of the
patient cohort was 1.14:1. A total of 26 patients (55.3%) had
supratentorial tumors, 18 patients (38.3%) had infratentorial
tumors, three patients (6.4%) had spinal ATRT and 14/47 patients
(29.8%) had metastatic tumors. Among the 14 patients with
metastatic tumors, nine patients (64.3%) were <3 years old,
while 5 patients (35.7%) were ≥3 years old. The median maximum
tumor dimension was 5.2 cm (range, 1.6–14.5 cm). Among all
patients, 22/47 patients (46.8%) had a preoperative maximum tumor
diameter <5 cm. In comparison, 23/47 patients (48.9%) had a
tumor diameter ≥5 cm, whilst two patient files did not contain
available initial imaging data. GTR/NTR was achieved in 22/47
patients (46.8%), STR was achieved in 8/47 patients (17.0%), and
16/47 patients (34.0%) underwent a biopsy only. A single (2.1%)
patient was not assessed postoperatively but no indication
regarding the reasoning was found in the patient's medical file. No
patients underwent second-look surgery.
 | Table I.Characteristics of the 47 patients
with atypical teratoid rhabdoid tumor in the present study. |
Table I.
Characteristics of the 47 patients
with atypical teratoid rhabdoid tumor in the present study.
| Patient
characteristic | n (%) |
|---|
| Sex |
|
|
Male | 25 (53.2) |
|
Female | 22 (46.8) |
| Age, years |
|
|
<3 | 30 (63.8) |
| ≥3 | 17 (36.2) |
| Primary tumor
site |
|
|
Supratentorial | 26 (55.3) |
|
Infratentorial | 18 (38.3) |
| Spinal
cord | 3 (6.4) |
| Maximum tumor
diameter, cm |
|
|
<5 | 22 (46.8) |
| ≥5 | 23 (48.9) |
|
Unknown | 2 (4.3) |
| Extent of tumor
resection |
|
| Gross
total resection/near-total resection | 22 (46.8) |
|
Subtotal resection | 8 (17.0) |
|
Debulking or biopsy | 16 (34.0) |
|
Unknown | 1 (2.1) |
| Metastasis |
|
| No | 33 (70.2) |
|
Yes | 14 (29.8) |
| Chemotherapy |
|
| No | 2 (4.3) |
|
Yes | 45 (95.7) |
| Full
dose | 40 (88.9) |
| Reduced
dose | 5 (11.1) |
| Radiotherapy |
|
| No | 18 (38.3) |
|
Yes | 29 (61.7) |
|
Focal | 23 (79.3) |
|
Craniospinal | 4 (13.8) |
|
Unknown | 2 (6.9) |
A total of 40/45 patients (88.9%) received full-dose
chemotherapy, while 5/45 patients (11.1%) received chemotherapy
with dose reduction due to chemotherapy toxicity. Two patients did
not receive chemotherapy due to patients' guardian refusal. A total
of 11/45 patients (24.4%) reached the end of the treatment protocol
and the rest died at different phases of treatment, whilst 33
patients (73.3%) completed the pre-irradiation phase of the
treatment and were evaluated at week 6. At week 6, 9/33 patients
(27.3%) achieved CR, 15/33 patients (45.5%) achieved PR, 3/33
patients (9.1%) had SD, 4/33 patients (12.1%) had PD and 2/33
patients (6.1%) did not undergo imaging.
A total of 29/47 patients (61.7%) received radiation
therapy. A total of 23/29 patients (79.3%) received focal
radiation, whilst 4/29 patients (13.8%) received CSI. The
radiotherapy details were missing for two patients. Not all
patients received radiation therapy either due to poor general
condition or due to PD before radiotherapy. The median duration of
the treatment protocol was 66.9 weeks (range, 60.1–84.4 weeks). The
long duration of therapy compared to the number of weeks of the
protocol is related to the encountered toxicity with prolonged
supportive care.
Prognostic factors
There was no significant impact of the following
prognostic factors on the response at week 6: Age (P=0.185), sex
(P=0.615), tumor size (P=0.289), surgery (P=0.498), extent of
resection (P=0.616) and initial metastatic status (P=0.717)
(Table II). The primary tumor site
(supratentorial vs. infratentorial vs. spinal) significantly
affected the response at week 6 (P=0.008). After excluding the 2
cases of spinal ATRT, there was no significant difference between
the supratentorial and infratentorial sites (P=0.6). Various
prognostic factors such as age, sex, tumor size, extent of
resection and metastatic status had no significant impact on the
response at the end of treatment. The median EFS for patients <3
years old was 9.3 months (95% CI, 2.3–16.4 months), while it was
8.1 months (95% CI, 0.0–16.4 months) for patients ≥3 years old. The
1-year EFS and OS for patients <3 years old were 33.3% (95% CI,
16.4–50.2) and 36.7% (95% CI, 19.5–53.9), respectively, and these
were 35.3% (95% CI, 12.6–58.0) and 41.2% (95% CI, 17.9–64.5),
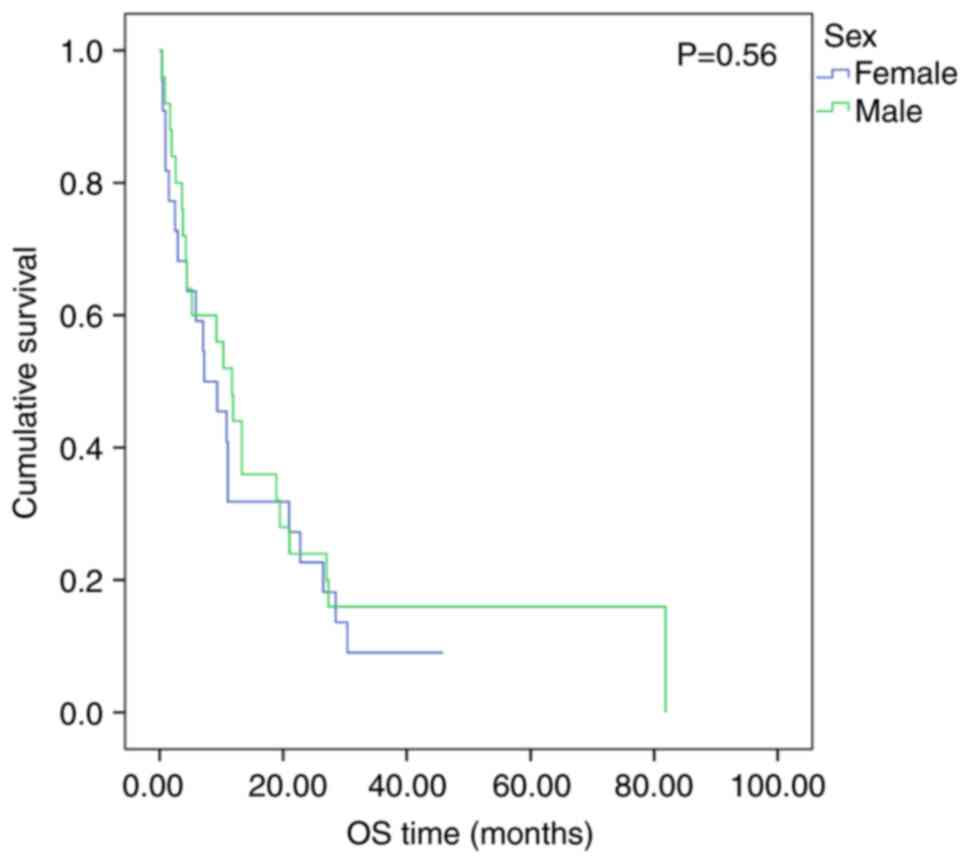
respectively, for patients ≥3 years old (EFS, P=0.75; Fig. 1; OS, P=0.56; Fig. 2). The 2-year OS was 20.0% (95% CI,
5.7–34.3) for patients <3 years old, compared with 29.4% (95%
CI, 7.6–51.2), respectively, for patients ≥3 years old.
| Table II.Association between different
prognostic factors and response of patients with atypical teratoid
rhabdoid tumor after 6 weeks of treatment. |
Table II.
Association between different
prognostic factors and response of patients with atypical teratoid
rhabdoid tumor after 6 weeks of treatment.
| Prognostic
factor | No. | Complete response,
stable disease or partial response, n (%) | Disease progression
or recurrence, n (%) | P-value |
|---|
| Age, years |
|
|
| 0.185 |
|
<3 | 19 | 17 (89.5) | 2 (10.5) |
|
| ≥3 | 14 | 10 (71.4) | 4 (28.6) |
|
| Sex |
|
|
|
|
|
Male | 19 | 15 (78.9) | 4 (21.1) | 0.615 |
|
Female | 14 | 12 (85.7) | 2 (14.3) |
|
| Primary tumor
site |
|
|
| 0.008 |
|
Supratentorial | 18 | 16 (88.9) | 2 (11.1) |
|
|
Infratentorial | 13 | 11 (84.6) | 2 (15.4) |
|
| Spinal
cord | 2 | 0 (0.0) | 2 (100.0) |
|
| Maximum tumor
diameter, cm |
|
|
| 0.289 |
|
<5 | 18 | 15 (83.3) | 3 (16.7) |
|
| ≥5 | 15 | 11 (73.3) | 4 (26.7) |
|
| Location surgery
was performed |
|
|
|
|
|
Children's Cancer Hospital of
Egypt | 31 | 25 (80.6) | 6 (19.4) | 0.498 |
| Other
institutions | 2 | 2 (100.0) | 0 (0.0) |
|
| Extent of tumor
resection |
|
|
|
|
| Gross
total resection or near-total resection | 16 | 14 (87.5) | 2 (12.5) | 0.616 |
|
Subtotal resection | 6 | 5 (83.3) | 1 (16.7) |
|
|
Debulking or biopsy | 11 | 8 (72.7) | 3 (27.3) |
|
| Metastasis |
|
|
| 0.717 |
| No | 24 | 20 (83.3) | 4 (16.7) |
|
|
Yes | 9 | 7 (77.8) | 2 (22.2) |
|
Patient sex (EFS, P=0.42; Fig. 3; OS, P=0.56; Fig. 4), primary tumor site (EFS, P=0.35;
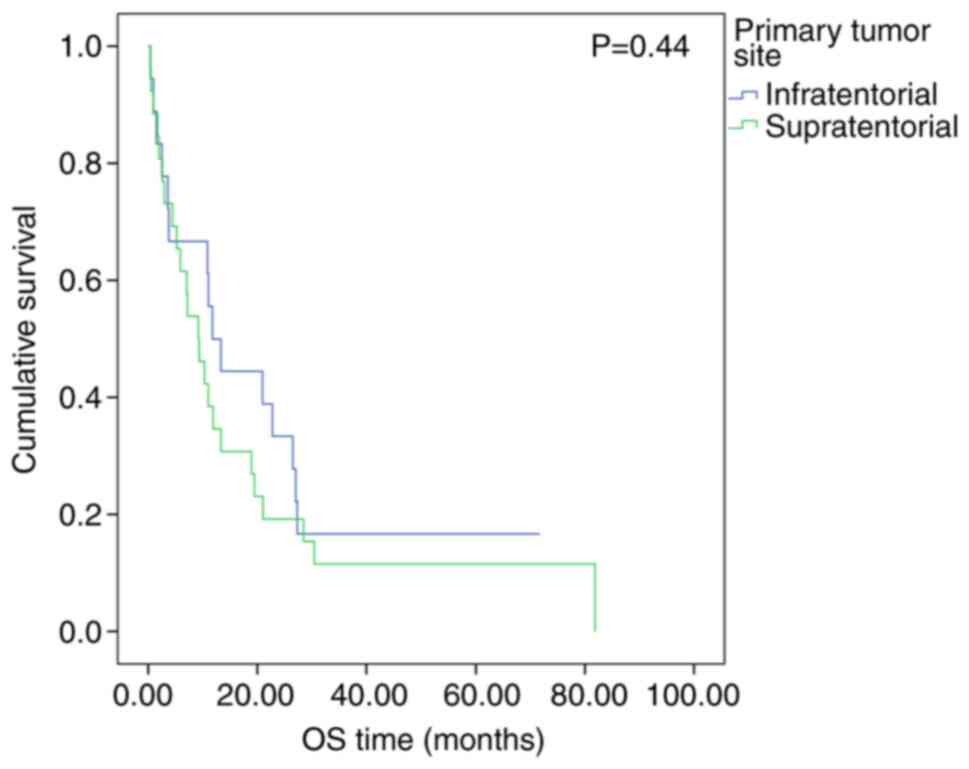
Fig. 5; OS, P=0.44; Fig. 6), metastatic disease (EFS, P=0.21;
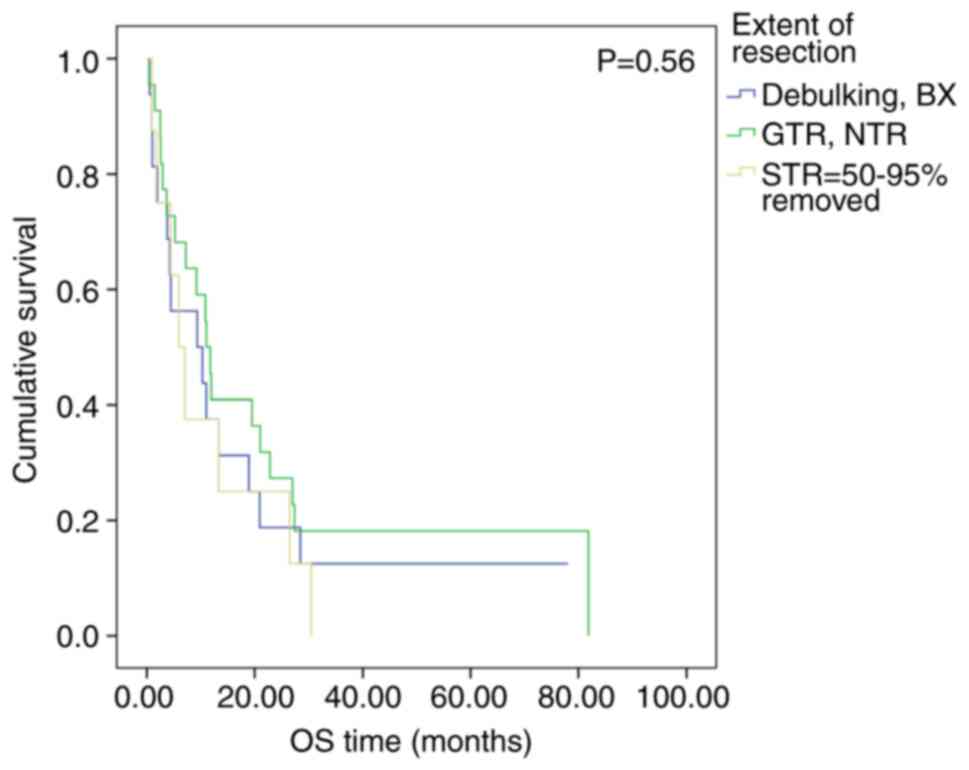
Fig. 7; OS, P=0.25; Fig. 8) and extent of resection (EFS,
P=0.58; Fig. 9; OS, P=0.56;
Fig. 10) did not have a
significant impact on the treatment outcome. The median EFS and OS
for patients <3 years old with metastatic disease were 9.4
months (95% CI, 6.8–12.8 months) and 7.7 months (95% CI, 3.6–15.8
months), respectively, and those for patients ≥3 years old were
10.6 months (95% CI, 8.1–13.2 months) and 8.3 months (95% CI,
2.1–14.5 months), respectively (EFS, P=0.498; OS, P=0.993; data not
shown).
Preoperative tumor size significantly affected both
EFS and OS. The median EFS for tumors <5 cm was 11.7 months (95%
CI, 3.2–20.3 months), while the median EFS for tumors ≥5 cm was 6.0
months (95% CI, 0.3–11.6 months). The 1-year EFS for patients with
tumor size <5 or ≥5 cm was 45.5% (95% CI, 24.7–66.3) and 21.7%
(95% CI, 4.8–38.6), respectively (P=0.03; Fig. 11).
The median OS for tumors <5 or ≥5 cm was 11.9
months (95% CI, 2.8–21.0 months) and 7.2 months (95% CI, 1.0–13.4
months), respectively. The 1-year and 2-year OS for patients with
tumors <5 cm was 50.0% (95% CI, 29.0–71.0) and 36.4% (95% CI,
16.2–56.6), respectively, compared with 26.1% (95% CI, 8.1–44.1)
and 8.7% (95% CI, 0.0–20.3), respectively, for patients with tumors
≥5 cm (P=0.04; Fig. 12).
Chemotherapy dose reduction did not have a
significant negative impact on the outcome of treatment. The median
EFS for patients who received a full dose of chemotherapy was 8.1
months (95% CI, 2.1–14.0 months), while the median EFS for patients
who received a reduced dose was 20.7 months (95% CI, 6.3–35.1
months). The 1-year EFS for patients who received the full or the
reduced dose was 30.0% (95% CI, 15.9–44.1) and 80.0% (95% CI,
44.9–100.0), respectively, which had clinical significance,
although no statistical significance was observed (P=0.08; Fig. 13).
The median OS for patients who received a full dose
of chemotherapy was 9.2 months (95% CI, 4.1–14.3 months), while the
median OS for patients who received a reduced dose was 28.5 months
(95% CI, 9.2–47.7 months). The 1- and 2-year OS was 35.0% (95% CI,
20.3–49.7) and 20.0% (95% CI, 7.7–32.3), respectively, for patients
who received the full dose, compared with 80.0% (95% CI,
44.9–100.0) and 60.0% (95% CI, 17.1–100.0), respectively, for
patients who received the reduced dose (P=0.06; Fig. 14).
Radiotherapy administration significantly impacted
the treatment outcome. The median EFS and OS for patients who
received radiotherapy were 13.9 months (95% CI, 2.8–25.0 months)
and 19.5 months (95% CI, 6.1–32.8 months), respectively. The median
EFS and OS for patients who did not receive radiotherapy were 1.5
months (95% CI, 0.6–2.5 months) and 2.0 months (95% CI, 0.3–3.6
months), respectively. The 1-year EFS for patients who received
radiotherapy and those who did not was 55.2% (95% CI, 37.2–73.2)
and 0.0%, respectively (P<0.001; Fig. 15). The 1- and 2-year OS were 62.1%
(95% CI, 44.5–79.7) and 37.9% (95% CI, 20.3–55.5) for patients who
received radiotherapy, vs. 0% for those who did not receive
radiotherapy (P<0.001; Fig.
16). The radiotherapy field had no statistically significant
effect on the treatment outcome (EFS, P=0.74; Fig. 17; OS, P=0.71; Fig. 18).
Treatment-related toxicity
A total of 43/47 patients (91.5%) patients reported
≥1 treatment-related toxicity. These toxicities were reported in
all 40 patients who received full dose chemotherapy and in 3/5
patients who received a reduced dose of chemotherapy. All patients
who received focal or CSI radiotherapy developed treatment-related
toxicity. A total of 3/47 patients (6.4%) patients had no available
data about their toxicity profile, whilst one patient (2.1%) did
not develop any treatment-related toxicity. Blood cultures were
performed for 39/47 patients (83.0%) patients due to shortage of
blood culture bottles in the hospital. The majority of the
bacterial species recovered from these cultures were gram-negative
(33/39; 84.6%). The most commonly isolated bacteria were
Escherichia coli (n=18), Klebsiella pneumoniae (n=8),
Pseudomonas aeruginosa (n=4) and Acinetobacter
baumannii (n=3). Gram-positive bacteria were isolated in 6/39
patients (15.4%), and the most commonly isolated gram-positive
bacterium was Staphylococcus aureus. Central line-associated
bloodstream gram-negative infections were reported in 12/33
patients (36.4%) as follows: Escherichia coli (n=5),
Klebsiella pneumoniae (n=4), Pseudomonas aeruginosa
(n=2) and Acinetobacter baumannii (n=1).
Survival outcome
The median follow-up period of the patients was 71.7
months. The median EFS and OS for all patients were 9.3 months (95%
CI, 2.9–15.8 months) and 10.3 months (95% CI, 0.4–81.4 months),
respectively, with an estimated 1-year and 2-year EFS of 34.0% (95%
CI, 20.5–47.5) and 19.1% (95% CI, 7.8–30.2), respectively (Fig. 19). The 1-, 2- and 3-year OS of the
patient cohort was 38.3% (95% CI, 24.4–52.2), 23.4% (95% CI,
11.2–35.6) and 12.8% (95% CI, 3.2–22.4), respectively (Fig. 20). A total of 24/47 patients
(51.1%) exhibited disease progression or recurrence. The
progression site was either local (n=6), metastatic (n=9) or both
local and metastatic (n=9).
Causes of mortality
Toxicity-related mortality occurred in 14/47
patients (29.8%). A total of 8/14 patients (57.1%) died from
gram-negative septicemia due to Escherichia coli (n=2),
Klebsiella pneumoniae (n=5) or Pseudomonas aeruginosa
(n=1) infections. A total of 5/14 patients (35.7%) died from severe
pneumonia, whilst 1 (7.1%) patient developed grade V platinum
toxicity with electrolyte disturbance, which led to acute renal
failure.
Discussion
ATRT is a rare disease, and clinical trials
investigating the treatment modalities are largely non-randomized,
single-arm, collaborative, multi-centric studies from Europe and
North America. Real-world data on this rare disease are scarce,
especially from low middle-income countries (11). The present study reported the
treatment outcomes of patients with ATRT from a large pediatric
cancer center in Egypt (Children's Cancer Hospital of Egypt,
Cairo), using a unified treatment protocol with an intensive
multimodality approach of maximal safe resection, systemic and
intrathecal chemotherapy, and age-adjusted radiation therapy.
In the present study, 30 (63.8%) patients were <3
years old at the time of presentation. The median age of the
patients was 28.8 months (2.4 years), which was consistent with a
number of previous reports, including the North American ATRT
registry (24 months) (11), the
European Rhabdoid Registry (EU-RHAB; 29.5 months) (12) and the DFCI cohort (26 months)
(6). The percentage of patients
<3 years old was higher in the Children's Oncology Group (COG)
ACNS0333 study cohort (83%) (13).
The present study demonstrated that patient age did
not have a significant impact on the treatment outcome. This
finding is in accordance with the study published by Upadhyaya
et al (14) who analyzed 74
patients newly diagnosed with ATRT, who were treated either in the
SJYC07 trial (age, <3 years; n=52) or SJMB03 trial (age, 3–21
years; n=22). In the SJYC07 trial, patients with non-metastatic
(M0) disease (n=34) had a 5-year progression-free survival (PFS) of
39.1±11.5% and OS of 51.8±12.0%, whereas the 5-year PFS and OS for
patients with M+ disease (n=18) were both 0.0% (14). Additionally, survival did not differ
by age at diagnosis, sex, primary tumor site and extent of
resection for those with intermediate risk disease, which is
consistent with the results of the present study (14). According to the SJMB03 clinical
trial data, children with average risk (M0 disease and <1.5
cm2 residual tumor; n=11) had a 5-year PFS and OS of
72.7±12.7 and 81.8±11%, respectively, whereas those with high-risk
disease (M+ or >1.5 cm residual) (n=11) had a 5-year PFS and OS
of 18.2±9.5% (14). Additionally,
Chi et al (6) reported that
age did not have an impact on the survival outcome (6). By contrast, the EU-RHAB study reported
that age was the most important prognostic factor of survival with
a 5-year OS of 16.7±5.7% for patients <12 months old at
diagnosis vs. 45.3±6% for those ≥12 months old (12).
In the present study, the primary tumor site did not
have a significant impact on the survival of patients. This result
differed from that reported by Chi et al who indicated that
the tumor location markedly affected OS. The median OS for patients
with supratentorial tumors was 24 months, whereas the median OS was
not reached for patients with posterior fossa tumors (P=0.04)
(6). The absence of an association
between the primary tumor site and the outcome in the present study
was consistent with the COG ACNS0333 results and the St. Jude
trials data (13,14).
We hypothesized that Egyptian patients with ATRT may
be diagnosed later compared with cases in high-income countries due
to the long waiting lists and poor parental awareness about this
disease, which leads to more prevalent metastatic disease at the
time of presentation. However, in the present study cohort, 70.2%
of patients were M0, similar to other published data, including the
DFCI (70%), EU-RHAB (70.0%) and ACNS0333 (63.0%) studies (6,12,13).
The present results demonstrated that the presence
of metastasis did not significantly impact the treatment outcome.
This result was similar to previous studies published from the DFCI
and ACNS0333 (6,13). The lack of association may be
partially attributed to unpowered, unadjusted subgroup analysis. By
contrast, the EU-RHAB study reported a superior survival for 100 M0
cases with a 5-year OS of 43.0% compared with 16.9% for M+ patients
(P<0.0001) (12).
The negative impact of metastatic disease on
survival was demonstrated in the SJYC07 trial, where patients with
M0 disease had a 5-year PFS of 39.1±11.5% and OS of 51.8±12.0%,
compared with 0.0% for both PFS and OS for those with M+ disease
(P<0.001) (14).
In the present cohort, large tumor diameters (≥5 cm)
were associated with significantly decreased EFS (P=0.03) and OS
(P=0.04). To the best of our knowledge, this factor is a vital
prognostic variable that has not currently been reported in the
literature.
The extent of tumor resection did not significantly
affect the survival of patients in the present study. This finding
is supported by a previously published individual patient pooled
meta-analysis of 332 cases with ATRT and results obtained from the
EU-RHAB study (12,15). The DFCI study reported contradictory
results compared with the present study data; GTR/NTR was
associated with a 2-year OS of 91% (6). Furthermore, the Canadian Pediatric
Brain Tumor Consortium reported that patients with GTR had a 2-year
OS of 60.0±12.6% compared with 21.7±8.5% for those who underwent
alternative treatment to GTR (P=0.03) (16).
In the present cohort, patients who received
radiotherapy showed a significantly superior median EFS
(P<0.001) and OS (P<0.001) compared with those who did not
receive radiotherapy (13.9 vs. 1.5 months and 19.5 vs. 2.0 months,
respectively). Similarly, Schrey et al (15) reported higher EFS and OS rates for
patients receiving irradiation, in addition to improved
radiological response at the end of treatment (15).
The median EFS and OS for all patients included in
the present study were 9.3 months (95% CI, 2.9–15.8 months) and
10.3 months (95% CI, 0.4–81.4 months), respectively, which was
lower than those reported in a previous report with a 4-year OS of
50% (6). A total of 24 (89.4%)
patients either exhibited disease progression or recurrence. The
progression sites were local, metastatic or both local and
metastatic. In the SJYC07 trial, the pattern of failure for IR
patients (n=23) was local (n=9), distant (n=8) or combined (n=6)
(14).
The present study reported an inferior treatment
outcome compared with a previous study by Chi et al
(6), due to higher mortality rate
in the present study (29.8 vs. 5%). This could potentially be
attributed to the majority of the patients in the present study
living in low socioeconomic areas. These patients were more likely
to be infected by gram-negative bacteria, which caused severe
septicemia during the repeated and prolonged period of
myelosuppression. Furthermore, poor patient hygiene and made
patients susceptible to severe pneumonia, especially during the
immunosuppression period. Certain patients could not maintain a
high chemotherapy dose intensity due to a prolonged supportive
period, as highlighted by the high median therapy duration reported
in the present study. Therefore, administering a patient-specific
chemotherapy dose intensity could decrease infectious complications
and maintain dose intensity without treatment interruption.
Furthermore, a short, intensified induction followed
by intensified consolidation, either single as Head Start or tandem
transplant, as reported in the COG ACNS0333 study, may enhance CNS
prophylaxis using high-dose methotrexate. The long treatment
protocol duration (51 weeks) may increase the probability of
community-acquired infections observed in the patients of the
present study who live in low socioeconomic areas, which can
contribute to higher rates of toxicity-related mortality.
The limitations of the present study included its
retrospective nature and incomplete data on detailed toxicities and
chemotherapy protocol modifications. Furthermore, the frequency of
neurocognitive deficits was not measured. The present study also
did not investigate the role of the SHH, TYR or MYC molecular
subgroups or germline SMARCB1/A4 mutations in the patient
cohort.
In previous years, the importance of epigenetic
markers in carcinogenesis has been reported in relation to
understanding the mechanisms of metastatic tumor progression. Both
the upregulation and downregulation of microRNAs result in
increased cell proliferation, tumor invasion and interaction with
various driver markers. A number of different microRNAs have been
shown to be useful at both diagnostic and prognostic levels
(17).
Further research is required into targeted therapy
of ATRT specific to each molecular subtype (TYR, SHH and MYC), as
the outcome is still poor using the currently available therapeutic
options of standard chemotherapy, radiation therapy, surgery and
high-dose chemotherapy. Torchia et al (18) reported that cell lines derived from
ATRT-SHH tumors were highly sensitive to enhancer of zeste homolog
2 inhibitors (18). Although
subtype-specific targeted therapy is still in the early phases of
clinical trials, the preliminary results may indicate improved
outcomes of this aggressive disease. Delineating prognostic factors
affecting the survival of ATRT is necessary for adequate disease
management.
The treatment regimen should be dose-adjusted
according to each patient to maintain the treatment intensity and
to avoid toxicity-related mortality. The use of high-dose
chemotherapy may be associated with improved outcomes and higher
toxicity rates, necessitating timely supportive care, especially in
lower- and middle-income settings. Further clinical trials
incorporating novel targeted therapies are required in the
future.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
AEHe conceived the present study. MSZ and AEHe
confirmed the authenticity of all the raw data MS collected data
and participated in designing the study. EM and MSZ performed data
analysis and validation. AEHa drafted the manuscript and
interpreted the data. HT, AR and MEB analyzed the data. AEHa and
MSZ gave the final approval of the version to be published. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was conducted in accordance with
The Declaration of Helsinki. Ethics approval was provided by the
institutional review board of the Children's Cancer Hospital of
Egypt 57357 (approval no. 28-7-2021; Cairo, Egypt) and the
requirement for obtaining written consent was waived due to the
retrospective nature of the present study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ATRT
|
atypical teratoid rhabdoid tumor
|
|
CNS
|
central nervous system
|
|
COG
|
Children's Oncology Group
|
|
CR
|
complete response
|
|
CTC
|
Common Toxicity Criteria
|
|
CSI
|
craniospinal irradiation
|
|
FLAIR
|
fluid-attenuated inversion
recovery
|
|
GTR
|
gross total resection
|
|
INI1
|
integrase interactor 1
|
|
NTR
|
near-total resection
|
|
OS
|
overall survival
|
|
PD
|
progressive disease
|
|
SD
|
stable disease
|
|
SMARCB1
|
SWI/SNF-related matrix-associated
actin-dependent regulator of chromatin subfamily B member 1
|
|
STR
|
subtotal resection
|
References
|
1
|
Nesvick CL, Lafay-Cousin L, Raghunathan A,
Bouffet E, Huang AA and Daniels DJ: Atypical teratoid rhabdoid
tumor: Molecular insights and translation to novel therapeutics. J
Neurooncol. 150:47–56. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sultan I, Qaddoumi I, Rodríguez-Galindo C,
Nassan AA, Ghandour K and Al-Hussaini M: Age, stage, and
radiotherapy, but not the primary tumor site, affects the outcome
of patients with malignant rhabdoid tumors. Pediatr Blood Cancer.
54:35–40. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zin F, Cotter JA, Haberler C, Dottermusch
M, Neumann J, Schüller U, Schweizer L, Thomas C, Nemes K, Johann
PD, et al: Histopathological patterns in atypical teratoid/rhabdoid
tumors are related to the molecular subgroup. Brain Pathol.
31:e129672021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cacciotti C, Fleming A and Ramaswamy V:
Advances in the molecular classification of pediatric brain tumors:
A guide to the galaxy. J Pathol. 251:249–261. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ginn KF and Gajjar A: Atypical teratoid
rhabdoid tumor: Current therapy and future directions. Front Oncol.
2:1142012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chi SN, Zimmerman MA, Yao X, Cohen KJ,
Burger P, Biegel JA, Rorke-Adams LB, Fisher MJ, Janss A, Mazewski
C, et al: Intensive multimodality treatment for children with newly
diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol.
27:385–389. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Freites-Martinez A, Santana N,
Arias-Santiago S and Viera A: Using the common terminology criteria
for adverse events (CTCAE-Version 5.0) to evaluate the severity of
adverse events of anticancer therapies. Actas Dermosifiliogr (Engl
Ed). 112:90–92. 2019.(In English, Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Thompson EM, Bramall A, Herndon JE II,
Taylor MD and Ramaswamy V: The clinical importance of
medulloblastoma extents of resection: A systematic review. J
Neurooncol. 139:523–539. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Louis DN, Perry A, Wesseling P, Brat DJ,
Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM,
Reifenberger G, et al: The 2021 WHO Classification of Tumors of the
Central Nervous System: A summary. Neuro Oncol. 23:1231–1251. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chukwueke UN and Wen PY: Use of the
Response Assessment in Neuro-Oncology (RANO) criteria in clinical
trials and clinical practice. CNS Oncol. 8:28–44. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hilden JM, Meerbaum S, Burger P, Finlay J,
Janss A, Scheithauer BW, Walter AW, Rorke LB and Biegel JA: Central
nervous system atypical teratoid/rhabdoid tumor: Results of therapy
in children enrolled in a registry. J Clin Oncol. 22:2877–2884.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Frühwald MC, Hasselblatt M, Nemes K, Bens
S, Steinbügl M, Johann PD, Kerl K, Hauser P, Quiroga E, Solano-Paez
P, et al: Age and DNA methylation subgroup as potential independent
risk factors for treatment stratification in children with atypical
teratoid/rhabdoid tumors. Neuro Oncol. 22:1006–1017. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reddy AT, Strother DR, Judkins AR, Burger
PC, Pollack IF, Krailo MD, Buxton AB, Williams-Hughes C, Fouladi M,
Mahajan A, et al: Efficacy of high-dose chemotherapy and
three-dimensional conformal radiation for atypical
teratoid/rhabdoid tumor: A Report From the Children's Oncology
Group Trial ACNS0333. J Clin Oncol. 38:1175–1185. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Upadhyaya SA, Robinson GW, Onar-Thomas A,
Orr BA, Johann P, Wu G, Billups CA, Tatevossian RG, Dhanda SK,
Srinivasan A, et al: Relevance of molecular groups in children with
newly diagnosed atypical teratoid rhabdoid tumor: Results from
prospective St. Jude multi-institutional trials. Clin Cancer Res.
27:2879–2889. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schrey D, Carceller Lechón F, Malietzis G,
Moreno L, Dufour C, Chi S, Lafay-Cousin L, Von Hoff K, Athanasiou
T, Marshall LV and Zacharoulis S: Multi-modal therapy in children
and adolescents with newly diagnosed atypical teratoid rhabdoid
tumor: Individual pooled data analysis and review of the
literature. J Neurooncol. 126:81–90. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lafay-Cousin L, Hawkins C, Carret AS,
Johnston D, Zelcer S, Wilson B, Jabado N, Scheinemann K, Eisenstat
D, Fryer C, et al: Central nervous system atypical teratoid
rhabdoid tumours: The canadian paediatric brain tumour consortium
experience. Eur J Cancer. 48:353–359. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pekarek L, Torres-Carranza D,
Fraile-Martinez O, García-Montero C, Pekarek T, Saez MA,
Rueda-Correa F, Pimentel-Martinez C, Guijarro LG, Diaz-Pedrero R,
et al: An Over view of the role of micoRNA on carcinogenesis: A
focus on cell cyle, angionesis and metastasis. Int J Mol Sci.
24:72682023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Torchia J, Golbourn B, Feng S, Ho KC,
Sin-Chan P, Vasiljevic A, Norman JD, Guilhamon P, Garzia L, Agamez
NR, et al: Integrated (epi)-genomic analyses identify
subgroup-specific therapeutic targets in CNS rhabdoid tumors.
Cancer Cell. 30:891–908. 2016. View Article : Google Scholar : PubMed/NCBI
|