The diagnosis of GC involves histological
examination through endoscopic biopsy and staging with CT,
endoscopic ultrasound, positron emission tomography and laparoscopy
(10). GC is anatomically
classified as true gastric adenocarcinoma (gADC; refers
specifically to cancer originating in the stomach itself) or
gastroesophageal-junction adenocarcinoma (refers to cancer that
occurs at the junction where the esophagus meets the stomach), with
histological categorization into diffuse and intestinal types
(12). Intestinal-type GC, mainly
caused by H. pylori infection, is characterized by tubular
or glandular structures, and is associated with intestinal
metaplasia (13). Diffuse-type GC
is characterized by poorly differentiated tumor cells with
decreased adhesion, leading to infiltration of the stroma as small
subgroups or cellular forms (13).
Adenocarcinoma accounts for the majority of GC cases (90–95%),
followed by less frequent types such as lymphoma (4%),
gastrointestinal stromal tumors (<1%), carcinoid tumors (3%) and
hereditary diffuse GC (1–3%) according to the American Cancer
Society (9).
GC treatment necessitates a multidisciplinary
approach. For early-stage cases with low lymph node metastasis
(LNmet) risk, endoscopic therapy or surgery alone can be curative
(14). Innovations such as sentinel
lymph node biopsy can improve the quality of life without
compromising oncologic outcomes, yet their use outside East Asia is
limited and long-term studies are ongoing (14). Later-stage localized GC requires
extensive lymphadenectomy and multimodality therapy to prevent the
occurrence of nodal and distant metastases (14). Targeted therapies have also been
implemented for treatment, including the use of trastuzumab, an
anti-HER2 antibody, and ramucirumab, a VEGFR-2 antibody (12). Despite these advancements, the
prognosis and recurrence rates of GC remain discouraging due to its
complex heterogeneity and the specific tumor microenvironment (TME)
that promotes tumor progression and metastasis (15).
The field of single-cell omics has experienced
remarkable advancements since its inception. Technologies of
milestone significance in the field of single-cell sequencing are
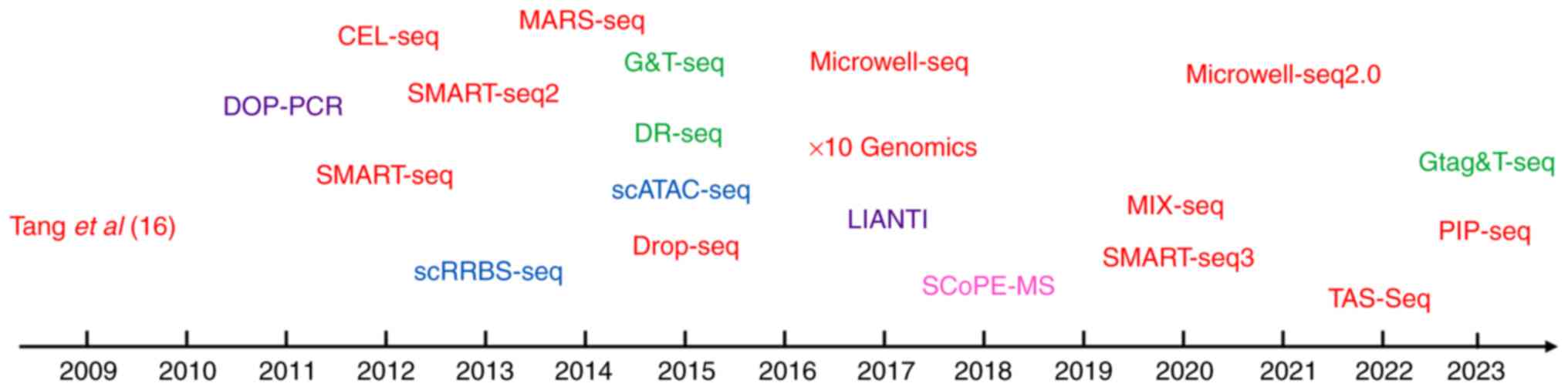
discussed subsequently. In 2009, the first single-cell mRNA
sequencing method was proposed (16), followed by its application to human
cancer cells in 2011 (46).
Subsequently, in 2012, the first single-cell exon was sequenced
(47). Building upon these
developments, Picelli et al (48) introduced Smart-seq2 in 2013, a
method that improved coverage and sensitivity compared with
previous techniques. In 2017, Zheng et al (49) introduced a novel scRNA-seq method
referred to as 10× Genomics, which revolutionized the study of
cellular communication, the TME and tumor heterogeneity (Fig. 1). These advancements in single-cell
omics technologies have paved the way for investigating genomic,
transcriptomic and epigenomic heterogeneity at the single-cell
level. Table I presents an overview
of single-cell technologies and their respective
characteristics.
scRNA-seq offers a deeper understanding of cellular
heterogeneity compared with traditional bulk cell analysis
(50). It provides insights into
gene expression at the single-cell level. The process involves
isolating and lysing individual cells, followed by reverse
transcribing mRNA and then amplifying it (50). However, previous single-cell
isolation methods, such as manual picking (31,51–53),
FACS-sorting (54–56) and integrated microfluidic circuits
(57–59), have limitations in scalability due
to cost, time and labor constraints. To address these limitations,
advancements have been made to enhance efficiency and reduce costs.
For example, Seq-Well (60),
developed in 2017, is a portable and straightforward platform for
massively-parallel scRNA-seq. Its working principle involves
confining mRNA capture beads with unique barcodes and cells within
small pores (pinholes), followed by sealing with a semipermeable
membrane. This setup is conducive to efficient cell lysis and mRNA
capture. After lysis, the beads are removed for parallel
sequencing. The unique barcode on each bead allows for the
identification of the originating cell of each transcript. Due to
its simplicity and portability, Seq-Well can be implemented in a
variety of settings, making it a versatile tool for single-cell
genomics and transcriptomics research. It uses selective chemical
functionalization (mRNA capture beads) to enable rapid cell lysis
and efficient transcript capture while minimizing
cross-contamination. Another scalable method, split-pool
ligation-based transcriptome sequencing (SPLiT-seq), was proposed
in 2018. SPLiT-seq enables efficient sample multiplexing without
the need for specialized equipment, and is compatible with fixed
cells or nuclei (61). In 2019,
massively parallel RNA single cell sequencing (version 2.0) was
introduced, combining sub-microliter reaction volumes, optimized
enzymatic mixtures and an enhanced analytical pipeline (62). These methods substantially reduced
costs, improved reproducibility and decreased well-to-well
contamination (62). Unlike most
single-cell transcriptomic profiling methods that focus on the
3′-end of polyadenylated transcripts, C1 Cap Analysis of Gene
Expression, developed in 2019, detects transcript 5′-ends using an
original sample multiplexing strategy in the C1TM microfluidic
system. Analyzing transcript 5′-ends enhances the understanding of
gene expression by allowing precise mapping of transcription start
sites, which sheds light on the complexity of promoter usage and
regulatory mechanisms. This approach not only reveals the diversity
of transcript isoforms, contributing to protein variability, but
also improves the accuracy of gene expression profiling across
different conditions and cell types. Consequently, focusing on
5′-ends is crucial for unraveling the intricacies of gene
regulation and the functional diversity within cellular processes
(63).
Single-cell genome sequencing has allowed greater
examination of genetic diversity, making it easier to analyze both
de novo germline and somatic mutations in both normal and
cancerous cells (23,64). In 2001, a simple method using
rolling circle amplification was introduced for the amplification
of vector DNA from single colonies or plaques, removing the
requirement for lengthy growth periods and conventional DNA
isolation techniques (65). In
2011, Navin et al (46) used
flow-sorted nuclei, whole genome amplification and next-generation
sequencing to precisely measure genomic copy numbers within
individual nuclei. This method was used to explore the population
structure and evolutionary dynamics of tumors in human breast
cancer cases. Another method, multiple annealing and looping based
amplification cycles, introduced quasi-linear preamplification in
2012 (66), reducing biases
associated with nonlinear amplification (whole genome
amplification). Furthermore, linear amplification via transposon
insertion, proposed in 2017 (67),
overcame limitations of current whole genome amplification methods,
including amplification bias, amplification errors and limited
resolution for detecting variations, enabling micro-copy number
variation detection with kilobase resolution, while minimizing
amplification biases and errors. Multiplexed end-tagging
amplification of complementary strands (METACs) (68), developed in 2021, improved
single-cell whole-genome amplification by leveraging the
complementary strands of double-stranded DNA to filter out false
positives and reduce sequencing costs, achieving high accuracy in
detecting single-nucleotide variations and other genomic variants,
which improves single-cell whole-genome amplification by leveraging
both strands of the DNA. Unique tags are added to DNA ends before
amplification, allowing for the pairing of complementary strands
during sequencing. This method helps filter out false positives and
reduces sequencing costs by requiring less sequencing depth for
high accuracy. METACs is particularly effective in detecting
single-nucleotide variations and other genomic variants, differing
from traditional methods that often amplify only one DNA strand and
may have higher error rates. It is applicable in fields such as
single-cell genomics, clinical diagnostics and population genetics
(68).
Single-cell epigenome analysis provides valuable
insights into DNA methylation, histone modification and chromatin
states, which influence cellular activity (69). In 2013, the first single-cell method
for methylome analysis, single-cell reduced representation
bisulfite sequencing, was introduced (70,71).
This technique enables the measurement of the methylation state in
~10% of CpG sites through enrichment of CpG dense regions (70,71).
These sites predominantly cover most promoters, yet a limitation is
their relatively poor coverage of a number of crucial regulatory
regions, such as enhancers (70).
The post-bisulfite adapter tagging method involves bisulfite
conversion prior to library preparation, ensuring that DNA
degradation does not compromise the adapter-tagged fragments. This
allows for the measurement of methylation at up to 50% of CpG sites
in individual cells (53).
Chromatin immunoprecipitation (ChIP) followed by sequencing
demonstrates improved data compared with chromatin
immunoprecipitation combined with DNA microarray (ChIP combined
with DNA microarray technology is a method used to identify DNA
regions that interact with specific proteins, by precipitating
protein-DNA complexes and hybridizing the extracted DNA onto
microarrays) by providing higher resolution, greater genomic
coverage, increased sensitivity and cost-effectiveness, leading to
more precise and comprehensive analysis of DNA-protein
interactions, allowing genome-wide profiling of DNA-binding
proteins, histone modifications and nucleosomes (72,73).
Another method, single cell assay for transposase-accessible
chromatin with sequencing, utilizes a transposase enzyme to insert
sequencing adapters into open chromatin regions, revealing which
parts of the genome are active or accessible in each cell. This
technology is widely applied in epigenetics to understand
cell-to-cell variability, identify regulatory elements such as
enhancers and promoters, and explore the mechanisms of gene
regulation in diverse cell types (57).
Single-cell protein mass spectrometry allows for
comprehensive measurement of protein expression patterns in a cell
(74). Cytometry by time of flight
(75), a mass cytometry-based
method, has been used to analyze surface and intracellular proteins
using metal-labeled antibodies labeled with heavy metal isotopes,
allowing simultaneous detection of multiple proteins in cells with
minimal overlap and higher precision compared with fluorescent
labels used in traditional flow cytometry. SCoPE by mass
spectrometry (76), a
high-throughput method, was developed based on liquid
chromatography-tandem mass spectrometry techniques, and is a
high-throughput method for single-cell proteomic analysis that
allows for the isolation, enzymatic digestion, and subsequent
identification and quantification of proteins from individual
cells. This technique provides detailed insights into the protein
composition of single cells, revealing cellular functions and
heterogeneity at the proteomic level. Subsequent improvements led
to the development of SCoPE2 (77),
which offers enhanced quantitative accuracy, proteome coverage,
sample preparation ease and cost-effectiveness.
Single-cell CRISPR sequencing is a cutting-edge
technique that integrates CRISPR-Cas9, a powerful gene-editing
tool, with SCS methods. This innovative approach primarily focuses
on executing targeted gene edits at the single-cell level and then
analyzing the consequent changes in the cell transcriptome using
SCS, thereby allowing researchers to directly observe the effects
of specific genetic alterations on gene expression in individual
cells (78). Through this approach,
a deeper understanding of gene functions, cellular networks and
disease mechanisms is attainable. These techniques allow the use of
compiled CRISPR libraries for collective cellular interventions,
followed by high-throughput phenotypic analysis by using collective
cellular interventions via CRISPR libraries to simultaneously edit
multiple genes, followed by high-throughput phenotypic analysis,
allowing for a comprehensive study of the resulting changes in
cellular behavior and characteristics, revealing complex gene
functions and interactions within cellular networks (78). To date, ≥30 different single-cell
CRISPR techniques have been developed; the present review focuses
on introducing a few representative technologies. Perturb-seq
(79) and CRISP-seq (80) were among the first single-cell
CRISPR techniques to be developed. Perturb-seq combines
CRISPR-mediated gene perturbation with scRNA-seq for large-scale
gene function screening and studying the impact of gene expression
changes on cellular states (79).
CRISP-seq, similar to Perturb-seq, focuses on studying individual
genes or a small numbers of genes, assessing how specific
gene-editing events affect cell function (80). By introducing specific gene
alterations via CRISPR, CRISPR droplet sequencing (81), followed by scRNA-seq evaluates the
influence of gene perturbations on cellular states and behaviors,
which combines CRISPR technology with droplet-based single-cell
sequencing, allowing simultaneous editing and gene expression
profiling in individual cells. Unlike traditional CRISPR techniques
that focus primarily on gene editing, CRISPR droplet sequencing
integrates gene editing with detailed, single-cell level
transcriptomic analysis, revealing how edits affect cellular
functions (78). Mosaic-seq
(82) generates cellular mosaics (a
collection of cells in which each cell has a distinct genetic
alteration) with various genetic perturbations, and analyzes the
combined effects of these disruptions using scRNA-seq. This method
is used to study the interactions between different genes and their
impact on cellular functions. Direct-capture Perturb-seq (83), a variant of Perturb-seq, improves
data quality and analysis efficiency by directly capturing and
sequencing CRISPR-guided RNA, facilitating a more precise
association between gene-editing events (deliberate alterations
made to the genome using CRISPR-Cas9 technology, such as knocking
out, knocking in or modifying specific gene sequences) and
transcriptomic changes.
Single-cell CRISPR sequencing and its derivative
technologies hold potential in cancer research. These techniques
assess the complex molecular networks within tumor cells and aid
the understanding of the TME, drug responses and mechanisms of
treatment resistance (79). For
instance, Jun et al (84),
using in vitro experiments, explored all cytosine-to-thymine
mutations in the exon regions of three genes (MAP2K1, KRAS and
NRAS), revealing the insertions and deletions and transcriptomic
markers contributing to melanoma drug resistance. Roth et al
(85) developed pooled knockin
sequencing (PoKI-seq), a technology that measures cell abundance
and state both ex vivo and in vivo. This method
facilitates the barcoding and tracking of targeted integrations of
large non-viral DNA templates in primary human T cells. The
technology notably identified a novel TGF-β R2-41BB chimeric
receptor, enhancing the clearance of solid tumors. PoKI-seq enables
the parallelized rewriting of endogenous genetic sequences,
accelerating the identification of effective knockin programs for
cell therapies. However, specific applications of single-cell
CRISPR technology in GC research have yet to be discovered.
Notable advancements have been made in integrating
single cell multiple-omics analyses. For instance, Trio-seq allows
for the simultaneous analysis of the genome sequence, epigenome and
transcriptome in a single cell (86). Another technique, cellular indexing
of transcriptomes and epitopes by sequencing, combines surface
protein analysis with transcriptome sequencing (87), while Single Cell Methylome and
Transcriptome Sequencing enables the simultaneous analysis of both
the epigenome (specifically DNA methylation patterns) and
transcriptome (gene expression profiles) at the single-cell level,
and combines DNA methylation analysis with transcriptome sequencing
within a single cell (88).
In conclusion, single-cell omics technologies,
encompassing SCS, single-cell proteomics and multi-omics have
undergone advancements, enabling researchers to explore the
intricate details of cellular heterogeneity across various omics
layers. These technologies offer insights into gene expression,
genetic variations, epigenomic modifications and protein expression
patterns at the single-cell level, enhancing the understanding of
cellular dynamics and disease mechanisms.
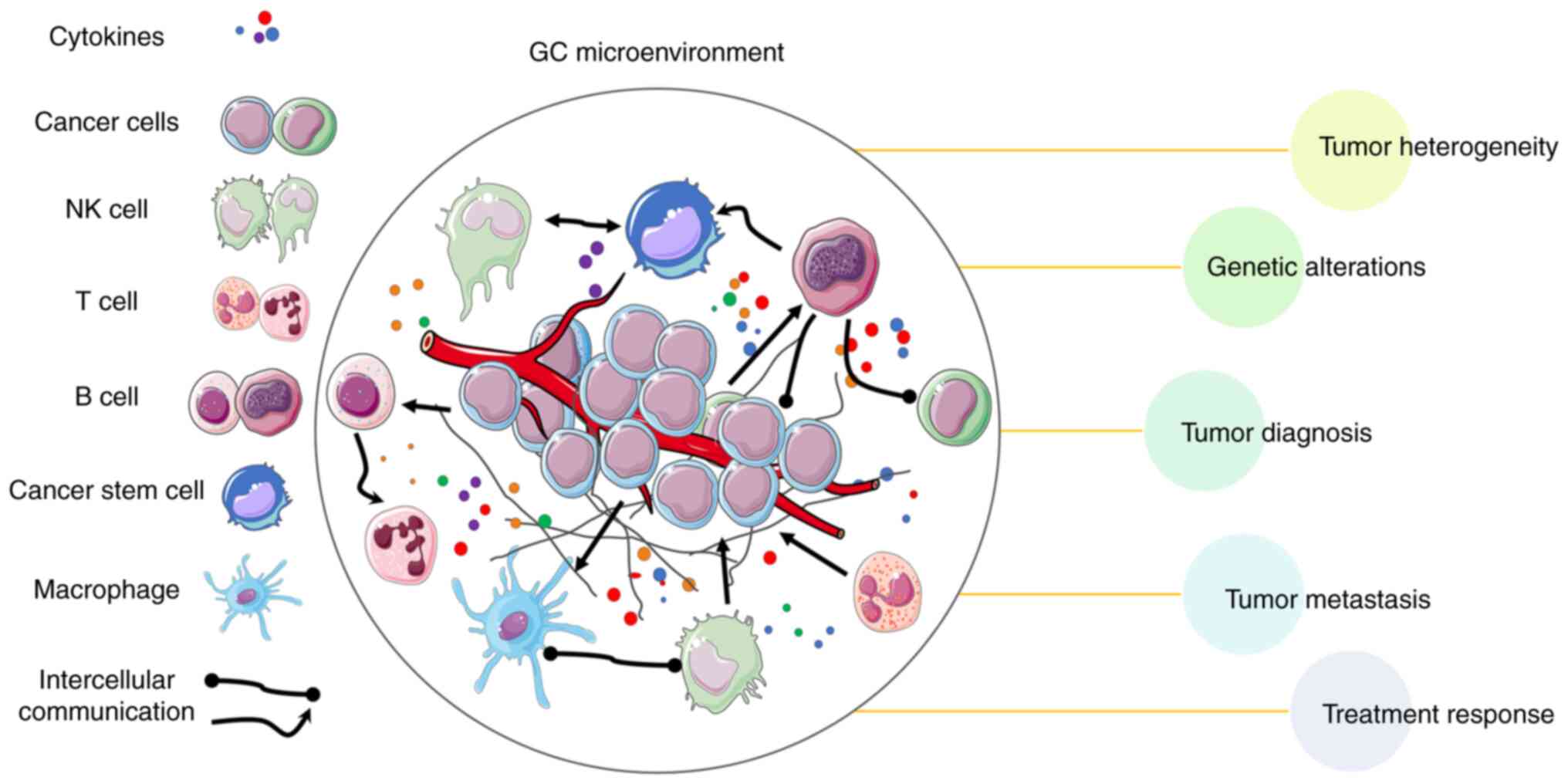
Single-cell omics technologies have transformed the
comprehension of GC by revealing cellular heterogeneity and
molecular landscapes. Numerous studies have used SCS methods to
investigate GC, providing insights into tumor heterogeneity,
metastasis, genetic alterations, diagnosis, treatment response and
the TME (47,89–92)
(Fig. 2). The present review
summarizes key findings of these studies, highlighting the
contributions of different SCS technologies in advancing the
knowledge of GC. Additionally, a summary of SCS technology
applications in GC is presented in Table II.
Early applications of SCS in GC focused on
transcriptome and single-cell genome analysis of GC cell lines and
reported marked genetic and transcriptional diversity (93). In one study, the identification of
24 notable mutated genes among tumor cells demonstrated the genetic
alterations underlying GC and potential therapeutic targets
(94). Analysis of circulating
tumor cells (CTCs) from patients with advanced GC demonstrated
numerous mutations in the genes associated with the KRAS and Rap1
pathways, as well as mutations in the genes associated with the
MET/PI3K/AKT pathway and the SMARCB1 gene in patients with large
multiploid CTCs (95), leading to
the development of resistance to either chemotherapy alone
(96) or chemotherapy combined with
targeted therapy (97) in patients
with GC.
In a study on GC lymph node metastasis, scRNA-seq
was performed on primary and metastatic tissues from 3 patients,
revealing intratumoural heterogeneity and distinct carcinoma
profiles. The results identified a subgroup of cells indicating a
transitional state in the metastasis process, and also revealed
potential marker genes (ERBB2, CLDN11 and CDK12) and genes driving
gastric cancer evolution (FOS and JUN), offering insights for GC
treatment (98). A panel of
biomarkers was identified for discriminating between benign and
malignant epithelial tissues, potentially aiding in early detection
and diagnosis of GC (99). Using
scRNA-seq analysis, subtypes of peritoneal carcinomatosis samples
from patients with GC were classified, and a 12-gene prognostic
signature was identified (100).
Investigation of metastatic GC using peritoneal ascite samples and
cerebrospinal fluid revealed that poor prognosis was associated
with M2-like characteristics in tumor-associated macrophages
(101). In a study of patients
with non-metastatic GC, immunosuppressive gene expression patterns
were enriched in regulatory T cells (Tregs) within gastric tumor
tissues, indicating an immunosuppressive TME. The absence of a
separate exhausted CD8+ T cell cluster and low
expression levels of exhaustion markers were also observed, and
ACKR1 was identified as a potential marker associated with poor
prognosis (102). Using scRNA-seq,
a broad spectrum of GC subtypes was assessed to create a
transcriptomic map of biomarkers from malignant epithelial cells
for the prediction of overall survival in patients with GC
(103). OR51E1 has been identified
as a key marker gene for unique endocrine cells in early-malignant
lesions of gastric cancer, offering a potential avenue for early
detection of malignancy (103).
Simultaneously, HES6 has been recognized for its potential utility
in identifying metaplasia at an early stage, demonstrating its
importance in the early diagnosis and intervention of precancerous
gastric conditions (104).
Furthermore, a panel of early GC-specific signatures was identified
using mucosa biopsies, which may be used in clinical applications
for early diagnosis (104).
Analysis of the TME in patients with GC revealed
increased stromal cells and Tregs, unique transcriptional cell
states in dendritic cells (DCs), exhausted cytotoxic T lymphocytes
and a specific extracellular matrix composition not found in normal
tissue (105). Furthermore, in
more advanced disease stages, a downregulation of interferon
regulatory factor 8 in CD8+ tumor-infiltrating
lymphocytes has been reported, revealing changes in the
immunological landscape in GC and its potential implications for
disease progression (106).
Another study demonstrated that monocyte-like DCs
and autophagy-related genes marking high-plasticity (ability of
certain cancer cells to adapt and change in response to different
environments or therapeutic pressures) GC were associated with poor
prognosis during GC peritoneal metastasis progression (107). In tumors with a high alternate
promoter burden (APB-high), characterized by increased use of
alternative gene promoters, distinct immunological populations were
observed along with a reduced proportion of T cells. These findings
shed light on the immunological aspects of GC and how the APB-high
status influences tumor progression and the immune response
(108).
Comparisons between GC and colorectal cancer
revealed distinct mutational landscapes and microbiomes,
contributing to differences in the TME, and thus, disease prognosis
(109). Furthermore, the
communication between cancer-associated fibroblasts (CAFs) within
the TME and other cells provides insights into their regulatory
functions (110).
Activated fibroblasts, endothelial cells,
immunosuppressive myeloid cell subsets and Tregs present in the TME
were associated with an unfavorable prognosis and resistance to
anti-programmed cell death 1 therapy in patients GC (91). Characteristics linked to a positive
response to platinum-based chemotherapy were defined, aiding
personalized treatment decisions. For example, response was
associated with on-treatment TME remodeling, including natural
killer cell recruitment, decreased tumor-associated macrophages,
M1-macrophage repolarization and increased effector T-cell
infiltration (91). In
non-responders to chemotherapy, Kim et al (111) observed low or no programmed
death-ligand 1 expression, an increase in Wnt signaling and B-cell
infiltration, a higher presence of lymphocyte activating
3-expressing T cells, and a reduction in dendritic cells. This
suggests a distinct pattern of immune changes associated with
chemotherapy resistance.
To assess the effects of combination therapy with
camrelizumab and 5-fluorouracil, leucovorin and oxaliplatin on GC
and its impact on the TME, Li et al (112) conducted single-cell RNA sequencing
on 10 GC samples both before and after neoadjuvant treatment. This
study highlighted that high expression of interferon-γ in
CD8+ T cells was associated with enhanced responses to
this combination therapy, indicating an immunological impact on the
tumor environment (112).
Additionally, a murine model suggested the potential therapeutic
approach of combining anti-IL-17 and anti-programmed death-1
monoclonal antibodies for GC tumor regression (113).
ST techniques have notably improved the
understanding of cellular function within multicellular organisms
by revealing the precise location of cells in tissue sections.
These techniques can be broadly categorized into two main types:
Imaging-based methods and sequencing-based methods (114). Imaging-based methods include in
situ hybridization (ISH) and in situ sequencing (ISS),
while sequencing-based methods include laser capture
microdissection (LCM) and in situ barcoding (ISB) (115). Table
III provides an overview of the key characteristics of ST
technologies.
ISB-based ST techniques provide transcriptome-wide
resolution at the cellular and subcellular levels, enabling
investigations into gene expression patterns within the tissue
context. Notable examples of ISB-based techniques include
NanoString Technologies, Inc. digital spatial profiling (DSP)
(137), High-Definition ST
(138), Visium (139), Stereo-seq (140) and Slide-seq (141). These techniques enable
simultaneous detection of multiple genes and offer valuable
insights into cellular spatial organization and tissue
heterogeneity (138,140,142,143).
By using ST techniques, researchers can gain a
comprehensive and detailed understanding of the spatial
distribution of gene expression within tissues. These advancements
have notable implications for various fields, including
developmental biology, disease research and regenerative
medicine.
Previous research has demonstrated the diverse
applications of ST technologies. These applications encompass in
situ cell typing (144,145),
spatial gene expression pattern acquisition (139), tumor trajectory mapping (146), exploration of tumor pathogenesis
(147–150), investigation of the TME (33,151–155) and prediction of disease prognosis
(156,157). In Table IV, a summary of ST technology
applications in GC is presented. For instance, Kumar et al
(90) pinpointed specific B-cell
sublineages exhibiting increased proportions in diffuse-type
gastric cancer and highlighted KLF12 expression in epithelial cells
as a potential driver of plasma cell recruitment. Furthermore, a
stepwise accumulation of CAF subpopulations characterized by high
co-expression of INHBA and FAP was identified.
Furthermore, immunohistochemistry (IHC) and duplex
ISH techniques were used to evaluate the distribution of major cell
types, and identified CCL2-expressing endothelial cells and
fibroblasts, thereby providing evidence of tumor invasion (158). Utilizing the NanoString
Technologies, Inc. ‘PanCancer Progression Panel’, Sundar et
al (159) conducted a
differential gene expression analysis and revealed that only 16% of
genes exhibited significant differences between primary tumor deep
(PTdeep) areas and corresponding LNmet samples. Notably, both LNmet
and PTdeep samples exhibited increased expression of several genes
with potential therapeutic significance, such as IGF1, PIK3CD and
TGFB1, compared with superficial primary tumors (159). In a separate study using 10×
Genomics Visium, Yamasaki et al (160) demonstrated the role of hypoxia
signaling in the metastatic progression of KRASG12V-expressing
gastric neoplasia-p53KO tumors, highlighting trametinib as a
promising therapeutic approach to curb metastasis in KRAS-mutated
GC. Furthermore, the application of DSP revealed upregulation of
genes related to triglyceride catabolism and endogenous sterols,
such as COL15A1, FABP2 and FABP4, particularly in cases positive
for stroma-reactive invasion front areas (161).
To summarize, the applications of ST technologies in
GC have yielded valuable insights into cell types, spatial gene
expression patterns, tumor invasion, metastatic progression and
potential therapeutic targets. These findings contribute to the
understanding of GC pathogenesis and open avenues for improved
treatment strategies.
scRNA-seq is a tool for identifying cell
subpopulations within tissues. However, it is unable to capture the
spatial arrangement of cells and the immediate networks of
intercellular communication in their native locations (41). ST technologies have not yet achieved
the same level of resolution as scRNA-seq in transcriptomic maps of
tissues (117). Therefore,
integrating both single-cell and ST data can provide a
comprehensive understanding of cell-type distribution and the
potential mechanisms of intercellular communication underlying
tissue architecture (41).
In summary, integrating single-cell and ST data
holds great potential for unraveling the complex architecture of
tissues and understanding intercellular communication networks. The
aforementioned studies (90,110,158,159)
provide valuable insights into the cellular heterogeneity, immune
response and spatial organization of GC, which contributes to the
knowledge of GC and demonstrates future research and therapeutic
strategies.
GC is an invasive disease associated with high
morbidity and characterized by notable heterogeneity. Single-cell
omics technologies and analytical tools have been identified as
resources for elucidating the complexity of the TME, and intra- and
intertumoral heterogeneity. In the present review, an overview of
current single-cell omics technologies and their applications in GC
research was provided. Discussing the rapidly advancing field of ST
and understanding the spatial organization of tumors is crucial for
evaluating tumorigenesis and disease progression, and how it can be
used with single-cell omics to gain deeper insights into the
characteristics of GC. This combined approach provides a method to
construct spatial histology information and assess the spatial
structure of tumors. By integrating these complementary approaches,
previously unknown mechanisms of tumor heterogeneity can be
assessed. This integrative effort holds great promise for defining
disease subtypes, predicting prognosis and enabling targeted
therapies to be delivered, based on the spatial distribution of
specific cell subtypes. It also allows the identification of
ligands and receptors involved in their mechanism of action.
However, SCS and ST research on GC has not yet been applied to the
clinical practice of treating GC. Ultimately, this comprehensive
approach will further the understanding of tumorigenesis and allow
the development of novel techniques for precision therapy in the
future.
In cancer research, the combination of
high-resolution data from single-cell omics and ST with the
analytical capabilities of artificial intelligence (AI) offers
insights into cellular heterogeneity and intercellular interactions
(25,164,165). AI, especially deep learning,
efficiently processes vast and complex data, automatically
identifying cell states and subgroups (166). This integration not only allows
the combination of results from a number of studies and data
classification, but also reveals intratumoral cell communication
and interactions, aiding in predicting tumor progression pathways,
identifying novel drug targets and providing decision support for
precision therapy and personalized strategies (167). This interdisciplinary fusion
markedly advances the exploration of tumor complexity, accelerates
research progress and promotes therapeutic innovation, marking a
notable advancement in the field of cancer medicine (168,169).
Not applicable.
The present study was supported by the Natural Science
Foundation of Sichuan Province (grant nos. 2022NSFSC1610,
2023NSFSC0678 and 2023NSFSC0569).
Not applicable.
Visualization of the data was performed by LR, LN,
PC and YiZ. Writing of the original draft of the manuscript was
performed by LR and DH. Reviewing and editing of the manuscript was
performed by LR, DH, HoL, LN, PC, XY, YaZ, HaL, JS, NL and YiZ, and
validation of the manuscript and supervision was provided by HaL,
NL and YiZ. Data authentication is not applicable. All authors read
and approved the final version of the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Machlowska J, Baj J, Sitarz M, Maciejewski
R and Sitarz R: Gastric Cancer: Epidemiology, risk factors,
classification, genomic characteristics and treatment strategies.
Int J Mol Sci. 21:40122020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wong MCS, Huang J, Chan PSF, Choi P, Lao
XQ, Chan SM, Teoh A and Liang P: Global incidence and mortality of
gastric cancer, 1980–2018. JAMA Netw Open. 4:e21184572021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
GBD 2017 Stomach Cancer Collaborators, .
The global, regional, and national burden of stomach cancer in 195
countries, 1990–2017: A systematic analysis for the Global Burden
of Disease study 2017. Lancet Gastroenterol Hepatol. 5:42–54. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bang YJ, Van Cutsem E, Feyereislova A,
Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T,
et al: Trastuzumab in combination with chemotherapy versus
chemotherapy alone for treatment of HER2-positive advanced gastric
or gastro-oesophageal junction cancer (ToGA): A phase 3,
open-label, randomised controlled trial. Lancet. 376:687–697. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cunningham D, Starling N, Rao S, Iveson T,
Nicolson M, Coxon F, Middleton G, Daniel F, Oates J and Norman AR;
Upper Gastrointestinal Clinical Studies Group of the National
Cancer Research Institute of the United Kingdom, : Capecitabine and
oxaliplatin for advanced esophagogastric cancer. N Engl J Med.
358:36–46. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Koizumi W, Narahara H, Hara T, Takagane A,
Akiya T, Takagi M, Miyashita K, Nishizaki T, Kobayashi O, Takiyama
W, et al: S-1 plus cisplatin versus S-1 alone for first-line
treatment of advanced gastric cancer (SPIRITS trial): A phase III
trial. Lancet. Oncol. 9:215–221. 2008.
|
|
8
|
Wilke H, Muro K, Van Cutsem E, Oh SC,
Bodoky G, Shimada Y, Hironaka S, Sugimoto N, Lipatov O, Kim TY, et
al: Ramucirumab plus paclitaxel versus placebo plus paclitaxel in
patients with previously treated advanced gastric or
gastro-oesophageal junction adenocarcinoma (RAINBOW): A
double-blind, randomised phase 3 trial. Lancet Oncol. 15:1224–1235.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Salvatori S, Marafini I, Laudisi F,
Monteleone G and Stolfi C: Helicobacter pylori and Gastric
cancer: Pathogenetic mechanisms. Int J Mol Sci. 24:28952023.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Smyth EC, Nilsson M, Grabsch HI, van
Grieken NC and Lordick F: Gastric cancer. Lancet. 396:635–648.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Polk DB and Peek RM Jr: Helicobacter
pylori: Gastric cancer and beyond. Nat Rev Cancer. 10:403–414.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Van Cutsem E, Sagaert X, Topal B,
Haustermans K and Prenen H: Gastric cancer. Lancet. 388:2654–2664.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Onoyama T, Ishikawa S and Isomoto H:
Gastric cancer and genomics: Review of literature. J Gastroenterol.
57:505–516. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li GZ, Doherty GM and Wang J: Surgical
management of gastric cancer: A review. JAMA Surg. 157:446–454.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu Z, Shi L, Dong Y, Zhang Y, Yang F, Wei
J, Huo M, Li P and Liu X: Effect of crosstalk among conspirators in
tumor microenvironment on niche metastasis of gastric cancer. Am J
Cancer Res. 12:5375–5402. 2022.PubMed/NCBI
|
|
16
|
Tang F, Barbacioru C, Wang Y, Nordman E,
Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al: mRNA-Seq
whole-transcriptome analysis of a single cell. Nat Methods.
6:377–382. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ou Z, Lin S, Qiu J, Ding W, Ren P, Chen D,
Wang J, Tong Y, Wu D, Chen A, et al: Single-nucleus RNA sequencing
and spatial transcriptomics reveal the immunological
microenvironment of cervical squamous cell carcinoma. Adv Sci
(Weinh). 9:e22030402022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun D, Guan X, Moran AE, Wu LY, Qian DZ,
Schedin P, Dai MS, Danilov AV, Alumkal JJ, Adey AC, et al:
Identifying phenotype-associated subpopulations by integrating bulk
and single-cell sequencing data. Nat Biotechnol. 40:527–538. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Casado-Pelaez M, Bueno-Costa A and
Esteller M: Single cell cancer epigenetics. Trends Cancer.
8:820–838. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hu W, Zeng H, Shi Y, Zhou C, Huang J, Jia
L, Xu S, Feng X, Zeng Y, Xiong T, et al: Single-cell transcriptome
and translatome dual-omics reveals potential mechanisms of human
oocyte maturation. Nat Commun. 13:51142022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ye J, Yang C, Xia L, Zhu Y, Liu L, Cao H
and Tao Y: Protoplast preparation for algal single-cell omics
sequencing. Microorganisms. 11:5382023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Y, Liu T, Hu X, Wang M, Wang J, Zou
B, Tan P, Cui T, Dou Y, Ning L, et al: CellCall: Integrating paired
ligand-receptor and transcription factor activities for cell-cell
communication. Nucleic Acids Res. 49:8520–8534. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kashima Y, Sakamoto Y, Kaneko K, Seki M,
Suzuki Y and Suzuki A: Single-cell sequencing techniques from
individual to multiomics analyses. Exp Mol Med. 52:1419–1427. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang Y, Wang J, Zhao Y, Wang H, Liu T, Li
Y, Cui T, Li W, Feng Y, Luo J, et al: cncRNAdb: A manually curated
resource of experimentally supported RNAs with both protein-coding
and noncoding function. Nucleic Acids Res. 49:D65–D70. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang YF, Wang YH, Gu ZF, Pan XR, Li J,
Ding H, Zhang Y and Deng KJ: Bitter-RF: A random forest machine
model for recognizing bitter peptides. Front Med (Lausanne).
10:10529232023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tan Z, Kan C, Sun M, Yang F, Wong M, Wang
S and Zheng H: Mapping breast cancer microenvironment through
single-cell omics. Front Immunol. 13:8688132022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gao B, Jiang B, Xing W, Xie Z, Luo Z and
Zou W: Discovery and application of postnatal nucleus pulposus
progenitors essential for intervertebral disc homeostasis and
degeneration. Adv Sci (Weinh). 9:e21048882022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Moline DC, Zenner ML, Burr A, Vellky JE,
Nonn L and Vander Griend DJ: Single-cell RNA-Seq identifies factors
necessary for the regenerative phenotype of prostate luminal
epithelial progenitors. Am J Clin Exp Urol. 10:425–439.
2022.PubMed/NCBI
|
|
29
|
Chen S, An G, Wang H, Wu X, Ping P, Hu L,
Chen Y, Fan J, Cheng CY and Sun F: Human obstructive
(postvasectomy) and nonobstructive azoospermia-Insights from
scRNA-Seq and transcriptome analysis. Genes Dis. 9:766–776. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tanemoto S, Sujino T, Miyamoto K, Moody J,
Yoshimatsu Y, Ando Y, Koya I, Harada Y, Tojo AO, Ono K, et al:
Single-cell transcriptomics of human gut T cells identifies
cytotoxic CD4+CD8A+ T cells related to mouse
CD4 cytotoxic T cells. Front Immunol. 13:9771172022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ning L, Abagna HB, Jiang Q, Liu S and
Huang J: Development and application of therapeutic antibodies
against COVID-19. Int J Biol Sci. 17:1486–1496. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ning L, Liu M, Gou Y, Yang Y, He B and
Huang J: Development and application of ribonucleic acid therapy
strategies against COVID-19. Int J Biol Sci. 18:5070–5085. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhang Y, Pan X, Shi T, Gu Z, Yang Z, Liu
M, Xu Y, Yang Y, Ren L, Song X, et al: P450Rdb: A manually curated
database of reactions catalyzed by cytochrome P450 enzymes. J Adv
Res. Oct 21–2023.doi: 10.1016/j.jare.2023.10.012 (Epub ahead of
print). View Article : Google Scholar
|
|
34
|
Williams CG, Lee HJ, Asatsuma T,
Vento-Tormo R and Haque A: An introduction to spatial
transcriptomics for biomedical research. Genome Med. 14:682022.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Anderson AC, Yanai I, Yates LR, Wang L,
Swarbrick A, Sorger P, Santagata S, Fridman WH, Gao Q, Jerby L, et
al: Spatial transcriptomics. Cancer Cell. 40:895–900. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang L, Chen D, Song D, Liu X, Zhang Y,
Xu X and Wang X: Clinical and translational values of spatial
transcriptomics. Signal Transduct Target Ther. 7:1112022.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Larsson L, Bergenstråhle L, He M,
Andrusivova Z and Lundeberg J: SnapShot: Spatial transcriptomics.
Cell. 185:2840–2840.e1. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang Y, Liu T, Wang J, Zou B, Li L, Yao
L, Chen K, Ning L, Wu B, Zhao X and Wang D: Cellinker: A platform
of ligand-receptor interactions for intercellular communication
analysis. Bioinformatics: btab036. 2021.doi:
10.1093/bioinformatics/btab036 (Epub ahead of print).
|
|
39
|
Ren L, Ning L, Yang Y, Yang T, Li X, Tan
S, Ge P, Li S, Luo N, Tao P and Zhang Y: MetaboliteCOVID: A
manually curated database of metabolite markers for COVID-19.
Comput Biol Med. 167:1076612023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ahmed R, Zaman T, Chowdhury F, Mraiche F,
Tariq M, Ahmad IS and Hasan A: Single-Cell RNA sequencing with
spatial transcriptomics of cancer tissues. Int J Mol Sci.
23:30422022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Longo SK, Guo MG, Ji AL and Khavari PA:
Integrating single-cell and spatial transcriptomics to elucidate
intercellular tissue dynamics. Nat Rev Genet. 22:627–644. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kijima Y, Evans-Yamamoto D, Toyoshima H
and Yachie N: A universal sequencing read interpreter. Sci Adv.
9:eadd27932023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ren L, Xu Y, Ning L, Pan X, Li Y, Zhao Q,
Pang B, Huang J, Deng K and Zhang Y: TCM2COVID: A resource of
anti-COVID-19 traditional Chinese medicine with effects and
mechanisms. Imeta. e422022.doi: 10.1002/imt2.42 (Epub ahead of
print). View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang Y, Liu C, Liu M, Liu T, Lin H, Huang
CB and Ning L: Attention is all you need: Utilizing attention in
AI-enabled drug discovery. Brief Bioinform. 25:bbad4672023.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ren L, Pan X, Ning L, Gong D, Huang J,
Deng K, Xie L and Zhang Y: Construction of a combined
hypoxia-related genes model for hepatocellular carcinoma prognosis.
Curr Comput Aided Drug Des. 19:150–161. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Navin N, Kendall J, Troge J, Andrews P,
Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, et
al: Tumour evolution inferred by single-cell sequencing. Nature.
472:90–94. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xie Z, Li J, Huang P, Zhang Y, Yang J, Liu
K and Jiang Y: Applications and achievements of single-cell
sequencing in gastrointestinal cancer. Front Oncol. 12:9055712022.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Picelli S, Björklund Å K, Faridani OR,
Sagasser S, Winberg G and Sandberg R: Smart-seq2 for sensitive
full-length transcriptome profiling in single cells. Nature
Methods. 10:1096–1098. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zheng GX, Terry JM, Belgrader P, Ryvkin P,
Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et
al: Massively parallel digital transcriptional profiling of single
cells. Nat Commun. 8:140492017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liang L, Yu J, Li J, Li N, Liu J, Xiu L,
Zeng J, Wang T and Wu L: Integration of scRNA-Seq and bulk RNA-Seq
to analyse the heterogeneity of ovarian cancer immune cells and
establish a molecular risk model. Front Oncol. 11:7110202021.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lohr JG, Adalsteinsson VA, Cibulskis K,
Choudhury AD, Rosenberg M, Cruz-Gordillo P, Francis JM, Zhang CZ,
Shalek AK, Satija R, et al: Whole-exome sequencing of circulating
tumor cells provides a window into metastatic prostate cancer. Nat
Biotechnol. 32:479–484. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Hashimshony T, Wagner F, Sher N and Yanai
I: CEL-Seq: Single-cell RNA-Seq by multiplexed linear
amplification. Cell Rep. 2:666–673. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Smallwood SA, Lee HJ, Angermueller C,
Krueger F, Saadeh H, Peat J, Andrews SR, Stegle O, Reik W and
Kelsey G: Single-cell genome-wide bisulfite sequencing for
assessing epigenetic heterogeneity. Nat Methods. 11:817–820. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Shalek AK, Satija R, Adiconis X, Gertner
RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C,
Lu D, et al: Single-cell transcriptomics reveals bimodality in
expression and splicing in immune cells. Nature. 498:236–240. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Shalek AK, Satija R, Shuga J, Trombetta
JJ, Gennert D, Lu D, Chen P, Gertner RS, Gaublomme JT, Yosef N, et
al: Single-cell RNA-seq reveals dynamic paracrine control of
cellular variation. Nature. 510:363–369. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Tirosh I, Izar B, Prakadan SM, Wadsworth
MH II, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G,
et al: Dissecting the multicellular ecosystem of metastatic
melanoma by single-cell RNA-seq. Science. 352:189–196. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Buenrostro JD, Wu B, Litzenburger UM, Ruff
D, Gonzales ML, Snyder MP, Chang HY and Greenleaf WJ: Single-cell
chromatin accessibility reveals principles of regulatory variation.
Nature. 523:486–490. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zeisel A, Muñoz-Manchado AB, Codeluppi S,
Lönnerberg P, La Manno G, Juréus A, Marques S, Munguba H, He L,
Betsholtz C, et al: Brain structure. Cell types in the mouse cortex
and hippocampus revealed by single-cell RNA-seq. Science.
347:1138–1142. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Treutlein B, Brownfield DG, Wu AR, Neff
NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA and Quake SR:
Reconstructing lineage hierarchies of the distal lung epithelium
using single-cell RNA-seq. Nature. 509:371–375. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gierahn TM, Wadsworth MH II, Hughes TK,
Bryson BD, Butler A, Satija R, Fortune S, Love JC and Shalek AK:
Seq-Well: Portable, low-cost RNA sequencing of single cells at high
throughput. Nat Methods. 14:395–398. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Rosenberg AB, Roco CM, Muscat RA, Kuchina
A, Sample P, Yao Z, Graybuck LT, Peeler DJ, Mukherjee S, Chen W, et
al: Single-cell profiling of the developing mouse brain and spinal
cord with split-pool barcoding. Science. 360:176–182. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Keren-Shaul H, Kenigsberg E, Jaitin DA,
David E, Paul F, Tanay A and Amit I: MARS-seq2.0: An experimental
and analytical pipeline for indexed sorting combined with
single-cell RNA sequencing. Nat Protoc. 14:1841–1862. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kouno T, Moody J, Kwon AT, Shibayama Y,
Kato S, Huang Y, Böttcher M, Motakis E, Mendez M, Severin J, et al:
C1 CAGE detects transcription start sites and enhancer activity at
single-cell resolution. Nat Commun. 10:3602019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lyu T, Lin Y, Wu K, Cao Z, Zhang Q and
Zheng J: Single-cell sequencing technologies in bladder cancer
research: Applications and challenges. Front Genet. 13:10279092022.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Dean FB, Nelson JR, Giesler TL and Lasken
RS: Rapid amplification of plasmid and phage DNA using Phi 29 DNA
polymerase and multiply-primed rolling circle amplification. Genome
Res. 11:1095–1099. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Zong C, Lu S, Chapman AR and Xie XS:
Genome-wide detection of single-nucleotide and copy-number
variations of a single human cell. Science. 338:1622–1626. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Chen C, Xing D, Tan L, Li H, Zhou G, Huang
L and Xie XS: Single-cell whole-genome analyses by Linear
Amplification via Transposon Insertion (LIANTI). Science.
356:189–194. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Xing D, Tan L, Chang CH, Li H and Xie XS:
Accurate SNV detection in single cells by transposon-based
whole-genome amplification of complementary strands. Proc Natl Acad
Sci USA. 118:e20131061182021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Weichenhan D, Lipka DB, Lutsik P, Goyal A
and Plass C: Epigenomic technologies for precision oncology. Semin
Cancer Biol. 84:60–68. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Clark SJ, Lee HJ, Smallwood SA, Kelsey G
and Reik W: Single-cell epigenomics: Powerful new methods for
understanding gene regulation and cell identity. Genome Biol.
17:722016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Guo H, Zhu P, Wu X, Li X, Wen L and Tang
F: Single-cell methylome landscapes of mouse embryonic stem cells
and early embryos analyzed using reduced representation bisulfite
sequencing. Genome Res. 23:2126–2135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Johnson DS, Mortazavi A, Myers RM and Wold
B: Genome-wide mapping of in vivo protein-DNA interactions.
Science. 316:1497–1502. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Park PJ: ChIP-seq: Advantages and
challenges of a maturing technology. Nat Rev Genet. 10:669–680.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Tajik M, Baharfar M and Donald WA:
Single-cell mass spectrometry. Trends Biotechnol. 40:1374–1392.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Iyer A, Hamers AAJ and Pillai AB:
CyTOF(®) for the Masses. Front Immunol. 13:8158282022.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Budnik B, Levy E, Harmange G and Slavov N:
SCoPE-MS: Mass spectrometry of single mammalian cells quantifies
proteome heterogeneity during cell differentiation. Genome Biol.
19:1612018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Specht H, Emmott E, Petelski AA, Huffman
RG, Perlman DH, Serra M, Kharchenko P, Koller A and Slavov N:
Single-cell proteomic and transcriptomic analysis of macrophage
heterogeneity using SCoPE2. Genome Biol. 22:502021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bock C, Datlinger P, Chardon F, Coelho MA,
Dong MB, Lawson KA, Lu T, Maroc L, Norman TM, Song B, et al:
High-content CRISPR screening. Nat Rev Methods Primers. 2:92022.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Adamson B, Norman TM, Jost M, Cho MY,
Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et
al: A multiplexed Single-Cell CRISPR screening platform enables
systematic dissection of the unfolded protein response. Cell.
167:1867–1882.e21. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Jaitin DA, Weiner A, Yofe I, Lara-Astiaso
D, Keren-Shaul H, David E, Salame TM, Tanay A, van Oudenaarden A
and Amit I: Dissecting immune circuits by linking CRISPR-Pooled
screens with Single-Cell RNA-Seq. Cell. 167:1883–1896.e15. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Datlinger P, Rendeiro AF, Schmidl C,
Krausgruber T, Traxler P, Klughammer J, Schuster LC, Kuchler A,
Alpar D and Bock C: Pooled CRISPR screening with single-cell
transcriptome readout. Nat Methods. 14:297–301. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Xie S, Duan J, Li B, Zhou P and Hon GC:
Multiplexed engineering and analysis of combinatorial enhancer
activity in single cells. Mol Cell. 66:285–299.e5. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Replogle JM, Norman TM, Xu A, Hussmann JA,
Chen J, Cogan JZ, Meer EJ, Terry JM, Riordan DP, Srinivas N, et al:
Combinatorial single-cell CRISPR screens by direct guide RNA
capture and targeted sequencing. Nat Biotechnol. 38:954–961. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Jun S, Lim H, Chun H, Lee JH and Bang D:
Single-cell analysis of a mutant library generated using
CRISPR-guided deaminase in human melanoma cells. Commu Biol.
3:1542020. View Article : Google Scholar
|
|
85
|
Roth TL, Li PJ, Blaeschke F, Nies JF,
Apathy R, Mowery C, Yu R, Nguyen MLT, Lee Y, Truong A, et al:
Pooled knockin targeting for genome engineering of cellular
immunotherapies. Cell. 181:728–744.e21. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hou Y, Guo H, Cao C, Li X, Hu B, Zhu P, Wu
X, Wen L, Tang F, Huang Y and Peng J: Single-cell triple omics
sequencing reveals genetic, epigenetic, and transcriptomic
heterogeneity in hepatocellular carcinomas. Cell Res. 26:304–319.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Stoeckius M, Hafemeister C, Stephenson W,
Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R and Smibert
P: Simultaneous epitope and transcriptome measurement in single
cells. Nat Methods. 14:865–868. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Angermueller C, Clark SJ, Lee HJ, Macaulay
IC, Teng MJ, Hu TX, Krueger F, Smallwood S, Ponting CP, Voet T, et
al: Parallel single-cell sequencing links transcriptional and
epigenetic heterogeneity. Nat Methods. 13:229–232. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Deng G, Zhang X, Chen Y, Liang S, Liu S,
Yu Z and Lü M: Single-cell transcriptome sequencing reveals
heterogeneity of gastric cancer: Progress and prospects. Front
Oncol. 13:10742682023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kumar V, Ramnarayanan K, Sundar R,
Padmanabhan N, Srivastava S, Koiwa M, Yasuda T, Koh V, Huang KK,
Tay ST, et al: Single-Cell atlas of lineage states, tumor
microenvironment, and subtype-specific expression programs in
gastric cancer. Cancer Discov. 12:670–691. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Kang B, Camps J, Fan B, Jiang H, Ibrahim
MM, Hu X, Qin S, Kirchhoff D, Chiang DY, Wang S, et al: Parallel
single-cell and bulk transcriptome analyses reveal key features of
the gastric tumor microenvironment. Genome Biol. 23:2652022.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zulfiqar H, Guo Z, Ahmad RM, Ahmed Z, Cai
P, Chen X, Zhang Y, Lin H and Shi Z: Deep-STP: A deep
learning-based approach to predict snake toxin proteins by using
word embeddings. Front Med (Lausanne). 10:12913522024. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Andor N, Lau BT, Catalanotti C, Sathe A,
Kubit M, Chen J, Blaj C, Cherry A, Bangs CD, Grimes SM, et al:
Joint single cell DNA-seq and RNA-seq of gastric cancer cell lines
reveals rules of in vitro evolution. NAR Genom Bioinform.
2:lqaa0162020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Peng L, Xing R, Liu D, Bao L, Cheng W,
Wang H, Yu Y, Liu X, Jiang L, Wu Y, et al: Characterization and
validation of somatic mutation spectrum to reveal heterogeneity in
gastric cancer by single cell sequencing. Sci Bull (Beijing).
64:236–244. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Chen Y, Li Y, Qi C, Zhang C, Liu D, Deng
Y, Fu Y, Khadka VS, Wang DD, Tan S, et al: Dysregulated KRAS
gene-signaling axis and abnormal chromatin remodeling drive
therapeutic resistance in heterogeneous-sized circulating tumor
cells in gastric cancer patients. Cancer Lett. 517:78–87. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Li Y, Zhang X, Ge S, Gong J, Lu M, Zhang
Q, Cao Y, Wang DD, Lin PP and Shen L: Clinical significance of
phenotyping and karyotyping of circulating tumor cells in patients
with advanced gastric cancer. Oncotarget. 5:6594–6602. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Li Y, Zhang X, Liu D, Gong J, Wang DD, Li
S, Peng Z, Li Y, Wang X, Lin PP, et al: Evolutionary expression of
HER2 conferred by chromosome aneuploidy on circulating gastric
cancer cells contributes to developing targeted and
chemotherapeutic resistance. Clin Cancer Res. 24:5261–5271. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Wang B, Zhang Y, Qing T, Xing K, Li J,
Zhen T, Zhu S and Zhan X: Comprehensive analysis of metastatic
gastric cancer tumour cells using single-cell RNA-seq. Sci Rep.
11:11412021. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zhang M, Hu S, Min M, Ni Y, Lu Z, Sun X,
Wu J, Liu B, Ying X and Liu Y: Dissecting transcriptional
heterogeneity in primary gastric adenocarcinoma by single cell RNA
sequencing. Gut. 70:464–475. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Wang R, Dang M, Harada K, Han G, Wang F,
Pool Pizzi M, Zhao M, Tatlonghari G, Zhang S, Hao D, et al:
Single-cell dissection of intratumoral heterogeneity and lineage
diversity in metastatic gastric adenocarcinoma. Nat Med.
27:141–151. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Eum HH, Kwon M, Ryu D, Jo A, Chung W, Kim
N, Hong Y, Son DS, Kim ST, Lee J, et al: Tumor-promoting
macrophages prevail in malignant ascites of advanced gastric
cancer. Exp Mol Med. 52:1976–1988. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Li Y, Hu X, Lin R, Zhou G, Zhao L, Zhao D,
Zhang Y, Li W, Zhang Y, Ma P, et al: Single-cell landscape reveals
active cell subtypes and their interaction in the tumor
microenvironment of gastric cancer. Theranostics. 12:3818–3833.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Huang Z, Wu C, Liu X, Lu S, You L, Guo F,
Stalin A, Zhang J, Zhang F, Wu Z, et al: Single-Cell and bulk RNA
sequencing reveal malignant epithelial cell heterogeneity and
prognosis signatures in gastric carcinoma. Cells. 11:25502022.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhang P, Yang M, Zhang Y, Xiao S, Lai X,
Tan A, Du S and Li S: Dissecting the Single-cell transcriptome
network underlying gastric premalignant lesions and early gastric
cancer. Cell Rep. 27:1934–1947.e5. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Sathe A, Grimes SM, Lau BT, Chen J, Suarez
C, Huang RJ, Poultsides G and Ji HP: Single-cell genomic
characterization reveals the cellular reprogramming of the gastric
tumor microenvironment. Clin Cancer Res. 26:2640–2653. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Fu K, Hui B, Wang Q, Lu C, Shi W, Zhang Z,
Rong D, Zhang B, Tian Z, Tang W, et al: Single-cell RNA sequencing
of immune cells in gastric cancer patients. Aging (Albany NY).
12:2747–2763. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Huang XZ, Pang MJ, Li JY, Chen HY, Sun JX,
Song YX, Ni HJ, Ye SY, Bai S, Li TH, et al: Single-cell sequencing
of ascites fluid illustrates heterogeneity and therapy-induced
evolution during gastric cancer peritoneal metastasis. Nat Commun.
14:8222023. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Sundar R, Huang KK, Kumar V, Ramnarayanan
K, Demircioglu D, Her Z, Ong X, Bin Adam Isa ZF, Xing M, Tan AL, et
al: Epigenetic promoter alterations in GI tumour immune-editing and
resistance to immune checkpoint inhibition. Gut. 71:1277–1288.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Yang W, Zhao Y, Ge Q, Wang X, Jing Y, Zhao
J, Liu G, Huang H, Cheng F, Wang X, et al: Genetic mutation and
tumor microbiota determine heterogenicity of tumor immune
signature: Evidence from gastric and colorectal synchronous
cancers. Front Immunol. 13:9470802022. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Li X, Sun Z, Peng G, Xiao Y, Guo J, Wu B,
Li X, Zhou W, Li J, Li Z, et al: Single-cell RNA sequencing reveals
a pro-invasive cancer-associated fibroblast subgroup associated
with poor clinical outcomes in patients with gastric cancer.
Theranostics. 12:620–638. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Kim R, An M, Lee H, Mehta A, Heo YJ, Kim
KM, Lee SY, Moon J, Kim ST, Min BH, et al: Early tumor-immune
microenvironmental remodeling and response to first-line
fluoropyrimidine and platinum chemotherapy in advanced gastric
cancer. Cancer Discov. 12:984–1001. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Li S, Li K, Tian F, Li H, Xia Q, Li T,
Dong B, Li D, Yu J, Zhang J, et al: A high interferon gamma
signature of CD8+ T cells predicts response to
neoadjuvant immunotherapy plus chemotherapy in gastric cancer.
Front Immunol. 13:10561442022. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Nagaoka K, Shirai M, Taniguchi K, Hosoi A,
Sun C, Kobayashi Y, Maejima K, Fujita M, Nakagawa H, Nomura S and
Kakimi K: Deep immunophenotyping at the single-cell level
identifies a combination of anti-IL-17 and checkpoint blockade as
an effective treatment in a preclinical model of data-guided
personalized immunotherapy. J Immunother Cancer. 8:e0013582020.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Rao A, Barkley D, França GS and Yanai I:
Exploring tissue architecture using spatial transcriptomics.
Nature. 596:211–220. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Yu Q, Jiang M and Wu L: Spatial
transcriptomics technology in cancer research. Front Oncol.
12:10191112022. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Femino AM, Fay FS, Fogarty K and Singer
RH: Visualization of single RNA transcripts in situ. Science.
280:585–590. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Asp M, Bergenstråhle J and Lundeberg J:
Spatially resolved transcriptomes-next generation tools for tissue
exploration. Bioessays. 42:e19002212020. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Shah S, Lubeck E, Zhou W and Cai L: In
situ transcription profiling of single cells reveals spatial
organization of cells in the mouse hippocampus. Neuron. 92:342–357.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Eng CL, Lawson M, Zhu Q, Dries R, Koulena
N, Takei Y, Yun J, Cronin C, Karp C, Yuan GC and Cai L:
Transcriptome-scale super-resolved imaging in tissues by RNA
seqFISH. Nature. 568:235–239. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Xia C, Fan J, Emanuel G, Hao J and Zhuang
X: Spatial transcriptome profiling by MERFISH reveals subcellular
RNA compartmentalization and cell cycle-dependent gene expression.
Proc Natl Acad Sci USA. 116:19490–19499. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Wang G, Moffitt JR and Zhuang X:
Multiplexed imaging of high-density libraries of RNAs with MERFISH
and expansion microscopy. Sci Rep. 8:48472018. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Wu C, Simonetti M, Rossell C, Mignardi M,
Mirzazadeh R, Annaratone L, Marchiò C, Sapino A, Bienko M, Crosetto
N and Nilsson M: RollFISH achieves robust quantification of
single-molecule RNA biomarkers in paraffin-embedded tumor tissue
samples. Commun Biol. 1:2092018. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Dar D, Dar N, Cai L and Newman DK: Spatial
transcriptomics of planktonic and sessile bacterial populations at
single-cell resolution. Science. 373:eabi48822021. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Goh JJL, Chou N, Seow WY, Ha N, Cheng CPP,
Chang YC, Zhao ZW and Chen KH: Highly specific multiplexed RNA
imaging in tissues with split-FISH. Nat Methods. 17:689–693. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Lee JH, Daugharthy ER, Scheiman J, Kalhor
R, Yang JL, Ferrante TC, Terry R, Jeanty SS, Li C, Amamoto R, et
al: Highly multiplexed subcellular RNA sequencing in situ. Science.
343:1360–1363. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Alon S, Goodwin DR, Sinha A, Wassie AT,
Chen F, Daugharthy ER, Bando Y, Kajita A, Xue AG, Marrett K, et al:
Expansion sequencing: Spatially precise in situ transcriptomics in
intact biological systems. Science. 371:eaax26562021. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Chen X, Sun YC, Church GM, Lee JH and
Zador AM: Efficient in situ barcode sequencing using padlock
probe-based BaristaSeq. Nucleic Acids Res. 46:e222018. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Wang X, Allen WE, Wright MA, Sylwestrak
EL, Samusik N, Vesuna S, Evans K, Liu C, Ramakrishnan C, Liu J, et
al: Three-dimensional intact-tissue sequencing of single-cell
transcriptional states. Science. 361:eaat56912018. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Wang Y, Liu B, Zhao G, Lee Y, Buzdin A, Mu
X, Zhao J, Chen H and Li X: Spatial transcriptomics: Technologies,
applications and experimental considerations. Genomics.
115:1106712023. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Elhanani O, Ben-Uri R and Keren L: Spatial
profiling technologies illuminate the tumor microenvironment.
Cancer Cell. 41:404–420. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Nichterwitz S, Chen G, Aguila Benitez J,
Yilmaz M, Storvall H, Cao M, Sandberg R, Deng Q and Hedlund E:
Laser capture microscopy coupled with Smart-seq2 for precise
spatial transcriptomic profiling. Nat Commun. 7:121392016.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Medaglia C, Giladi A, Stoler-Barak L, De
Giovanni M, Salame TM, Biram A, David E, Li H, Iannacone M, Shulman
Z and Amit I: Spatial reconstruction of immune niches by combining
photoactivatable reporters and scRNA-seq. Science. 358:1622–1626.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Junker JP, Noël ES, Guryev V, Peterson KA,
Shah G, Huisken J, McMahon AP, Berezikov E, Bakkers J and van
Oudenaarden A: Genome-wide RNA Tomography in the zebrafish embryo.
Cell. 159:662–675. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Chen J, Suo S, Tam PP, Han JJ, Peng G and
Jing N: Spatial transcriptomic analysis of cryosectioned tissue
samples with Geo-seq. Nat Protoc. 12:566–580. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Emmert-Buck MR, Bonner RF, Smith PD,
Chuaqui RF, Zhuang Z, Goldstein SR, Weiss RA and Liotta LA: Laser
capture microdissection. Science. 274:998–1001. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Nichterwitz S, Benitez JA, Hoogstraaten R,
Deng Q and Hedlund E: LCM-Seq: A method for spatial transcriptomic
profiling using laser capture microdissection coupled with
PolyA-Based RNA sequencing. Methods Mol Biol. 1649:95–110. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Merritt CR, Ong GT, Church SE, Barker K,
Danaher P, Geiss G, Hoang M, Jung J, Liang Y, McKay-Fleisch J, et
al: Multiplex digital spatial profiling of proteins and RNA in
fixed tissue. Nat Biotechnol. 38:586–599. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Vickovic S, Eraslan G, Salmén F,
Klughammer J, Stenbeck L, Schapiro D, Äijö T, Bonneau R,
Bergenstråhle L, Navarro JF, et al: High-definition spatial
transcriptomics for in situ tissue profiling. Nat Methods.
16:987–990. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Ståhl PL, Salmén F, Vickovic S, Lundmark
A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss
M, et al: Visualization and analysis of gene expression in tissue
sections by spatial transcriptomics. Science. 353:78–82. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Chen A, Liao S, Cheng M, Ma K, Wu L, Lai
Y, Qiu X, Yang J, Xu J, Hao S, et al: Spatiotemporal transcriptomic
atlas of mouse organogenesis using DNA nanoball-patterned arrays.
Cell. 185:1777–1792.e1721. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Rodriques SG, Stickels RR, Goeva A, Martin
CA, Murray E, Vanderburg CR, Welch J, Chen LM, Chen F and Macosko
EZ: Slide-seq: A scalable technology for measuring genome-wide
expression at high spatial resolution. Science. 363:1463–1467.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Cho CS, Xi J, Si Y, Park SR, Hsu JE, Kim
M, Jun G, Kang HM and Lee JH: Microscopic examination of spatial
transcriptome using Seq-scope. Cell. 184:3559–3572.e22. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Fazal FM, Han S, Parker KR, Kaewsapsak P,
Xu J, Boettiger AN, Chang HY and Ting AY: Atlas of subcellular RNA
localization revealed by APEX-Seq. Cell. 178:473–490.e26. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Wu SZ, Al-Eryani G, Roden DL, Junankar S,
Harvey K, Andersson A, Thennavan A, Wang C, Torpy JR, Bartonicek N,
et al: A single-cell and spatially resolved atlas of human breast
cancers. Nat Genet. 53:1334–1347. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Ji AL, Rubin AJ, Thrane K, Jiang S,
Reynolds DL, Meyers RM, Guo MG, George BM, Mollbrink A,
Bergenstråhle J, et al: Multimodal analysis of composition and
spatial architecture in human squamous cell carcinoma. Cell.
182:497–514.e22. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Pastushenko I and Blanpain C: EMT
transition states during tumor progression and metastasis. Trends
Cell Biol. 29:212–226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Saviano A, Henderson NC and Baumert TF:
Single-cell genomics and spatial transcriptomics: Discovery of
novel cell states and cellular interactions in liver physiology and
disease biology. J Hepatol. 73:1219–1230. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Sharma A, Seow JJW, Dutertre CA, Pai R,
Blériot C, Mishra A, Wong RMM, Singh GSN, Sudhagar S, Khalilnezhad
S, et al: Onco-fetal reprogramming of endothelial cells drives
immunosuppressive macrophages in hepatocellular carcinoma. Cell.
183:377–394.e21. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Massalha H, Bahar Halpern K, Abu-Gazala S,
Jana T, Massasa EE, Moor AE, Buchauer L, Rozenberg M, Pikarsky E,
Amit I, et al: A single cell atlas of the human liver tumor
microenvironment. Mol Syst Biol. 16:e96822020. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Ben-Moshe S, Shapira Y, Moor AE, Manco R,
Veg T, Bahar Halpern K and Itzkovitz S: Spatial sorting enables
comprehensive characterization of liver zonation. Nat Metab.
1:899–911. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Lei X, Lei Y, Li JK, Du WX, Li RG, Yang J,
Li J, Li F and Tan HB: Immune cells within the tumor
microenvironment: Biological functions and roles in cancer
immunotherapy. Cancer Lett. 470:126–133. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Andersson A, Larsson L, Stenbeck L, Salmén
F, Ehinger A, Wu SZ, Al-Eryani G, Roden D, Swarbrick A, Borg Å, et
al: Spatial deconvolution of HER2-positive breast cancer delineates
tumor-associated cell type interactions. Nat Commun. 12:60122021.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Nerurkar SN, Goh D, Cheung CCL, Nga PQY,
Lim JCT and Yeong JPS: Transcriptional Spatial Profiling of Cancer
Tissues in the Era of Immunotherapy: The potential and promise.
Cancers (Basel). 12:25722020. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Grauel AL, Nguyen B, Ruddy D, Laszewski T,
Schwartz S, Chang J, Chen J, Piquet M, Pelletier M, Yan Z, et al:
TGFβ-blockade uncovers stromal plasticity in tumors by revealing
the existence of a subset of interferon-licensed fibroblasts. Nat
Commun. 11:63152020. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Moncada R, Barkley D, Wagner F, Chiodin M,
Devlin JC, Baron M, Hajdu CH, Simeone DM and Yanai I: Integrating
microarray-based spatial transcriptomics and single-cell RNA-seq
reveals tissue architecture in pancreatic ductal adenocarcinomas.
Nat Biotechnol. 38:333–342. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Svedlund J, Strell C, Qian X, Zilkens KJC,
Tobin NP, Bergh J, Sieuwerts AM and Nilsson M: Generation of in
situ sequencing based OncoMaps to spatially resolve gene expression
profiles of diagnostic and prognostic markers in breast cancer.
EBioMedicine. 48:212–223. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Wu Y, Yang S, Ma J, Chen Z, Song G, Rao D,
Cheng Y, Huang S, Liu Y, Jiang S, et al: Spatiotemporal immune
landscape of colorectal cancer liver metastasis at Single-cell
level. Cancer Discov. 12:134–153. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Jeong HY, Ham IH, Lee SH, Ryu D, Son SY,
Han SU, Kim TM and Hur H: Spatially distinct reprogramming of the
tumor microenvironment based on tumor invasion in Diffuse-type
gastric cancers. Clin Cancer Res. 27:6529–6542. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Sundar R, Liu DH, Hutchins GG, Slaney HL,
Silva AN, Oosting J, Hayden JD, Hewitt LC, Ng CC, Mangalvedhekar A,
et al: Spatial profiling of gastric cancer patient-matched primary
and locoregional metastases reveals principles of tumour
dissemination. Gut. 70:1823–1832. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Yamasaki J, Hirata Y, Otsuki Y, Suina K,
Saito Y, Masuda K, Okazaki S, Ishimoto T, Saya H and Nagano O: MEK
inhibition suppresses metastatic progression of KRAS-mutated
gastric cancer. Cancer Sci. 113:916–925. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Grosser B, Glückstein MI, Dhillon C,
Schiele S, Dintner S, VanSchoiack A, Kroeppler D, Martin B, Probst
A, Vlasenko D, et al: Stroma AReactive invasion front areas
(SARIFA)-a new prognostic biomarker in gastric cancer related to
tumor-promoting adipocytes. J Pathol. 256:71–82. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Jia L, Wang T, Zhao Y, Zhang S, Ba T, Kuai
X, Wang B, Zhang N, Zhao W, Yang Z and Qiao H: Single-cell
profiling of infiltrating B cells and tertiary lymphoid structures
in the TME of gastric adenocarcinomas. Oncoimmunology.
10:19697672021. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Xie W, Cheng J, Hong Z, Cai W, Zhuo H, Hou
J, Lin L, Wei X, Wang K, Chen X, et al: Multi-transcriptomic
analysis reveals the heterogeneity and tumor-promoting role of
SPP1/CD44-mediated intratumoral crosstalk in gastric cancer.
Cancers (Basel). 15:1642022. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Yang Y, Gao D, Xie X, Qin J, Li J, Lin H,
Yan D and Deng K: DeepIDC: A prediction framework of injectable
drug combination based on heterogeneous information and deep
learning. Clin Pharmacokinet. 61:1749–1759. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Han YM, Yang H, Huang QL, Sun ZJ, Li ML,
Zhang JB, Deng KJ, Chen S and Lin H: Risk prediction of diabetes
and pre-diabetes based on physical examination data. Math Biosci
Eng. 19:3597–3608. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Chen J, Xu H, Tao W, Chen Z, Zhao Y and
Han JDJ: Transformer for one stop interpretable cell type
annotation. Nat Commun. 14:2232023. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Ma Q and Xu D: Deep learning shapes
single-cell data analysis. Nat Rev Mol Cell Biol. 23:303–304. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Yuan Z and Yao J: Harnessing computational
spatial omics to explore the spatial biology intricacies. Semin
Cancer Biol. 95:25–41. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Liu J, Fan Z, Zhao W and Zhou X: Machine
intelligence in Single-cell data analysis: Advances and new
Challenges. Front Genet. 12:6555362021. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Islam S, Kjällquist U, Moliner A, Zajac P,
Fan JB, Lönnerberg P and Linnarsson S: Characterization of the
single-cell transcriptional landscape by highly multiplex RNA-seq.
Genome Res. 21:1160–1167. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Ramsköld D, Luo S, Wang YC, Li R, Deng Q,
Faridani OR, Daniels GA, Khrebtukova I, Loring JF, Laurent LC, et
al: Full-length mRNA-Seq from single-cell levels of RNA and
individual circulating tumor cells. Nat Biotechnol. 30:777–782.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Klein AM, Mazutis L, Akartuna I,
Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA and Kirschner
MW: Droplet barcoding for single-cell transcriptomics applied to
embryonic stem cells. Cell. 161:1187–1201. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Han X, Wang R, Zhou Y, Fei L, Sun H, Lai
S, Saadatpour A, Zhou Z, Chen H, Ye F, et al: Mapping the mouse
cell atlas by microwell-seq. Cell. 172:1091–1107.e17. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Sasagawa Y, Nikaido I, Hayashi T, Danno H,
Uno KD, Imai T and Ueda HR: Quartz-Seq: A highly reproducible and
sensitive single-cell RNA sequencing method, reveals non-genetic
gene-expression heterogeneity. Genome Biol. 14:R312013. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Hayashi T, Ozaki H, Sasagawa Y, Umeda M,
Danno H and Nikaido I: Single-cell full-length total RNA sequencing
uncovers dynamics of recursive splicing and enhancer RNAs. Nat
Commun. 9:6192018. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Farlik M, Sheffield NC, Nuzzo A, Datlinger
P, Schönegger A, Klughammer J and Bock C: Single-cell DNA methylome
sequencing and bioinformatic inference of epigenomic cell-state
dynamics. Cell Rep. 10:1386–1397. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Han L, Wu HJ, Zhu H, Kim KY, Marjani SL,
Riester M, Euskirchen G, Zi X, Yang J, Han J, et al:
Bisulfite-independent analysis of CpG island methylation enables
genome-scale stratification of single cells. Nucleic Acids Res.
45:e772017.PubMed/NCBI
|
|
178
|
Litzenburger UM, Buenrostro JD, Wu B, Shen
Y, Sheffield NC, Kathiria A, Greenleaf WJ and Chang HY: Single-cell
epigenomic variability reveals functional cancer heterogeneity.
Genome Biol. 18:152017. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Buenrostro JD, Giresi PG, Zaba LC, Chang
HY and Greenleaf WJ: Transposition of native chromatin for fast and
sensitive epigenomic profiling of open chromatin, DNA-binding
proteins and nucleosome position. Nat Methods. 10:1213–1218. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Rotem A, Ram O, Shoresh N, Sperling RA,
Goren A, Weitz DA and Bernstein BE: Single-cell ChIP-seq reveals
cell subpopulations defined by chromatin state. Nat Biotechnol.
33:1165–1172. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Mooijman D, Dey SS, Boisset JC, Crosetto N
and van Oudenaarden A: Single-cell 5hmC sequencing reveals
chromosome-wide cell-to-cell variability and enables lineage
reconstruction. Nat Biotechnol. 34:852–856. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Kaya-Okur HS, Wu SJ, Codomo CA, Pledger
ES, Bryson TD, Henikoff JG, Ahmad K and Henikoff S: CUT&Tag for
efficient epigenomic profiling of small samples and single cells.
Nat Commun. 10:19302019. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Nagano T, Lubling Y, Stevens TJ,
Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A and Fraser P:
Single-cell Hi-C reveals cell-to-cell variability in chromosome
structure. Nature. 502:59–64. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Peterson VM, Zhang KX, Kumar N, Wong J, Li
L, Wilson DC, Moore R, McClanahan TK, Sadekova S and Klappenbach
JA: Multiplexed quantification of proteins and transcripts in
single cells. Nat Biotechnol. 35:936–939. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Macaulay IC, Haerty W, Kumar P, Li YI, Hu
TX, Teng MJ, Goolam M, Saurat N, Coupland P, Shirley LM, et al:
G&T-seq: Parallel sequencing of single-cell genomes and
transcriptomes. Nat Methods. 12:519–522. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Dey SS, Kester L, Spanjaard B, Bienko M
and van Oudenaarden A: Integrated genome and transcriptome
sequencing of the same cell. Nat Biotechnol. 33:285–289. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Rooijers K, Markodimitraki CM, Rang FJ, de
Vries SS, Chialastri A, de Luca KL, Mooijman D, Dey SS and Kind J:
Simultaneous quantification of protein-DNA contacts and
transcriptomes in single cells. Nat Biotechnol. 37:766–772. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Satpathy AT, Saligrama N, Buenrostro JD,
Wei Y, Wu B, Rubin AJ, Granja JM, Lareau CA, Li R, Qi Y, et al:
Transcript-indexed ATAC-seq for precision immune profiling. Nat
Med. 24:580–590. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Chen S, Lake BB and Zhang K:
High-throughput sequencing of the transcriptome and chromatin
accessibility in the same cell. Nat Biotechnol. 37:1452–1457. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Liu L, Liu C, Quintero A, Wu L, Yuan Y,
Wang M, Cheng M, Leng L, Xu L, Dong G, et al: Deconvolution of
single-cell multi-omics layers reveals regulatory heterogeneity.
Nat Commun. 10:4702019. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Meyer AR, Engevik AC, Madorsky T, Belmont
E, Stier MT, Norlander AE, Pilkinton MA, McDonnell WJ, Weis JA,
Jang B, et al: Group 2 innate lymphoid cells coordinate damage
response in the stomach. Gastroenterology. 159:2077–2091.e8. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Kwon M, An M, Klempner SJ, Lee H, Kim KM,
Sa JK, Cho HJ, Hong JY, Lee T, Min YW, et al: Determinants of
response and intrinsic resistance to PD-1 blockade in
microsatellite instability-high gastric cancer. Cancer Discov.
11:2168–2185. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Zhou X, Yang J, Lu Y, Ma Y, Meng Y, Li Q,
Gao J, Jiang Z, Guo L, Wang W, et al: Relationships of tumor
differentiation and immune infiltration in gastric cancers revealed
by single-cell RNA-seq analyses. Cell Mol Life Sci. 80:572023.
View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Jiang H, Yu D, Yang P, Guo R, Kong M, Gao
Y, Yu X, Lu X and Fan X: Revealing the transcriptional
heterogeneity of organ-specific metastasis in human gastric cancer
using single-cell RNA Sequencing. Clin Transl Med. 12:e7302022.
View Article : Google Scholar : PubMed/NCBI
|
|
195
|
Honda M, Oki S, Kimura R, Harada A,
Maehara K, Tanaka K, Meno C and Ohkawa Y: High-depth spatial
transcriptome analysis by photo-isolation chemistry. Nat Commun.
12:44162021. View Article : Google Scholar : PubMed/NCBI
|
|
196
|
Chen KH, Boettiger AN, Moffitt JR, Wang S
and Zhuang X: RNA imaging. Spatially resolved, highly multiplexed
RNA profiling in single cells. Science. 348:aaa60902015. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Kishi JY, Lapan SW, Beliveau BJ, West ER,
Zhu A, Sasaki HM, Saka SK, Wang Y, Cepko CL and Yin P: SABER
amplifies FISH: Enhanced multiplexed imaging of RNA and DNA in
cells and tissues. Nat Methods. 16:533–544. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Ke R, Mignardi M, Pacureanu A, Svedlund J,
Botling J, Wählby C and Nilsson M: In situ sequencing for RNA
analysis in preserved tissue and cells. Nat Methods. 10:857–860.
2013. View Article : Google Scholar : PubMed/NCBI
|