Introduction
Prostate carcinoma is the most frequently diagnosed
visceral cancer in men worldwide. An increasing prevalence has been
reported in recent decades (1).
Radiation therapy is one of the primary modalities in prostate
cancer treatment. Ionizing radiation damages cells through free
radicals from the radiolysis of water that cause DNA double-strand
breaks. However, the efficacy of the radiotherapy may be affected
by the cellular response to radiation. Radiotherapy is highly
effective in treating radiosensitive tumors and enhancing the
therapeutic efficacy can increase the overall survival rate.
However, the presence of radioresistant tumors leads to cancer
relapse and metastasis. Understanding the tumor-radiation-related
genes to predict the tumor response to radiotherapy may potentially
modulate the treatment outcome for prostate cancer patients.
MicroRNAs (miRNAs) are a family of small,
non-coding, single-stranded RNAs composed of ~22 nucleotides (nt)
that negatively regulate protein expression at the
post-transcriptional level (2).
They function as gene regulators by binding to partially
complementary sites of mRNAs and cause translation inhibition or
direct degradation of the target mRNA. It has been suggested that
miRNAs are responsible for controlling ~50% of all protein-coding
genes (3). The widespread
regulation of protein levels has been studied in cellular models
(4). Previous studies have
demonstrated that the expression of miRNAs is clearly involved in
cancer development, and the deregulation of several miRNAs has been
observed in various types of cancer, including prostate cancer.
Porkka et al (5) was the
first to identify a miRNA signature specific for prostate cancer by
systematically profiling prostate cancer cell lines. Numerous
studies have identified many dysfunctional miRNAs by using a
high-throughput approach, which contributed to prostate cancer
progression, including the let-7 family, miR-1, -20a, -21,-34a,
-106b, -125b, -205 and -521 (6–13).
Although several studies have investigated the role of these
dysfunctional miRNAs to develop prostate cancer therapy, few
studies have determined the roles of miRNAs in radiation response
in prostate cancer. The upregulation of miR-521 reduces the
response to radiation damage by specifically targeting a DNA repair
protein, the Cockayne syndrome protein A (13). Li et al (14) found that miR-106b was dysregulated
after radiation treatment and suppressed radiation-induced p21
activation, suggesting it may override radiation-induced cell cycle
arrest and cell growth inhibition. Radiation delivered in daily
fractions altered a greater number of miRNAs compared with
single-dose radiation, and involved the upregulation of miR-34a and
let-7 miRNAs (15).
Next-generation sequencing (NGS) is a
high-throughput screening technology, and NGS data can be applied
in investigating miRNA expression, miRNA isoforms (isomiRs) and the
arm selection preferences of miRNAs. Therefore, the purpose of the
present study was to comprehensively investigate the distribution
of miRNAs after radiation treatment in PC3 cells by using an NGS
approach. Furthermore, we explored the function of
radiation-associated miRNA by conducting an in silico
analysis.
Materials and methods
Cell culture and radiation treatment
A PC3 cell line was obtained from the American Type
Culture Collection and was maintained in RPMI-1640 and supplemented
with 10% inactivated fetal bovine serum (FBS; Invitrogen, Carlsbad,
CA, USA). The cells were exposed to various radiation dosages (0,
2, 6, 10, 14 and 18 Gy) and were subsequently cultured in fresh
medium. The total RNA was obtained at various time points (0, 5, 15
and 40 h after treatment) by using TRIzol (Invitrogen) according to
the manufacturer’s instructions. The concentration, purity and
amount of total RNA were determined using a NanoDrop 1000
spectrophotometer (NanoDrop Technologies, Inc., USA).
Collection and preprocessing of sequence
reads
PC3 cells were exposed to 10 Gy of radiation. After
radiation treatment, the cells were lysed at various time points
(0, 5, 15 and 40 h) for RNA extraction. The RNA samples were
prepared using an Illumina small RNA preparation kit, and were
subsequently sequenced using the Illumina HiSeq platform. The
generated sequence reads were first subjected to quality control to
remove low-quality reads. The sequence reads were then subjected to
3′ adaptor trimming to generate clean reads, as previously
described (15,16). To attain a high confidence level,
only the clean reads with a read count ≥2 and with a length ranging
from 15 to 27 nt were included in further analyses.
Mapping clean reads to pre-miRNAs
To investigate miRNA expression profiles in
different libraries, we mapped the qualified clean reads back to
human pre-miRNAs (miRBase 19). To eliminate ambiguous multiple hits
during the mapping procedure, no mismatch was allowed. Previous
studies reported that, when mapped back to pre-miRNAs, sequence
reads usually carried mismatches preferentially located at their
terminal 3′ ends (17–20). This mismatch was named the 3′ end
modification. To determine whether the 3′ end modification patterns
differed among libraries, as described in our previous studies
(21), we trimmed and collected the
terminal 3′ end mismatches one by one. In addition, the remaining
perfect match reads had to be at least 18 nt in length. As a
result, we kept reads with no less than 18-nt perfect alignment and
3′ end modification patterns.
Classifying non-miRNA reads into
different data sets
The sequence reads that may not be mapped back to
pre-miRNAs were classified into classes by mapping to acquire
different data sets with Bowtie (22) and allowing a single nucleotide
variation. The sequences of mRNAs and other ncRNAs were derived
from the NCBI RefSeq 47 (23). The
tRNA sequences were downloaded from the Genomic tRNA database
(24) and the rRNA sequences were
downloaded from the SILVA database (25). The snoRNA, scaRNA and snRNA
sequences were all downloaded from NONCODE (26). The sequence reads not belonging to
any of the described RNA classes were uploaded to the RepeatMasker
to identify repeat elements, which were classified as unknown.
miRNA expression level according to The
Cancer Genome Atlas (TCGA) data
TCGA project collects both cancer and corresponding
normal tissues from hundreds of prostate cancer patients. We
downloaded all level-3 miRNA expression data of prostate
adenocarcinoma from the TCGA Data Portal (https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htm).
These level-3 data included calculated expressions for each miRNA
derived from the Illumina HiSeq sequencing results. A total of 198
tumor samples and 50 normal samples were found at the time the data
were downloaded. We kept only the expression data of 50
participants who had both miRNA expression levels from both tumor
and normal tissues. Normalized quantification expression levels for
these 50 participants were further examined for each investigated
miRNA.
Pathway enrichment analysis
We attempted to determine the functions of the miRNA
target genes by investigating the pathways with which the miRNA
target genes were involved. Therefore, we first downloaded the
target genes of differentially expressed miRNAs from TargetScan
6.0, and then mapped the target genes onto the Kyoto Encyclopedia
of Genes and Genomes (KEGG) pathways based on the Enzyme Commission
(EC) numbers by using the R package SubPathwayMiner v.3.1 (27). Subsequently, the hypergeometric test
was performed to identify significantly enriched pathways and
calculate the false positive discovery rate in the FDR-corrected
q-value.
Results
miRNA profiling of radiation-treated
prostate cancer cells
To characterize the mechanism involved in the
radiation response of prostate cancer, we used NGS to
comprehensively analyze the distribution of miRNAs after radiation
treatment in PC3 cells. As indicated in Fig. 1A, PC3 cells were exposed to various
dosages of radiation (0, 2, 4, 6, 8, 10, 12, 14, 16 and 18 Gy) and
then subjected to a fresh culture medium for an additional 4 days.
We found that the growth of the PC3 cells obviously decreased when
exposed to 10 Gy of radiation. Therefore, we collected cell RNA at
various times (0, 5, 15 and 40 h) following the 10-Gy radiation
treatment. We confirmed the expression levels of Cox-2 and p21,
which may be induced by radiation at 24 h according to previous
studies (16,28). The expression levels of Cox-2 and
p21 may be upregulated by radiation treatment in PC3 cells
(Fig. 1B). We then performed the
comprehensive miRNA profile at various time-points in
radiation-treated PC3 cells by using the Illumina HiSeq
platform.
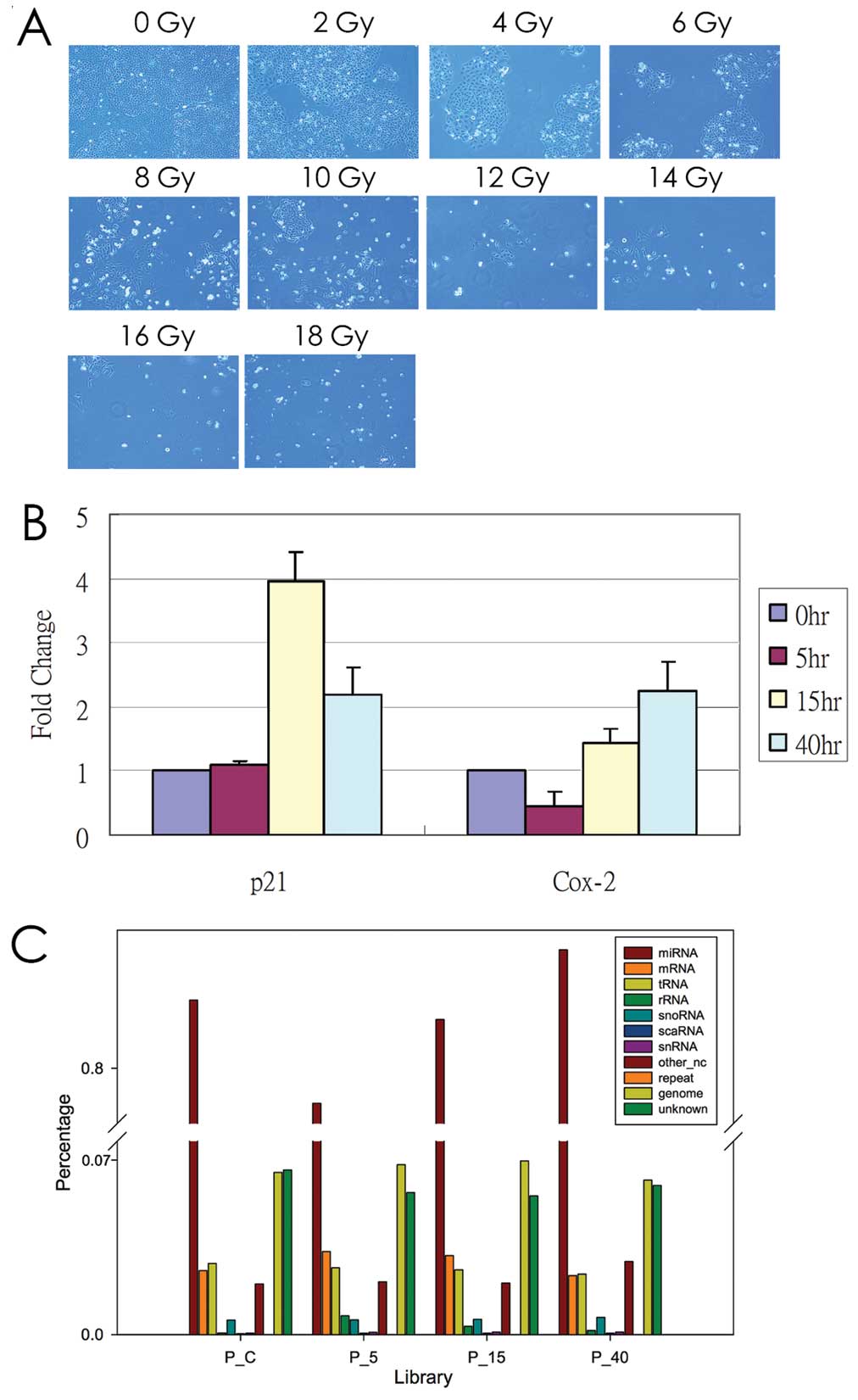 | Figure 1Radiation treatment of human prostate
cancer cells, PC3. (A) PC3 cells were treated with various
radiation doses (0, 2, 4, 6, 8, 10, 12, 14, 16 and 18 Gy) and were
subsequently subjected to fresh culture medium. After culturing for
an additional 4 days, the morphology was observed using light
microscopy (x40 magnification). (B) The expression pattern of COX-2
and p21 in radiation-treated PC3 cells was examined using a
real-time PCR method. S26 was used as an internal control. (C) The
distribution of small RNA reads in 11 categories was
classified. |
Analysis of miRNA sequence reads
Once the samples were sequenced, we collected >9
million clean reads in all libraries (Table I). In addition to miRNA, we also
determined which molecules were the remaining non-miRNA reads. By
mapping the non-miRNA reads back to a different data set, we
classified the reads into 11 categories. Fig. 1C demonstrates that miRNA accounted
for 80% of all clean reads in the prostate cell libraries. Other
categories accounted for relatively low proportions, which
indicated the high performance of the sample preparation protocol.
In addition, the proportions of the categories were considerably
similar among libraries, indicating that radiation treatment did
not alter the composition of RNA samples in the prostate cell
libraries.
 | Table ISummary of sequence reads and the
detected miRNAs. |
Table I
Summary of sequence reads and the
detected miRNAs.
| Library | Clean read (n) | miRNA read (%) | pre-miRNA
(n) | miRNA
(n) |
|---|
| P_C | 9,482,400 | 80.49 | 693 | 916 |
| P_5 | 9,748,570 | 79.75 | 687 | 915 |
| P_15 | 9,589,440 | 80.35 | 712 | 933 |
| P_40 | 10,589,934 | 80.86 | 739 | 964 |
After mapping the clean reads to the genome, most of
the miRNA reads tend to exist as isomiR. As demonstrated in
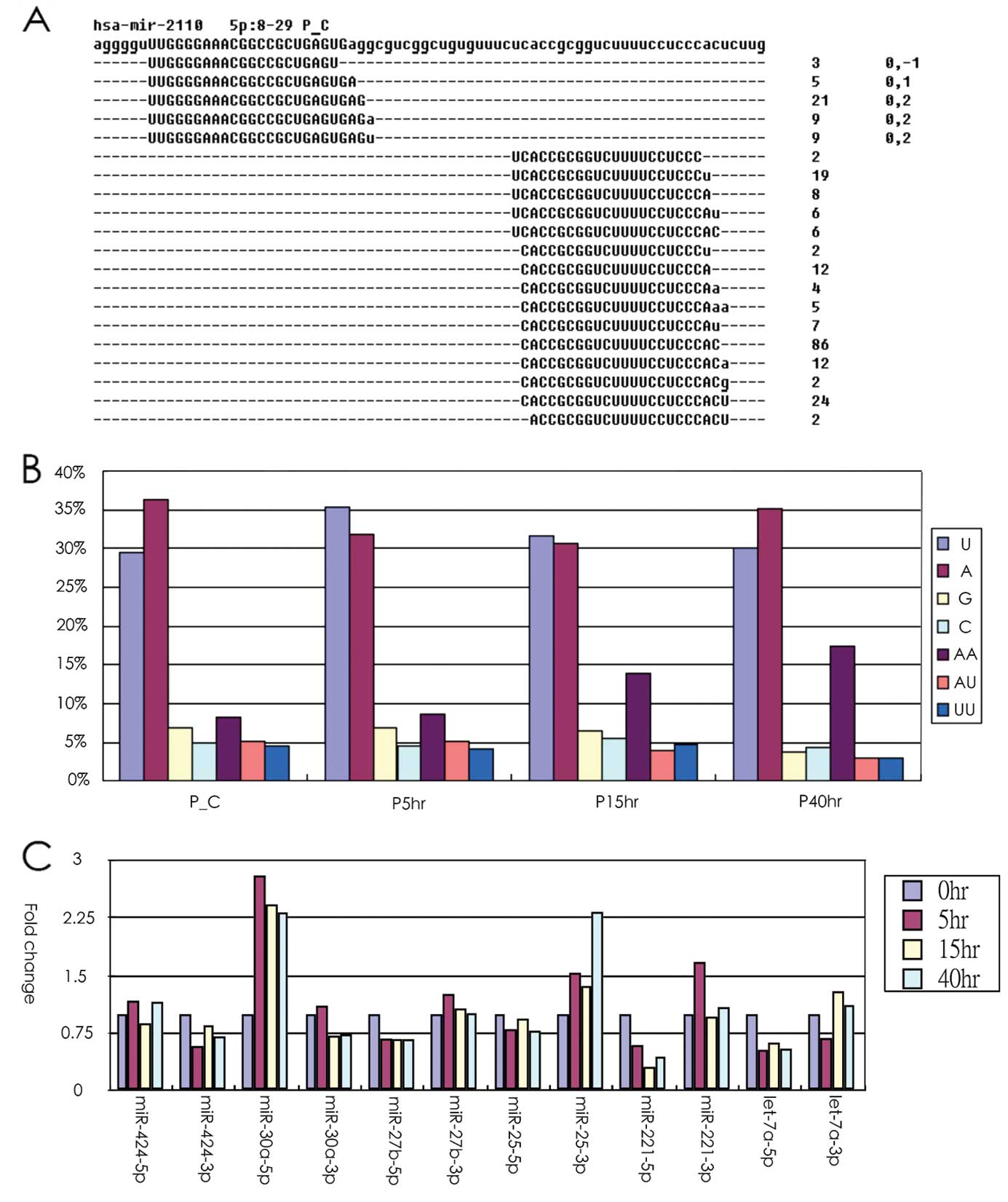
Fig. 2A, hsa-miR-2110-5p had 5
isomers, whereas the opposite-arm miRNA-3p had 15 isomiRs, which
demonstrated that abundant miRNAs tend to have more isomiRs. Our
data revealed that the isomiR quantity was highly correlated with
miRNA abundance (Pearson’s correlation coefficient, 0.91). In
addition, we observed that the modified nucleotides were
preferentially located at the 3′ end of the sequence read
(presented in lower case in Fig.
2A). The data indicated that one A nucleotide or one U
nucleotide was frequently added at the end of the read. Notably, we
found that the proportion of AA dinucleotides modified at the end
of the read was gradually increased in a time-dependent manner
after the PC3 cells were treated with radiation, which indicated
that the 3′ end modification may be altered by radiation treatment
in PC3 cells. Our previous studies indicated that the use of miR-5p
and -3p may be altered in human cancer (29–31).
In the present study, our data indicated that arm selection
preference was consistent across nearly all libraries. Only a few
cases were observed in which the use of -5p and -3p arm selection
had different preferences at various time-points after radiation
treatment (Fig. 2C). Further
research is required to support these findings.
Radiation-response miRNAs in prostate
cancer
By summarizing the read count of all the isomiRs
that belonged to the same mature miRNAs, we quantified the miRNA
expression abundances, and presented the result in transcript per
million (TPM). Twenty-two miRNAs were selected and are presented in
Table II, demonstrating that their
expression levels were altered >2-fold after being subjected to
radiation exposure (expression of 6 miRNAs increased, and
expression of 16 miRNAs decreased). To explore the putative role
contributing to prostate cancer progression, we examined the effect
of expression levels of radiation-associated miRNAs on prostate
cancer from the available TCGA dataset by using an in silico
analysis. We downloaded 100 miRNA expression profiles from 50
prostate cancer patients, including 50 cancer lesion and 50
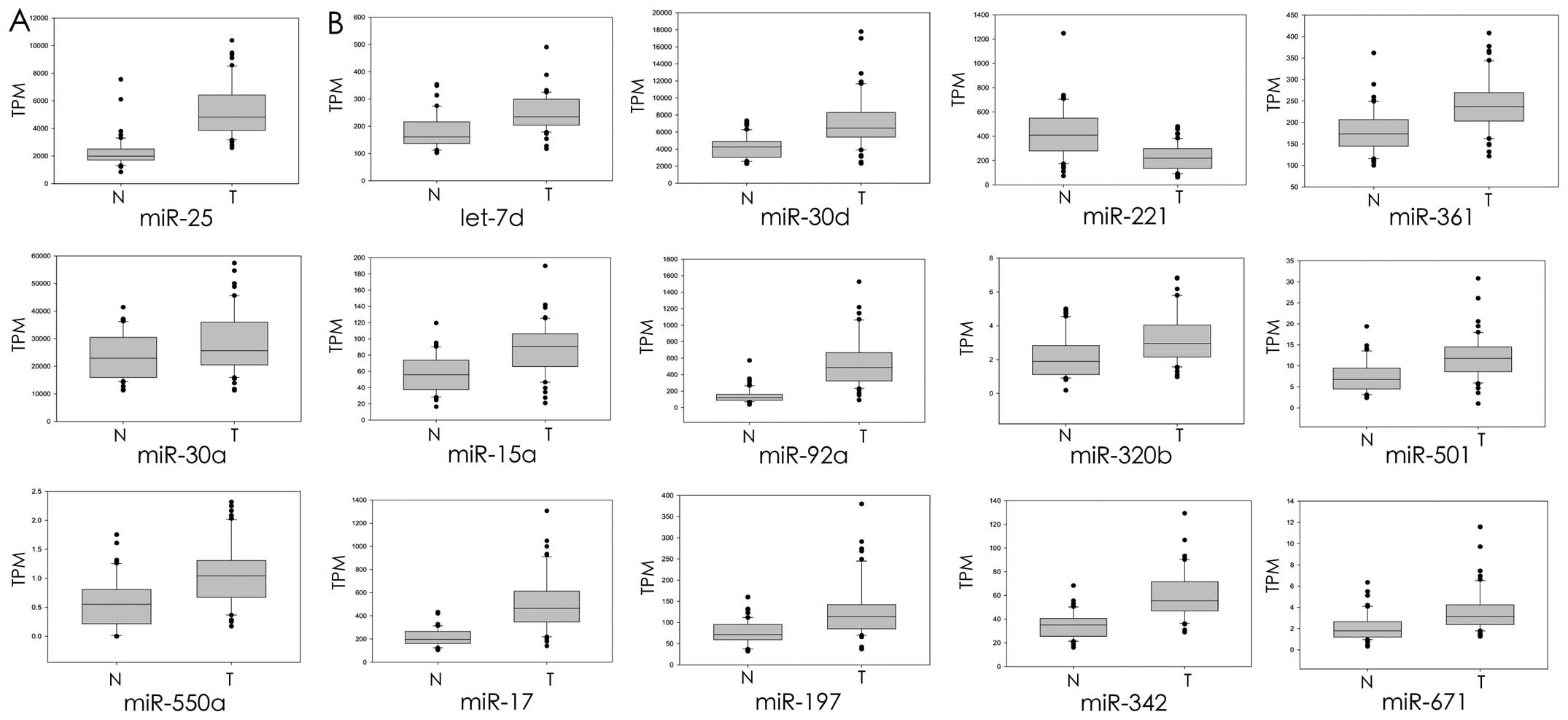
corresponding normal tissues. As demonstrated in Fig. 3A, the expression levels of
radiation-induced miRNAs, miR-25, miR-30a and miR-550a, were
significantly upregulated in the prostate cancer cells compared
with the corresponding normal tissue cells. Twelve
radiation-suppressed miRNAs were identified, i.e. let-7d, miR-15a,
miR-17, miR-30d, miR-92a, miR-197, miR-221, miR-320b, miR-342,
miR-361, miR-501 and miR-671, and a significantly different
expression between prostate cancer and the corresponding adjacent
part was found, including 11 upregulated and 1 downregulated
(Fig. 3B). Overall, the data
indicated that most of the radiation-response miRNAs were
identified as dysregulated in prostate cancer according to an in
silico analysis (15/22; 1 downregulated, 14 upregulated and the
rest demonstrated no change in expression in prostate cancer).
| Table IImiRNAs with altered expression in
response to radiation in PC3 cells using next-generation
sequencing. |
Table II
miRNAs with altered expression in
response to radiation in PC3 cells using next-generation
sequencing.
| 0 h | 5 h | 15 h | 40 h | Expression data for
TCGA |
|---|
|
Upregulationa |
| hsa-miR-9-5p | 1 | 2.82 | 0.90 | 2.63 | |
| hsa-miR-22-3p | 1 | 1.60 | 2.85 | 2.73 | |
| hsa-miR-25-3p | 1 | 1.54 | 1.39 | 2.33 |
Upregulationd |
|
hsa-miR-30a-5p | 1 | 2.81 | 2.43 | 2.33 |
Upregulationc |
|
hsa-miR-550a-3p | 1 | 1.88 | 1.46 | 2.09 |
Upregulationd |
|
hsa-miR-548h-5p | 1 | 0.50 | 0.44 | 2.56 | |
|
Downregulationb |
| hsa-let-7c | 1 | 0.93 | 0.78 | 0.45 | |
| hsa-let-7d-5p | 1 | 0.30 | 0.11 | 0.40 |
Upregulationd |
| hsa-let-7e-5p | 1 | 0.58 | 0.55 | 0.40 | |
|
hsa-miR-15a-5p | 1 | 0.78 | 0.67 | 0.45 |
Upregulationd |
| hsa-miR-17-3p | 1 | 0.52 | 0.49 | 0.47 |
Upregulationd |
|
hsa-miR-30d-3p | 1 | 0.92 | 0.75 | 0.41 |
Upregulationd |
|
hsa-miR-92a-5p | 1 | 0.65 | 0.52 | 0.50 |
Upregulationd |
|
hsa-miR-125a-3p | 1 | 0.42 | 0.42 | 0.32 | |
|
hsa-miR-197-3p | 1 | 0.77 | 0.79 | 0.44 |
Upregulationd |
|
hsa-miR-221-5p | 1 | 0.59 | 0.31 | 0.44 |
Downregulationd |
| hsa-miR-320b | 1 | 0.65 | 0.41 | 0.32 |
Upregulationd |
|
hsa-miR-342-5p | 1 | 0.59 | 0.63 | 0.47 |
Upregulationd |
|
hsa-miR-361-3p | 1 | 0.45 | 0.53 | 0.40 |
Upregulationd |
|
hsa-miR-374a-5p | 1 | 1.04 | 0.93 | 0.47 | |
|
hsa-miR-501-3p | 1 | 0.77 | 0.48 | 0.45 |
Upregulationd |
|
hsa-miR-671-3p | 1 | 0.77 | 0.62 | 0.41 |
Upregulationd |
Pathway enrichment analysis of
miRNAs
miRNAs can function as either oncogenes or tumor
suppressors depending on their target genes. Therefore, identifying
a target can facilitate elucidating the role of miRNAs in prostate
cancer treatment radiation (32,33).
Typically, one miRNA tends to have hundreds of target genes and a
group of miRNAs co-modulated as a biological function involved in
the regulation of a signaling pathway. Therefore, we further
explored the biological function of radiation-response miRNAs by
conducting a pathway-enrichment analysis. The putative target genes
of miRNAs were obtained from TargetScan 6.0; subsequently, these
target genes of the individual miRNAs were mapped onto KEGG
pathways. Our data indicated that the target genes of
radiation-response miRNAs were frequently significantly enriched in
several cancer- or radiation-related pathways, including the
mitogen-activated protein kinase (MAPK), ErbB, p53, Wnt,
transforming growth factor-β (TGF-β) and mTOR signaling pathways
with an FDR <0.05 (Table
III). We also subjected the target genes of the 2-gene set,
upregulated miRNA and downregulated miRNAs, to pathway-enrichment
analysis. Similar results were observed; their targets were
significantly enriched in the prostate cancer pathway (Fig. 4) and radiation-related pathways,
including the MAPK, ErbB, Wnt and TGF-β signaling pathways
(Tables IV and V).
| Table IIIThe enriched pathways of
radiation-induced miRNA target genes. |
Table III
The enriched pathways of
radiation-induced miRNA target genes.
| microRNA | Cancer-relative
pathway (FDR<0.05) |
|---|
| Upregulation |
| hsa-miR-9-5p | Focal adhesion,
pathways in cancer, ErbB signaling pathway,
MAPK signaling
pathway, prostate cancer |
| hsa-miR-22-3p | Chronic myeloid
leukemia, MAPK
signaling pathway, ErbB signaling pathway,
pathways in cancer, glioma, prostate cancer, phosphatidylinositol signaling
system, colorectal cancer |
| hsa-miR-25-3p | N.D |
|
hsa-miR-30a-5p | N.D |
|
hsa-miR-550a-3p | N.D |
|
hsa-miR-548h-5p | N.D |
| Downregulation |
| hsa-let-7c | MAPK signaling pathway,
pathways in cancer, p53 signaling pathway, melanoma, chronic
myeloid leukemia, glioma, pancreatic cancer, focal adhesion, small
cell lung cancer, bladder cancer, prostate cancer |
| hsa-let-7d-5p | MAPK signaling pathway,
pathways in cancer, p53 signaling pathway, melanoma, chronic
myeloid leukemia, glioma, pancreatic cancer, focal adhesion, small
cell lung cancer, bladder cancer, prostate cancer |
| hsa-let-7e-5p | MAPK signaling pathway,
pathways in cancer, p53 signaling pathway, melanoma, chronic
myeloid leukemia, glioma, pancreatic cancer, focal adhesion, small
cell lung cancer, bladder cancer, prostate cancer |
|
hsa-miR-15a-5p | Pathways in cancer,
regulation of actin cytoskeleton, renal cell carcinoma,
MAPK signaling
pathway, focal adhesion, melanoma, prostate cancer,
Wnt signaling
pathway, p53 signaling pathway, mTOR signaling pathway,
non-small cell lung cancer, pancreatic cancer, cell cycle |
| hsa-miR-17-3p | MAPK signaling pathway,
pathways in cancer, chronic myeloid leukemia, pancreatic cancer,
melanoma, bladder cancer, TGF-β signaling pathway,
prostate
cancer, mTOR
signaling pathway, non-small cell lung cancer, renal
cell carcinoma, cell cycle, p53 signaling
pathway |
|
hsa-miR-30d-3p | N.D |
|
hsa-miR-92a-5p | N.D |
|
hsa-miR-125a-3p | MAPK signaling pathway,
adherens junction, pancreatic cancer, TGF-β signaling
pathway |
|
hsa-miR-197-3p | N.D |
|
hsa-miR-221-5p | Wnt signaling pathway,
ErbB signaling
pathway hsa-miR-320b Chronic myeloid leukemia, non-small
cell lung cancer, glioma, pathways in cancer, focal adhesion,
pancreatic cancer, melanoma, ErbB signaling pathway,
colorectal cancer, TGF-β signaling pathway,
prostate cancer, MAPK
signaling pathway, mTOR signaling pathway |
|
hsa-miR-342-5p | N.D |
|
hsa-miR-361-3p | Pathways in cancer,
mTOR signaling
pathway, melanogenesis, renal cell carcinoma,
nucleotide excision
repair |
|
hsa-miR-374a-5p | Pathways in cancer,
prostate cancer, TGF-β
signaling pathway, endometrial cancer, non-small cell
lung cancer, basal cell carcinoma, MAPK signaling pathway |
|
hsa-miR-501-3p | N.D |
|
hsa-miR-671-3p | N.D |
| Table IVThe enriched pathways of
radiation-upregulated miRNA target genes. |
Table IV
The enriched pathways of
radiation-upregulated miRNA target genes.
| Pathway Id | Pathway name | FDR |
|---|
| Path:04360 | Axon guidance | 7.88E-08 |
| Path:04722 | Neurotrophin
signaling pathway | 1.11E-07 |
| Path:04010 | MAPK signaling pathway | 1.11E-07 |
| Path:05200 | Pathways in
cancer | 3.25E-07 |
| Path:04012 | ErbB signaling pathway | 3.14E-06 |
| Path:04120 | Ubiquitin mediated
proteolysis | 8.82E-06 |
| Path:04144 | Endocytosis | 1.24E-05 |
| Path:04520 | Adherens
junction | 5.86E-05 |
| Path:05412 | Arrhythmogenic
right ventricular cardiomyopathy (ARVC) | 7.26E-05 |
| Path:04510 | Focal adhesion | 0.000105 |
| Path:04916 | Melanogenesis | 0.000245 |
| Path:05220 | Chronic myeloid
leukemia | 0.000245 |
| Path:04810 | Regulation of actin
cytoskeleton | 0.000261 |
| Path:05214 | Glioma | 0.000261 |
| Path:05414 | Dilated
cardiomyopathy | 0.000268 |
| Path:04910 | Insulin signaling
pathway | 0.000282 |
| Path:04720 | Long-term
potentiation | 0.000294 |
| Path:04070 | Phosphatidylinositol signaling
system | 0.000294 |
| Path:05410 | Hypertrophic
cardiomyopathy (HCM) | 0.000802 |
| Path:05100 | Bacterial invasion
of epithelial cells | 0.001018 |
| Path:05215 | Prostate cancer | 0.001096 |
| Path:04310 | Wnt signaling pathway | 0.001153 |
| Path:04914 |
Progesterone-mediated oocyte
maturation | 0.002548 |
| Path:05211 | Renal cell
carcinoma | 0.003135 |
| Path:04920 | Adipocytokine
signaling pathway | 0.003902 |
| Path:04350 | TGF-β signaling
pathway | 0.003902 |
| Path:04666 | Fc γ R-mediated
phagocytosis | 0.004314 |
| Path:04141 | Protein processing
in endoplasmic reticulum | 0.005738 |
| Path:05210 | Colorectal
cancer | 0.005747 |
| Path:04130 | SNARE interactions
in vesicular transport | 0.007624 |
| Path:05216 | Thyroid cancer | 0.008679 |
| Path:00562 | Inositol phosphate
metabolism | 0.008679 |
| Path:00532 | Glycosaminoglycan
biosynthesis-chondroitin sulfate | 0.008679 |
| Path:05014 | Amyotrophic lateral
sclerosis (ALS) | 0.008679 |
| Path:05212 | Pancreatic
cancer | 0.008679 |
| Path:04020 | Calcium signaling
pathway | 0.008679 |
| Path:05218 | Melanoma | 0.010147 |
| Path:04930 | Type II diabetes
mellitus | 0.018227 |
| Path:04962 |
Vasopressin-regulated water
reabsorption | 0.018227 |
| Path:04530 | Tight junction | 0.019289 |
| Path:04512 | ECM-receptor
interaction | 0.019289 |
| Path:04912 | GnRH signaling
pathway | 0.019289 |
| Path:05223 | Non-small cell lung
cancer | 0.023349 |
| Path:05222 | Small cell lung
cancer | 0.032425 |
| Path:05213 | Endometrial
cancer | 0.035464 |
| Path:04662 | B cell receptor
signaling pathway | 0.035464 |
| Path:05131 | Shigellosis | 0.035464 |
| Path:05221 | Acute myeloid
leukemia | 0.03804 |
| Path:04540 | Gap junction | 0.041068 |
| Path:00250 | Alanine, aspartate
and glutamate metabolism | 0.045237 |
| Path:04114 | Oocyte meiosis | 0.049286 |
| Table VThe enriched pathways of
radiation-downregulated miRNA target genes. |
Table V
The enriched pathways of
radiation-downregulated miRNA target genes.
| Pathway Id | Pathway name | FDR |
|---|
| Path:04010 | MAPK signaling pathway | 0 |
| Path:04360 | Axon guidance | 0 |
| Path:05200 | Pathways in cancer | 0 |
| Path:04722 | Neurotrophin
signaling pathway | 1.18E-12 |
| Path:04310 | Wnt signaling pathway | 1.50E-10 |
| Path:04510 | Focal adhesion | 4.27E-10 |
| Path:04144 | Endocytosis | 1.40E-09 |
| Path:04810 | Regulation of actin
cytoskeleton | 2.34E-09 |
| Path:05215 | Prostate cancer | 3.58E-09 |
| Path:05211 | Renal cell
carcinoma | 3.58E-09 |
| Path:04720 | Long-term
potentiation | 1.14E-08 |
| Path:05220 | Chronic myeloid
leukemia | 3.41E-08 |
| Path:04120 | Ubiquitin mediated
proteolysis | 7.40E-08 |
| Path:04020 | Calcium signaling
pathway | 1.10E-07 |
| Path:05212 | Pancreatic
cancer | 1.57E-07 |
| Path:05214 | Glioma | 1.59E-07 |
| Path:05218 | Melanoma | 2.36E-07 |
| Path:05223 | Non-small cell lung
cancer | 3.64E-07 |
| Path:04916 | Melanogenesis | 4.67E-07 |
| Path:04520 | Adherens
junction | 4.67E-07 |
| Path:04910 | Insulin signaling
pathway | 6.79E-07 |
| Path:04350 | TGF-β signaling
pathway | 7.72E-07 |
| Path:05210 | Colorectal
cancer | 2.50E-06 |
| Path:04012 | ErbB signaling pathway | 3.89E-06 |
| Path:05222 | Small cell lung
cancer | 3.20E-05 |
| Path:04730 | Long-term
depression | 5.13E-05 |
| Path:05221 | Acute myeloid
leukemia | 7.50E-05 |
| Path:04150 | mTOR signaling pathway | 8.68E-05 |
| Path:05213 | Endometrial
cancer | 8.68E-05 |
| Path:05217 | Basal cell
carcinoma | 9.44E-05 |
| Path:04710 | Circadian
rhythm-mammal | 9.64E-05 |
| Path:04070 |
Phosphatidylinositol signaling system | 9.64E-05 |
| Path:04540 | Gap junction | 9.83E-05 |
| Path:04115 | p53 signaling pathway | 0.000113 |
| Path:04141 | Protein processing
in endoplasmic reticulum | 0.000137 |
| Path:04114 | Oocyte meiosis | 0.000196 |
| Path:05014 | Amyotrophic lateral
sclerosis (ALS) | 0.000353 |
| Path:04970 | Salivary
secretion | 0.000414 |
| Path:04660 | T cell receptor
signaling pathway | 0.000449 |
| Path:04666 | Fc γ R-mediated
phagocytosis | 0.000526 |
| Path:04512 | ECM-receptor
interaction | 0.000526 |
| Path:05142 | Chagas disease | 0.000563 |
| Path:04062 | Chemokine signaling
pathway | 0.000812 |
| Path:04210 | Apoptosis | 0.000844 |
| Path:04914 |
Progesterone-mediated oocyte
maturation | 0.001465 |
| Path:04110 | Cell cycle | 0.001527 |
| Path:04662 | B cell receptor
signaling pathway | 0.00169 |
| Path:04530 | Tight junction | 0.001692 |
| Path:04930 | Type II diabetes
mellitus | 0.002128 |
| Path:05412 | Arrhythmogenic
right ventricular cardiomyopathy (ARVC) | 0.002128 |
| Path:04920 | Adipocytokine
signaling pathway | 0.002184 |
| Path:04664 | Fc ɛ RI signaling
pathway | 0.002621 |
| Path:04960 |
Aldosterone-regulated sodium
reabsorption | 0.002802 |
| Path:04912 | GnRH signaling
pathway | 0.002955 |
| Path:04320 | Dorso-ventral axis
formation | 0.00373 |
| Path:05219 | Bladder cancer | 0.00373 |
| Path:04340 | Hedgehog signaling
pathway | 0.005218 |
| Path:04971 | Gastric acid
secretion | 0.005633 |
| Path:04130 | SNARE interactions
in vesicular transport | 0.006926 |
| Path:00532 | Glycosaminoglycan
biosynthesis-chondroitin sulfate | 0.006926 |
| Path:05131 | Shigellosis | 0.008185 |
| Path:05160 | Hepatitis C | 0.008185 |
| Path:04630 | Jak-STAT signaling
pathway | 0.008368 |
| Path:05410 | Hypertrophic
cardiomyopathy (HCM) | 0.009939 |
| Path:05414 | Dilated
cardiomyopathy | 0.010733 |
| Path:00534 | Glycosaminoglycan
biosynthesis-heparan sulfate | 0.011359 |
| Path:04670 | Leukocyte
transendothelial migration | 0.011852 |
| Path:04370 | VEGF signaling
pathway | 0.013265 |
| Path:00562 | Inositol phosphate
metabolism | 0.013625 |
| Path:04270 | Vascular smooth
muscle contraction | 0.014557 |
| Path:00512 | O-Glycan
biosynthesis | 0.016472 |
| Path:04330 | Notch signaling
pathway | 0.026769 |
| Path:04142 | Lysosome | 0.038882 |
| Path:00533 | Glycosaminoglycan
biosynthesis-keratan sulfate | 0.038882 |
| Path:05145 | Toxoplasmosis | 0.048785 |
Discussion
Our previous studies indicated that the
distributions of 3′ end modifications and the arm selection
preference of miRNAs were different between normal and tumor
tissues (29–31). The -5p and -3p of miRNA play a
distant role by suppressing the different target genes. It was
previously reported that, in contrast to the oncogenic effect of
miR-17 (-5p), miR-17*(-3P) plays a tumor suppressive
role in prostate cancer (9,34,35).
The miR-28-5p and miR-28-3p also play opposite roles in colon
cancer cell proliferation and migration (36). In the present study, our data showed
that the -5p and -3p of particular miRNAs were differently
regulated by radiation (shown in Fig.
2C). Several studies have demonstrated that miRNAs contain
various ends, which were caused by either RNA editing or
non-template nucleotide additions (17,18,20).
These miRNA isoforms (isomiRs) contribute to increased miRNA
stability or strengthened miRNA-target gene interaction and are
differentially expressed in different cellular conditions,
including cancer (16,37,38).
Our data revealed that the proportion of AA dinucleotide
modifications at the end of the read gradually increased in a
time-dependent manner after the PC3 cells were treated with
radiation, suggesting that radiation may influence the particular
miRNA stability or efficiency of silencing targets by regulating
the 3′ end modifications, which warrants further research.
miRNAs are known to function as gene silencers and
are involved in modulating biological functions, including cell
growth, apoptosis, the cell cycle and the metastasis of cancer
(39). Comprehensive miRNA
profiling of prostate cancer has indicated that several miRNAs are
differentially expressed between prostate cancer and the adjacent
normal, which contributes to prostate cancer progression (40–42).
In the present study, we analyzed miRNA expression from TCGA
database and found that the expression levels of radiation-induced
miRNAs were frequently dysregulated in prostate cancer (Fig. 3). Our results are consistent with
those of previous studies and demonstrated that miR-25, miR-17,
miR-30d and miR-92a are overexpressed, and miR-221 is downregulated
in prostate cancer (9,42–44).
However, the expression levels of let-7d and miR-15a decreased
according to TCGA, which contradicted the results of previous
studies (45–47). These dysfunctional miRNAs have
potential to be used as biomarkers for prostate cancer prognosis or
diagnosis. Therefore, understanding the function of miRNAs may
provide practical benefits for clinical applications. Predicting
the outcome of cancer treatment is the most promising application
of miRNAs. Gonzales et al found miR-141 to be consistent
with changes in other conventional biomarkers and to the clinical
outcomes, suggesting that miR-141 can be used as a marker for
monitoring therapeutic response in prostate cancer patients
(48). The prognostic value of
miRNA expression profiling in prostate cancer has also been
demonstrated by Hulf et al. They demonstrated that DNA
methylation and histone H3K9-deacetylation of the miR-205 locus is
associated with miRNA silencing and deregulation of MED1, which is
predictive of a poor prognosis in localized prostate cancer
(49).
Ionizing radiation is one of the 3 primary
modalities used in cancer therapy. Radiation induces considerable
DNA damages, which, if not repaired, cause cancer cells to progress
to apoptosis and cell cycle arrest. Some cancer cells are resistant
to radiation treatment due to activation of complex signaling
pathways that counteract these damages, including ErbB, nuclear
factor κB (NFκB), MAPK, PI3K/AKT and transforming growth factor-β
(TGF-β) signaling pathways (50–52).
Several radiation-related miRNAs have been identified that
contribute to the radiosensitivity of cancer cells by modulating
the radiation-response signaling pathway (51,52).
Since miRNAs are generally slightly repressed by their target
genes, the alteration of an individual miRNA is insufficient for
accomplishing a biological function. Previous studies have
introduced the concept of miRNA regulatory modules (MRMs), which
potentially serve as a model for understanding the detailed
influences of miRNAs in cellular biological functions (53–55).
Therefore, in the present study, we were particularly interested in
the consequences of changes in a group of radiation-induced miRNAs
in prostate cancer. Our data indicated that targets of the
co-expressed miRNAs were enriched in a radiation-related signaling
pathway, suggesting that they co-modulated an abundance of target
genes in the same pathway (Table
III).
miRNAs regulate various factors in radiation-related
biological pathways and may affect the radiosensitivity of tumor
cells (51). Radiation-response
miRNAs have been identified in prostate cancer by using a
microarray approach (13–15). By comparing these data, we
identified known and unknown radiation-response miRNAs in prostate
cancer by using an NGS approach. Li et al reported that the
expression levels of miR-9, miR-22 and miR-30a decreased in
radiation-treated PC3 cells (14).
Radiation reduced the expression level of an miR-17-92a cluster and
the let-7 family in prostate cancer (15). In the present study, we also
identified radiation-response miRNAs that had been reported in
other types of cancer but not in prostate cancer, such as miR-25,
miR-15a, miR-30d, miR-125a, miR-221 and miR-342 (21,56–63).
In addition, we identified a group of radiation-response miRNAs
that have not been reported in any type of cancer. The
pathway-enrichment analysis revealed that their targets are
frequently enriched in the radiation-response signaling
pathway.
In summary, in the present study, we thoroughly
investigated radiation-response miRNAs, which may be involved in
the radiosensitivity of prostate cancer, by modulating
radiation-related signaling pathways using an NGS approach. These
miRNA candidates may be effective targets for improving the
efficacy of radiation treatment in future prostate cancer therapy.
In addition, we observed that 3′ end modifications and the -5p/-3p
arm selection of miRNAs were altered in prostate cancer after
radiation treatment. These finding require further research.
Acknowledgements
This study was supported by grants from Kaohsiung
Veterans General Hospital (VGHKS 102-005 and VGHKS 102-074). The
authors thank Genomics and Proteomics Core Laboratory, Department
of Medical Research, Kaohsiung Chang Gung Memorial Hospital, for
the assistance with NGS data analysis.
References
|
1
|
Crawford ED: Epidemiology of prostate
cancer. Urology. 62(Suppl 1): 3–12. 2003. View Article : Google Scholar
|
|
2
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Krol J, Loedige I and Filipowicz W: The
widespread regulation of microRNA biogenesis, function and decay.
Nat Rev Genet. 11:597–610. 2010.PubMed/NCBI
|
|
4
|
Selbach M, Schwanhäusser B, Thierfelder N,
Fang Z, Khanin R and Rajewsky N: Widespread changes in protein
synthesis induced by microRNAs. Nature. 455:58–63. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Porkka KP, Pfeiffer MJ, Waltering KK,
Vessella RL, Tammela TL and Visakorpi T: MicroRNA expression
profiling in prostate cancer. Cancer Res. 67:6130–6135. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sevli S, Uzumcu A, Solak M, Ittmann M and
Ozen M: The function of microRNAs, small but potent molecules, in
human prostate cancer. Prostate Cancer Prostatic Dis. 13:208–217.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li T, Li D, Sha J, Sun P and Huang Y:
MicroRNA-21 directly targets MARCKS and promotes apoptosis
resistance and invasion in prostate cancer cells. Biochem Biophys
Res Commun. 383:280–285. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ambs S, Prueitt RL, Yi M, et al: Genomic
profiling of microRNA and messenger RNA reveals deregulated
microRNA expression in prostate cancer. Cancer Res. 68:6162–6170.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sylvestre Y, De Guire V, Querido E, et al:
An E2F/miR-20a autoregulatory feedback loop. J Biol Chem.
282:2135–2143. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fujita Y, Kojima K, Hamada N, et al:
Effects of miR-34a on cell growth and chemoresistance in prostate
cancer PC3 cells. Biochem Biophys Res Commun. 377:114–119. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi XB, Xue L, Yang J, et al: An
androgen-regulated miRNA suppresses Bak1 expression and induces
androgen-independent growth of prostate cancer cells. Proc Natl
Acad Sci USA. 104:19983–19988. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gandellini P, Folini M, Longoni N, et al:
miR-205 exerts tumor-suppressive functions in human prostate
through down-regulation of protein kinase Cɛ. Cancer Res.
69:2287–2295. 2009.PubMed/NCBI
|
|
13
|
Josson S, Sung SY, Lao K, Chung LW and
Johnstone PA: Radiation modulation of microRNA in prostate cancer
cell lines. Prostate. 68:1599–1606. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li B, Shi XB, Nori D, et al:
Down-regulation of microRNA 106b is involved in p21-mediated cell
cycle arrest in response to radiation in prostate cancer cells.
Prostate. 71:567–574. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
John-Aryankalayil M, Palayoor ST, Makinde
AY, et al: Fractionated radiation alters oncomir and tumor
suppressor miRNAs in human prostate cancer cells. Radiat Res.
178:105–117. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cloonan N, Wani S, Xu Q, et al: MicroRNAs
and their isomiRs function cooperatively to target common
biological pathways. Genome Biol. 12:R1262011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ebhardt HA, Tsang HH, Dai DC, Liu Y,
Bostan B and Fahlman RP: Meta-analysis of small RNA-sequencing
errors reveals ubiquitous post-transcriptional RNA modifications.
Nucleic Acids Res. 37:2461–2470. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Landgraf P, Rusu M, Sheridan R, et al: A
mammalian microRNA expression atlas based on small RNA library
sequencing. Cell. 129:1401–1414. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Reid JG, Nagaraja AK, Lynn FC, et al:
Mouse let-7 miRNA populations exhibit RNA editing that is
constrained in the 5′-seed/cleavage/anchor regions and stabilize
predicted mmu-let-7a:mRNA duplexes. Genome Res. 18:1571–1581.
2008.PubMed/NCBI
|
|
20
|
Morin RD, O’Connor MD, Griffith M, et al:
Application of massively parallel sequencing to microRNA profiling
and discovery in human embryonic stem cells. Genome Res.
18:610–621. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chaudhry MA, Omaruddin RA, Brumbaugh CD,
Tariq MA and Pourmand N: Identification of radiation-induced
microRNA transcriptome by next-generation massively parallel
sequencing. J Radiat Res. 54:808–822. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Langmead B, Trapnell C, Pop M and Salzberg
SL: Ultrafast and memory-efficient alignment of short DNA sequences
to the human genome. Genome Biol. 10:R252009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pruitt KD, Tatusova T, Klimke W and
Maglott DR: NCBI Reference Sequences: current status, policy and
new initiatives. Nucleic Acids Res. 37:D32–D36. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chan PP and Lowe TM: GtRNAdb: a database
of transfer RNA genes detected in genomic sequence. Nucleic Acids
Res. 37:D93–D97. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pruesse E, Quast C, Knittel K, et al:
SILVA: a comprehensive online resource for quality checked and
aligned ribosomal RNA sequence data compatible with ARB. Nucleic
Acids Res. 35:7188–7196. 2007.PubMed/NCBI
|
|
26
|
Liu C, Bai B, Skogerbø G, et al: NONCODE:
an integrated knowledge database of non-coding RNAs. Nucleic Acids
Res. 33:D112–D115. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li C, Li X, Miao Y, et al:
SubpathwayMiner: a software package for flexible identification of
pathways. Nucleic Acids Res. 37:e1312009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
John-Aryankalayil M, Palayoor ST, Cerna D,
et al: Fractionated radiation therapy can induce a molecular
profile for therapeutic targeting. Radiat Res. 174:446–458. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chang HT, Li SC, Ho MR, et al:
Comprehensive analysis of microRNAs in breast cancer. BMC Genomics.
13(Suppl 7): S182012.PubMed/NCBI
|
|
30
|
Li SC, Liao YL, Ho MR, Tsai KW, Lai CH and
Lin WC: miRNA arm selection and isomiR distribution in gastric
cancer. BMC Genomics. 13(Suppl 1): S132012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li SC, Tsai KW, Pan HW, Jeng YM, Ho MR and
Li WH: MicroRNA 3′ end nucleotide modification patterns and arm
selection preference in liver tissues. BMC Syst Biol. 6(Suppl 2):
S142012.
|
|
32
|
Krek A, Grün D, Poy MN, et al:
Combinatorial microRNA target predictions. Nat Genet. 37:495–500.
2005. View
Article : Google Scholar
|
|
33
|
Rehmsmeier M, Steffen P, Hochsmann M and
Giegerich R: Fast and effective prediction of microRNA/target
duplexes. RNA. 10:1507–1517. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xu Y, Fang F, Zhang J, Josson S, St Clair
WH and St Clair DK: miR-17*suppresses tumorigenicity of
prostate cancer by inhibiting mitochondrial antioxidant enzymes.
PLoS One. 5:e143562010.PubMed/NCBI
|
|
35
|
Zhang X, Ladd A, Dragoescu E, Budd WT,
Ware JL and Zehner ZE: MicroRNA-17-3p is a prostate tumor
suppressor in vitro and in vivo, and is decreased in high grade
prostate tumors analyzed by laser capture microdissection. Clin Exp
Metastasis. 26:965–979. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Almeida MI, Nicoloso MS, Zeng L, et al:
Strand-specific miR-28-5p and miR-28-3p have distinct effects in
colorectal cancer cells. Gastroenterology. 142:886–896. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fernandez-Valverde SL, Taft RJ and Mattick
JS: Dynamic isomiR regulation in Drosophila development.
RNA. 16:1881–1888. 2010. View Article : Google Scholar
|
|
38
|
Guo L, Li H, Liang T, et al: Consistent
isomiR expression patterns and 3′ addition events in miRNA gene
clusters and families implicate functional and evolutionary
relationships. Mol Biol Rep. 39:6699–6706. 2012.
|
|
39
|
Pan HW, Li SC and Tsai KW: MicroRNA
dysregulation in gastric cancer. Curr Pharm Des. 19:1273–1284.
2013.PubMed/NCBI
|
|
40
|
Leite KR, Tomiyama A, Reis ST, et al:
MicroRNA expression profiles in the progression of prostate cancer
- from high-grade prostate intraepithelial neoplasia to metastasis.
Urol Oncol. 31:796–801. 2013. View Article : Google Scholar
|
|
41
|
Schubert M, Spahn M, Kneitz S, et al:
Distinct microRNA expression profile in prostate cancer patients
with early clinical failure and the impact of let-7 as
prognostic marker in high-risk prostate cancer. PLoS One.
8:e650642013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Walter BA, Valera VA, Pinto PA and Merino
MJ: Comprehensive microRNA Profiling of Prostate Cancer. J Cancer.
4:350–357. 2013. View Article : Google Scholar
|
|
43
|
Kobayashi N, Uemura H, Nagahama K, et al:
Identification of miR-30d as a novel prognostic maker of prostate
cancer. Oncotarget. 3:1455–1471. 2012.PubMed/NCBI
|
|
44
|
Poliseno L, Salmena L, Riccardi L, et al:
Identification of the miR-106b~25 microRNA cluster as a
proto-oncogenic PTEN-targeting intron that cooperates with its host
gene MCM7 in transformation. Sci Signal. 3:ra292010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ramberg H, Alshbib A, Berge V, Svindland A
and Taskén KA: Regulation of PBX3 expression by androgen and Let-7d
in prostate cancer. Mol Cancer. 10:502011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bonci D, Coppola V, Musumeci M, et al: The
miR-15a-miR-16-1 cluster controls prostate cancer by
targeting multiple oncogenic activities. Nat Med. 14:1271–1277.
2008.
|
|
47
|
Porkka KP, Ogg EL, Saramaki OR, et al: The
miR-15a-miR-16-1 locus is homozygously deleted in a subset of
prostate cancers. Genes Chromosomes Cancer. 50:499–509. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gonzales JC, Fink LM, Goodman OB Jr,
Symanowski JT, Vogelzang NJ and Ward DC: Comparison of circulating
microRNA 141 to circulating tumor cells, lactate dehydrogenase, and
prostate-specific antigen for determining treatment response in
patients with metastatic prostate cancer. Clin Genitourin Cancer.
9:39–45. 2011. View Article : Google Scholar
|
|
49
|
Hulf T, Sibbritt T, Wiklund ED, et al:
Epigenetic-induced repression of microRNA-205 is associated with
MED1 activation and a poorer prognosis in localized prostate
cancer. Oncogene. 32:2891–2899. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Runkle EA, Zhang H, Cai Z, et al:
Reversion of the ErbB malignant phenotype and the DNA damage
response. Exp Mol Pathol. 93:324–333. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhao L, Bode AM, Cao Y and Dong Z:
Regulatory mechanisms and clinical perspectives of miRNA in tumor
radiosensitivity. Carcinogenesis. 33:2220–2227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhao L, Lu X and Cao Y: MicroRNA and
signal transduction pathways in tumor radiation response. Cell
Signal. 25:1625–1634. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Joung JG, Hwang KB, Nam JW, Kim SJ and
Zhang BT: Discovery of microRNA-mRNA modules via population-based
probabilistic learning. Bioinformatics. 23:1141–1147. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Tran DH, Satou K and Ho TB: Finding
microRNA regulatory modules in human genome using rule induction.
BMC Bioinformatics. 9(Suppl 12): S52008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yoon S and De Micheli G: Prediction of
regulatory modules comprising microRNAs and target genes.
Bioinformatics. 21(Suppl 2): ii93–ii100. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chaudhry MA, Sachdeva H and Omaruddin RA:
Radiation-induced micro-RNA modulation in glioblastoma cells
differing in DNA-repair pathways. DNA Cell Biol. 29:553–561. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang Q, Li P, Li A, et al: Plasma specific
miRNAs as predictive biomarkers for diagnosis and prognosis of
glioma. J Exp Clin Cancer Res. 31:972012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chaudhry MA and Omaruddin RA: Differential
regulation of microRNA expression in irradiated and bystander
cells. Mol Biol. 46:634–643. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Vincenti S, Brillante N, Lanza V, et al:
HUVEC respond to radiation by inducing the expression of
pro-angiogenic microRNAs. Radiat Res. 175:535–546. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Chun-Zhi Z, Lei H, An-Ling Z, et al:
MicroRNA-221 and microRNA-222 regulate gastric carcinoma cell
proliferation and radioresistance by targeting PTEN. BMC Cancer.
10:3672010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xu Y, Zhou B, Wu D, Yin Z and Luo D:
Baicalin modulates microRNA expression in UVB irradiated mouse
skin. J Biomed Res. 26:125–134. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wagner-Ecker M, Schwager C, Wirkner U,
Abdollahi A and Huber PE: MicroRNA expression after ionizing
radiation in human endothelial cells. Radiat Oncol. 5:252010.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang Y, Scheiber MN, Neumann C, Calin GA
and Zhou D: MicroRNA regulation of ionizing radiation-induced
premature senescence. Int J Radiat Oncol Biol Phys. 81:839–848.
2011. View Article : Google Scholar : PubMed/NCBI
|