Introduction
Castration-resistant prostate cancer (CRPC) develops
over time as a consequence of androgen deprivation therapy
(1,2) and this is enhanced by
epithelial-mesenchymal transition and cancer stem cells (reviewed
in ref. 3). Unchecked CRPC followed
by metastasis is lethal (4) due to
limited options for treatment. The mechanisms by which prostate
cancer cells acquire castration resistance are numerous and include
activation of alternative pathways with dominant PI3K/AKT
signaling, androgen receptor (AR) gene mutations leading to
promiscuous activations (5,6), and recently identified AR splice
variants among other factors (7,8).
DNA methylation is one of the most intensely studied
epigenetic modifications that appears to be a decisive event in the
initiation and development of advanced CaP with the process of DNA
hypermethylation preceding to global hypomethylation (9,10).
Friedlander et al (4)
compared CRPC and benign tissue methylation profiles with the
finding of hypermethylated genomic DNA in CRPC patients. Moreover,
in these patients, DNA methylations at individual CpG loci both
within and outside CpG islands were found much more frequently than
common copy number changes. Methylated DNA could thus be a
treatment target for delaying the progression of the
castration-resistant disease using PI3K/AKT inhibitors and
hypomethylating agents (4).
AR plays an essential role in advanced CaP (11). Some CRPC expresses the AR
gene in autocrine pathways and remains dependent on AR while other
cancer cells show decreased AR gene expression attributed to
impaired AR protein stability, X chromosome loss or DNA methylation
silencing and are independent of AR (7). Tian et al (12) showed a link between AR gene
methylation and prostate cancer progression. These authors found
that AR gene methylation in promoter regions was likely
related to prostate stem/progenitor cell stemness and
differentiation. Low expression of the AR in prostate cancer stem
cells and LNCaP progenitor/stem cells were found to be due to high
DNA methyltransferase 1/3 level and MBD2 promoter binding.
Moreover, treatment of prostate cancer cells with 5 µM
5′5-Aza-2′-deoxycytidine (Aza-dC) resulted in the inhibition of
self-renewal/growth of prostate stem/progenitor cells in
vitro, reduced prostate tumorigenicity in vivo followed
by induction of the AR gene and functional protein
expression in a time-dependent manner following 6 days incubation
with 5 µM Aza-dC.
The hypothesis that epigenetically induced AR
expression in CRPC with AR methylated pattern might revert some
deleterious pathways and to some extent reduce the aggressiveness
of the cancer cells, is supported by several studies. McCabe et
al (13) for example found that
Aza-dC inhibited aberrant de novo DNA methylation in the
TRAMP mouse model and prevented CaP development during the drug
administration. Zorn et al (14) reported the delayed emergence of
androgen-independent CaP in castrated TRAMP mice after Aza-dC
treatment. Moreover, combined treatment by castration and Aza-dC
administration showed statistically significant longer survival
than single treatment. In a preclinical study, Gravina et al
(15) used 5-azacytidine for
reactivation of AR gene expression, silenced by DNA methylation in
the PC3 cell line. This led to resensitisation to bicalutamide
(BCLT) responsiveness and subsequent apoptosis. In detail,
5-azacytidine treatment increased the effect of BCLT therapy in
AR-expressing and AR-deficient prostate cancer, both in
vitro and in vivo. Co-treatment with both agents led to
synergistic/additive effects in nude male mice xenografted with
22rv1 and PC3 cells (AR-expressing or AR-deficient cell lines,
respectively) followed by significantly reduced tumor mass and
delayed cancer progression. In the co-treated cell lines (22rv1 and
PC3), increased cell cycle and apoptosis proteins expression were
observed.
This study describes the epigenetic consequences of
the combined treatment of two inhibitors, sodium butyrate (NaB) as
a histone deacetylase (HDAC) inhibitor and 5′-Aza-2′-deoxycytidine
(Aza-dC) as a DNA methyltransferase (DNMT) inhibitor, in both
cancer and normal prostate cells. DNA methyltransferases (DNMTs)
and histone deacetylases (HDACs) are co-regulators of the AR and
could imply potential targets for affecting androgen receptor
function and stability. These are now an option in epigenetic
treatment (16) with the aim of
overcoming the mechanism of hormonal resistance and in its
consequences, of target therapy to regulation of the AR without
therapy based on hormone treatment. Our previous results (17) showed demethylation of specific CpG
sites in the AR gene in the DU145 prostate cancer cell line
following co-treatment with Aza-dC and NaB. The focus of this study
was the methylated AR gene with subsequent epigenetic modulation
leading to targeted histone acetylation and AR gene re-expression.
Our results imply that the epigenetic drugs used, depending on the
concentration, affected the acetylation level of histones H3 and H4
in a vicinity of the AR gene promoter. In addition, the used
epigenetic agents induced activation and maintaining of G2/M cell
cycle arrest in RWPE-1 cells. Better survival in normal RWPE-1
cells compared to cancer DU145 cells implies the selective toxic
effects of the used inhibitors.
Materials and methods
Cell culture, treatment conditions and
viability assay
The androgen-independent human prostate cancer cell
line DU145 was purchased from ATCC (Rockville, MD, USA), maintained
in DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS),
0.01% antibiotics, 2 mM L-glutamine. Non-tumorigenic, immortalized
human prostate cell line RWPE-1 was kindly provided by the
Department of Experimental Biology, Masaryk University (Brno, Czech
Republic). The normal cell line RWPE-1 was cultivated in
keratinocyte-SFM medium (kit) with L-glutamine, human recombinant
epidermal growth factor (EGF) and bovine pituitary extract (BPE)
(Gibco) supplemented with a final 0.01% concentration combination
of penicillin and streptomycin, 0.01% concentration of amfomycin
and 0.005% concentration of gentamycin. All cells were maintained
at 37°C and 5% CO2 atmosphere. Both cell lines were
treated with NaB (Sigma-Aldrich), Aza-dC (Sigma-Aldrich) and their
combinations for 2 and 6 days when medium and agents were changed
after 2 days. Cell viability assays of cells were performed using
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide agent
(MTT) as described (17). The
cancer cell line DU145 was seeded on 96-well plates at 4,500 cells
per well and RWPE-1 cells at 6,000 cells per well and increased to
40–50% confluent prior to the first treatment. The cell culture
medium with inhibitors was changed after 2 days for 6-day
treatments. All treatments were performed in triplicate. The
percentage of viable cells was calculated as follows: average
absorbance of treated cells/average absorbance of control cells ×
100.
mRNA analysis using RT-qPCR
The cells were seeded in 100-mm dishes
(1×106 cells for RWPE-1 and 7×105 for DU145
cells) and treated with 5 µM Aza-dC, 5 mM NaB and their
combinations (0.5 µM Aza-dC + 1 mM NaB, 0.5 µM Aza-dC
+ 5 mM NaB and 5 µM Aza-dC + 5 mM NaB) for 2 and 6 days when
medium and agents were exchanged after 2 days in prolonged 6-days
culture. Total RNA from both cell lines was isolated with High Pure
RNA Isolation kit (Roche) and 1,000–1,500 ng of total RNA was
converted to cDNA using Transcriptor First Strand cDNA synthesis
kit (Roche), in both cases according to the manufacturer's
instructions. For following quantitative real-time PCR analyses,
the total amount of 100 ng of cDNA from each sample was amplified
using Taq-Man probes labelled with hexafluorescein and Thermo-Start
DNA Polymerase (AB gene) using the real-time PCR analyser
Rotor-Gene RG-3000 (Corbett Research). Primers and probe sequences
for AR, PSA (prostate specific antigen) and GAPDH genes are
summarized in Table I. The
experiments were performed in triplicate with a similar pattern of
results.
 | Table IPrimer sequences and probes used for
mRNA and ChIP-qPCR assays. |
Table I
Primer sequences and probes used for
mRNA and ChIP-qPCR assays.
| GOI | Forward
(5′-3′) | Reverse
(5′-3′) | Probe (5′-3′) | Product (bp) |
|---|
| AR |
ATCCCAGTCCCACTTGTGTC |
GGTCTTCTGGGGTGGAAAGT |
AAGCGAAATGGGCCCTGGA | 137 |
| PSA |
CGGAGAGCTGTGTCACCAT |
CACAATCCGAGACAGGATGA |
CGTGGATTGGTGCTGCACCC | 95 |
| GAPDH |
GAAGATGGTGGGGATTTC |
GAAGGTGAAGGTCGGAGT |
CAAGCTTCCCGTTCTCAGCC | 226 |
| AR (ChIP) |
CAGGAGCTATTCAGGAAGCAG |
GGCTTTGGAGAAACAAGTGC | CTCCTGCC | 93 |
Chromatin immunoprecipitation coupled
with quantitative PCR (ChIP-qPCR)
ChIP was performed on both cell lines as described
(17) with the following
modifications: briefly, the cells were seeded in 100-mm dishes
(1×106 cells for RWPE-1) and in 150-mm dishes
(1.9×106 cells for DU145). After a formaldehyde
cross-linking terminated by glycine, RWPE-1 cells were washed with
Dulbecco's phosphate-buffered saline (D-PBS) instead of standard
PBS used for DU145 line. Cells were immunoprecipitated with 2
µg anti-acetyl histone H3 rabbit polyclonal antibody (cat.
06-599), and anti-acetyl histone H4 rabbit polyclonal antibody
(cat. 06-866, both from Millipore), and normal mouse IgG polyclonal
antibody (cat. 12-371, Millipore). For input, 100 µg of each
chromatin lysates and 1 µl of proteinase K (100
µg/ml) were added to each tube, incubation at 55°C for 3 h
with gentle agitation was performed followed by DNA purification
with QIAquick® PCR Purification kit (Qiagen).
Quantitative PCR analyses were performed on LightCycler 480 (Roche)
with probe no 51 (Universal Probe Library cat. no. 04688481001)
using ProbeFinder assay design software and primers amplifying AR
gene promoter region (Table I). DNA
from each input sample was diluted 10× and primer efficiency was
tested. All experiments were repeated three times independently and
all measurements were performed in triplicate.
Cell cycle analysis
Briefly, the cell lines were seeded in 6-well plates
(2.2×105 cells for RWPE-1 and 1.5×105 cells
for DU145) and treated with the same inhibitors and their
combinations for 2 and 6 days as described above. Cells were
harvested at indicated times after treatment, also detached cells
were collected. Samples were fixed in cold 70% ethanol. After
treatment with RnaseA, samples were stained with propidium iodide
(PI). Cellular DNA content was analyzed using flow cytometry (BD
FACSVerse, BD, USA) and collected data were processed using BD
FACSuite (BD). At least 10,000 cells per sample were analyzed.
Statistical analysis
Multifactoral analysis of variance (ANOVA) with
post-hoc two-tailed Dunnett's t-test and Bonferroni multiple
comparison test were used for cell viability assays and
quantitative experiments. All statistical analyses were performed
with the SPSS software version 15 (SPSS, Inc. Chicago, Il, USA) and
the significance level was set at p<0.05 (two-tailed).
Results
Aza-dC decreases cytotoxicity of NaB in
prostate cancer cells
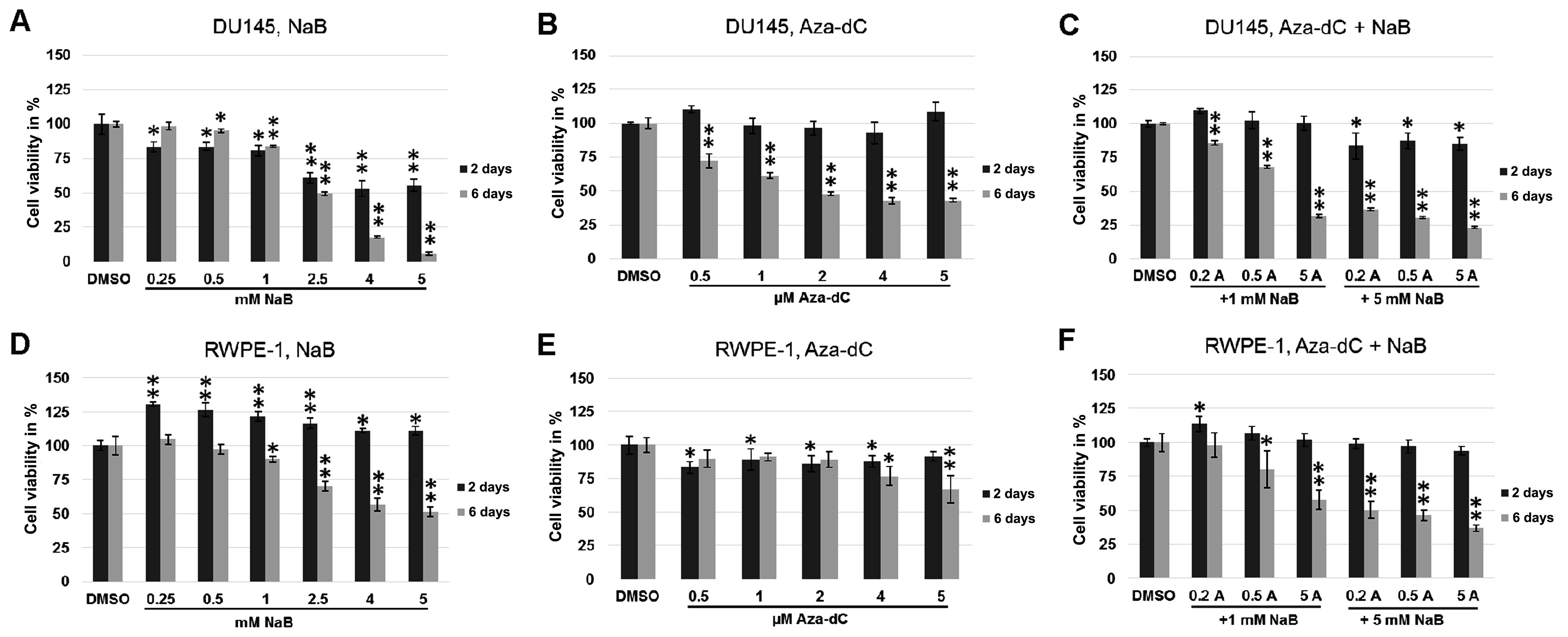
Cell viability was analysed using MTT assay to
compare cell cytotoxicity after treatment with one of the
inhibitors used or inhibitor combinations. In DU145 cells treated
with NaB or Aza-dC, or combinations of different concentrations of
Aza-dC with NaB (Fig. 1), the cell
viability was significantly lower following all treatments after 6
days than in 2-day experiments (p≤0.0004 for Aza-dC treatment,
p≤0.0001 for co-treatment, Fig. 1B and
C, respectively). The 6-day treatment with NaB (Fig. 1A) showed significantly lower cell
viability, ranging from 2.5 mM concentration (p=0.002 and
p<0.0001 for 4 and 5 mM NaB), and higher cell viability after
treatment with 0.25 and 0.5 mM concentrations (p=0.0001 for both
treatments) compared with the same conditions in the 2-day
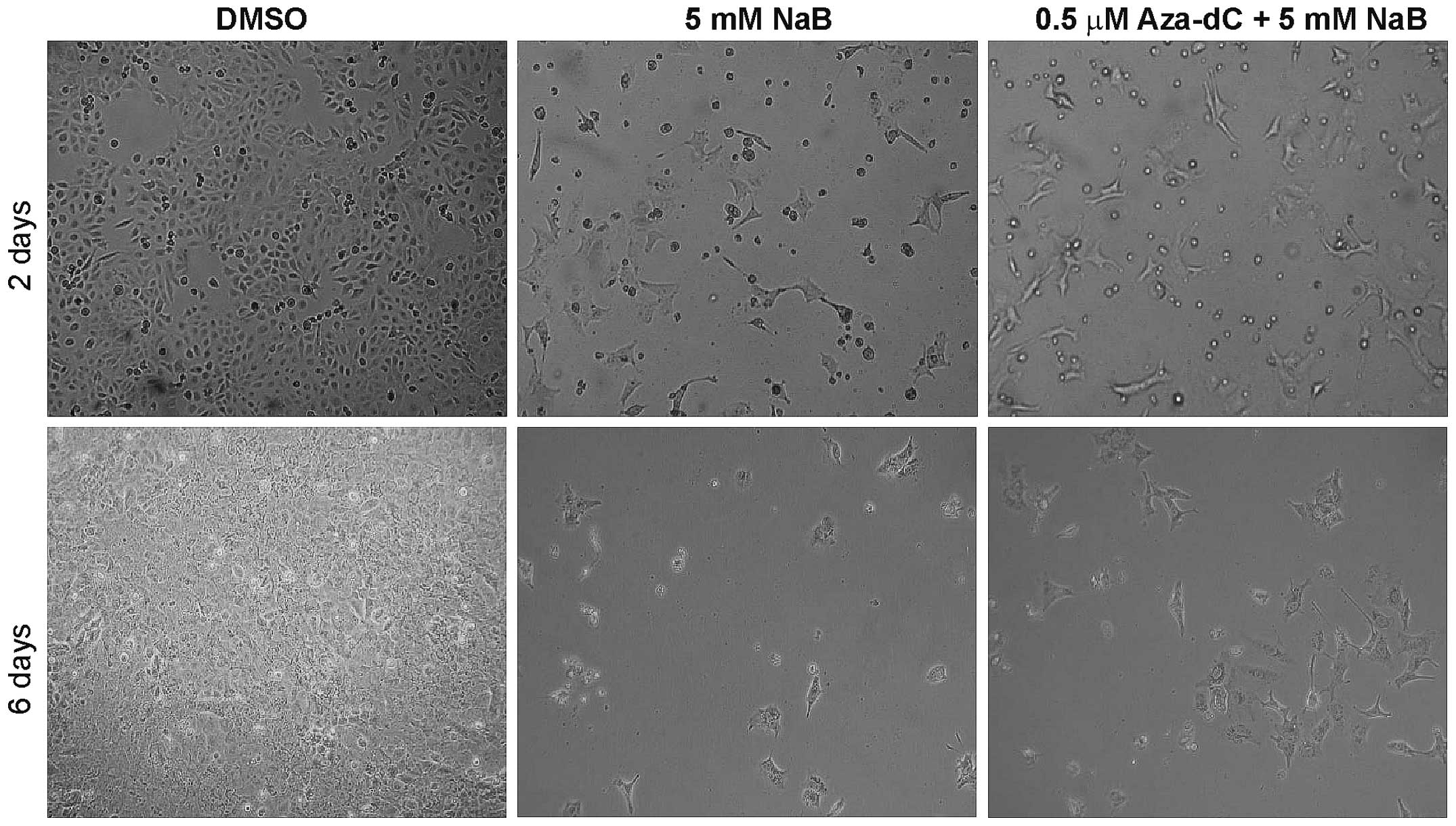
experiment. The images of prostate cancer cells DU145 (Fig. 2) demonstrate changes in cell
viability after 2 and 6 days of cultivation with 5 mM NaB and with
0.5 µM Aza-dC + 5 mM NaB co-treatment compare with DMSO.
Changes of cell viability in DU145 cells are comparable with the
results of MTT assay (Fig. 1A and
C) and suggest a lower cytotoxicity of 0.5 µM Aza-dC + 5
mM NaB combination used than 5 mM NaB treatment alone.
In normal RWPE-1 cells, the 6-day incubation led to
significantly lower cell viability on treatment with NaB alone
(p<0.0001; Fig. 1D) and NaB
co-treatment with Aza-dC (p≤0.008; Fig.
1F) in all concentrations compared to 2 days. The highest 4 and
5 µM Aza-dC concentrations caused higher toxicity after 6
days than the 2-day experiment (Fig.
1E, p=0.028 and 0.007, respectively). In contrast, no
differences in cell viability were observed on treatment with 0.5–2
µM Aza-dC concentrations between 2- and 6-day
experiments.
A combination of the Aza-dC and NaB
treatment induces AR gene re-expression in prostate cancer
cells
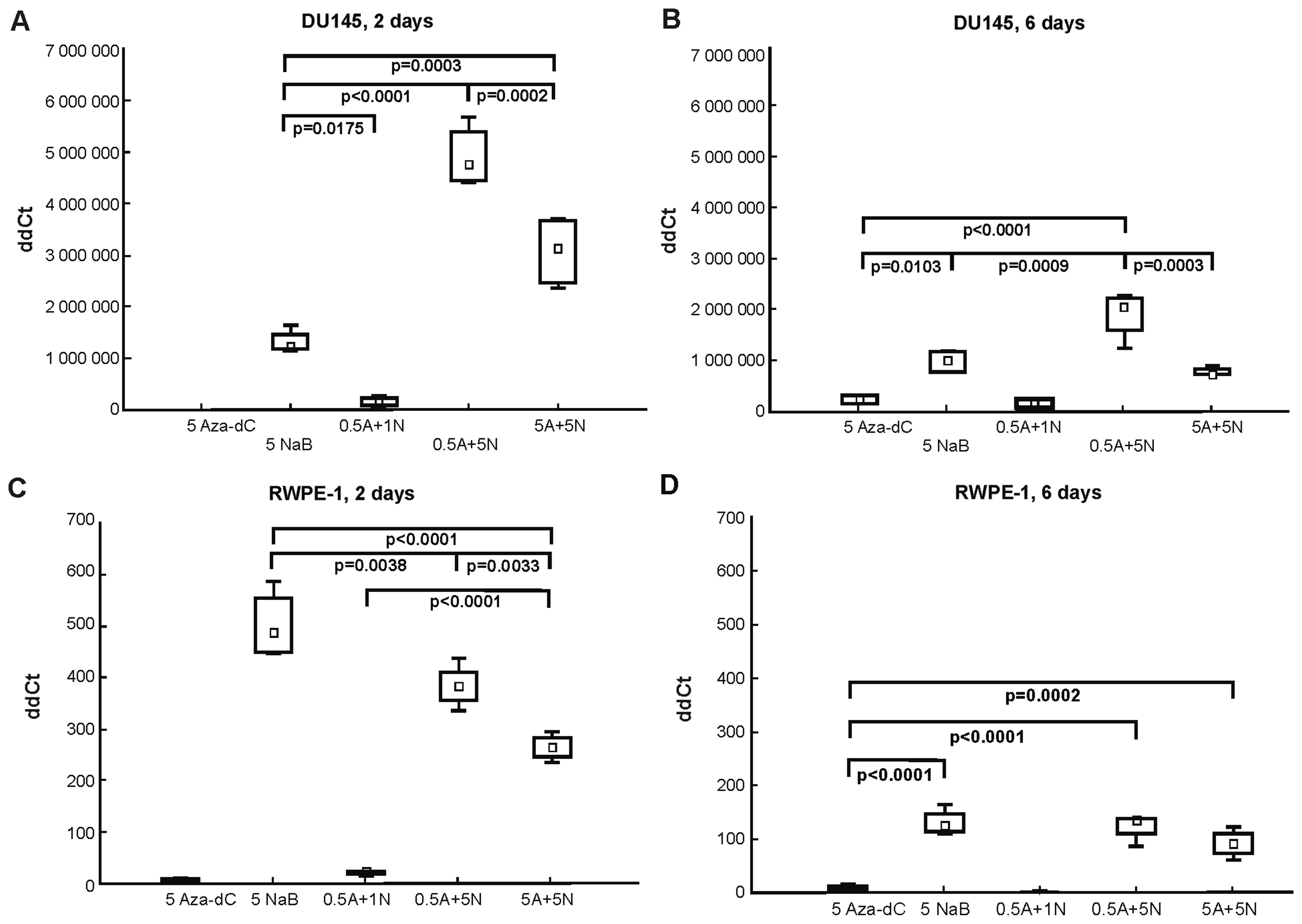
When designing the RT-qPCR, we followed results from
the MTT assay (Fig. 1) and from our
previous bisulfite sequencing results in DU145 cells (17), where the most effective DNA
demethylation effect was after 2-day treatment with 0.5 µM
Aza-dC + 5 mM NaB in the AR gene. Here in DU145 cells (Fig. 3A and B), co-treatment with the
combination of 0.5 µM Aza-dC + 5 mM NaB led to most
significantly higher AR gene re-expression after both 2- and 6-day
incubations. The 5 µM Aza-dC cell treatment had no effect on
AR gene re-expression after 2 days (Ct undetectable; Fig. 3A) as was further confirmed by mRNA
analysis, although a weak signal detection was observed after the
6-day exposure (Fig. 3B). As the
0.5 µM Aza-dC + 5 mM NaB co-treatment, the 5 mM NaB
treatment and 5 µM Aza-dC + 5 mM NaB combination were also
effective in inducing significant AR mRNA re-expression.
In normal RWPE-1 cells (Fig. 3C and D), we found that AR gene
expression was enhanced by all treatments with the highest effect
after 5 mM NaB treatment, where only the 2-day experiment showed
statistically significant differences between 5 mM NaB, 0.5
µM Aza-dC + 5 mM NaB and 5 µM Aza-dC + 5 mM NaB
co-treatments.
Comparing the results from the mRNA analysis in both
cell lines used (Fig. 3), a
tendency to overall decrease in AR mRNA expression after 6-day
treatment was observed. The treatments with 5 µM Aza-dC and
0.5 µM Aza-dC + 1 mM NaB in DU145 and RWPE-1 cells were weak
or ineffective in re-expressing the AR. However, no PSA gene
expression at either time course or in either cell line was
detected (Ct undetectable).
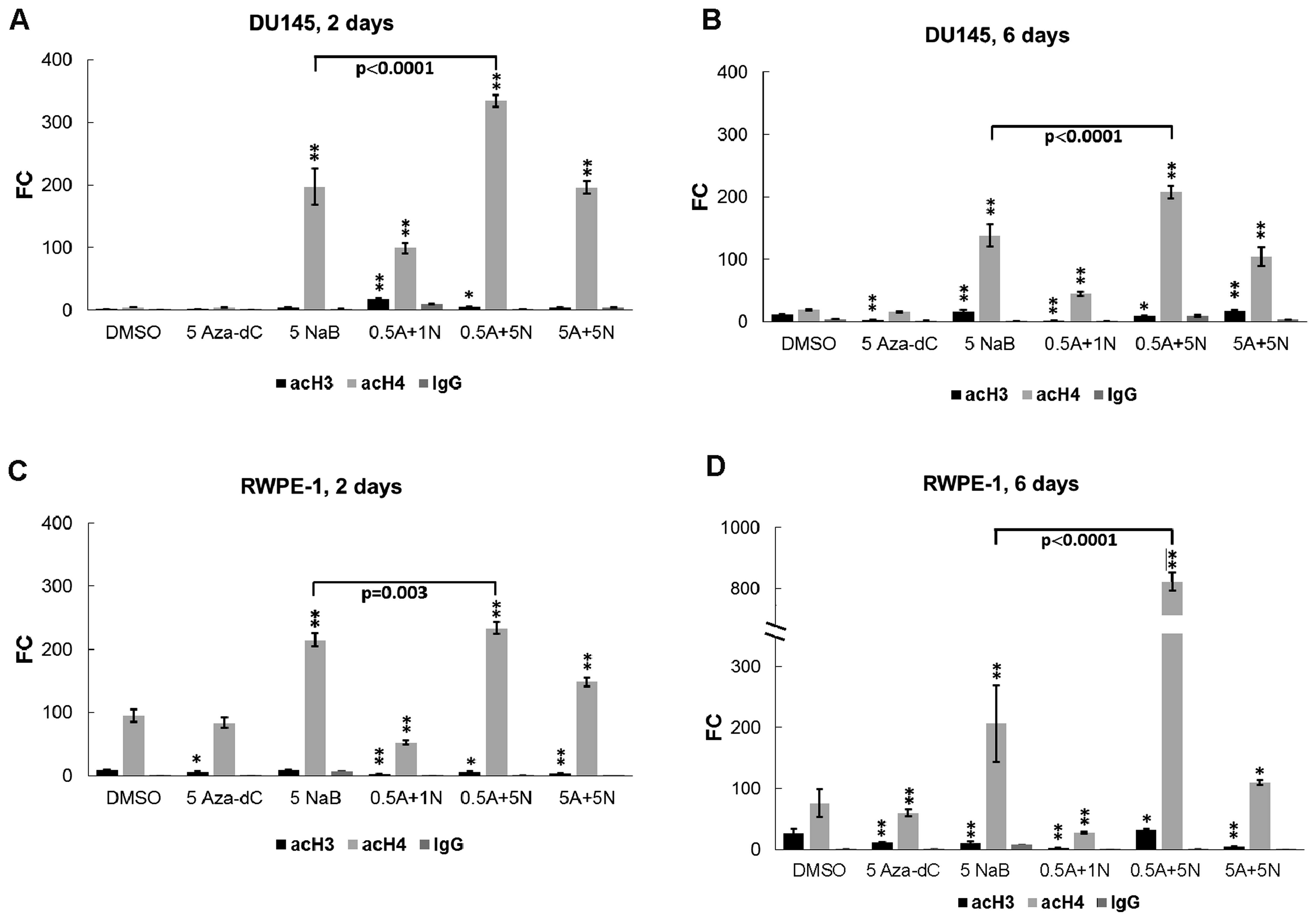
The NaB increases histone H4 acetylation,
but not histone H3 in prostate cancer cells affected with
epigenetic modulators
Since histone deacetylation is a hallmark of silent
condensed chromatin, to explore whether single NaB or its
combinations with Aza-dC could be effective in initiating the AR
gene re-expression through histone re-acetylation, we used the same
treatment as described for mRNA analysis followed by chromatin
immunoprecipitation coupled with qPCR. Sonicated chromatin samples
were immunoprecipitated with anti-acetyl histone H3-lysine 4,
-lysine 9, -lysine 14 and -lysine 18 (H3K4, H3K9, H3K14 and H3K18,
respectively) and anti-acetyl histone H4-lysine 5, -lysine 8,
-lysine 12 and -lysine 16 (H4K5, H4K8, H4K12 and H4K16,
respectively) antibodies. Normal IgG antibody served as a negative
control, and sonicated chromatin samples were processed with no
added antibody (NoAb sample) as a mock control (or noise control).
The NoAb sample was processed as the standard sample used for IP
without antibody and served as an internal control to minimize
noise of the manual workflow. ChIP-qPCR results were analyzed by
evaluating the signal of enrichment over noise normalized to input
(18).
In DU145 cells treated for 2 days with 0.5 µM
Aza-dC + 1 mM NaB and the 0.5 µM Aza-dC + 5 mM NaB (Fig. 4A), co-treatments were significantly
more effective for histone H3 acetylation than the DMSO control.
Monitored histone H4 sites were acetylated following all treatments
except for the 5 µM Aza-dC treatment (Fig. 4A). In 6 days (Fig. 4B), the histone H3 acetylation
increased after cell treatment with 5 mM NaB alone and with the 5
µM Aza-dC + 5 mM NaB combination, and decreased after
treatments with 5 µM Aza-dC, 0.5 µM Aza-dC + 1 mM NaB
and 0.5 µM Aza-dC + 5 mM NaB in comparison with untreated
DMSO. We observed significant increase in histone H4 acetylation in
the AR gene promoter region for all treatments, while the 5
µM Aza-dC treatment was ineffective (Fig. 4B).
In the RWPE-1, after 2-day treatment (Fig. 4C), we found significant decrease in
the histone H3 acetylation targeted to the AR gene promoter for all
used treatments except for 5 mM NaB. Significant upregulation of
histone H4 acetylation was observed in cells treated with 5 mM NaB
alone and with subsequent combinations of 0.5 µM Aza-dC + 5
mM NaB and 5 µM Aza-dC + 5 mM NaB. Downregulation of histone
H4 acetylation was observed following treatment with 0.5 µM
Aza-dC + 1 mM NaB combination but no significant change was
detected after treatment with 5 µM Aza-dC alone. After 6
days (Fig. 4D), the RWPE-1 cells
showed lower levels of histone H3 acetylation for all used
treatments except 0.5 µM Aza-dC + 5 mM NaB application
compare to DMSO.
Comparing histone H3 and H4 acetylations between 2-
and 6-day experiments, we found a decrease in histone H3
acetylation in DU145 cells treated with 0.5 µM Aza-dC + 1 mM
NaB combination (p=0.049; Fig. 4A and
B) while control DMSO treatment and subsequent applications of
5 µM Aza-dC alone and 5 µM Aza-dC + 5 mM NaB
combination induced significant time-dependent enrichment (p=0.001,
0.039 and 0.012, respectively). The DU145 cell line showed
significantly decreased histone H4 acetylation after all treatments
except the control DMSO and 5 µM Aza-dC applications that
led to increased histone H4 acetylation. In normal RWPE-1 cells
(Fig. 4C and D), the control DMSO,
5 µM Aza-dC and 0.5 µM Aza-dC + 5 mM NaB treatment
showed higher levels of histone H3 acetylation in comparison with
shorter 2-day incubation (p=0.036, p=0.025 and p=0.037,
respectively). We found no significant changes in histone H3
acetylation for the other treatments. For increasing histone H4
acetylation in normal prostate cells effective was the 0.5
µM Aza-dC + 5 mM NaB combination. Other treatments were
significantly less effective after 6-day exposure.
Cell cycle distribution explains the NaB
induced cell death in prostate cancer cells
To determine the influence of Aza-dC and NaB on cell
cycle regulation, exponentially growing RWPE-1 and DU145 cells were
treated with the same treatment scheme as for RT-PCR and ChIP
analysis. Cell cultures, including detached cells, were collected,
stained with PI and DNA content was assessed by flow cytometry
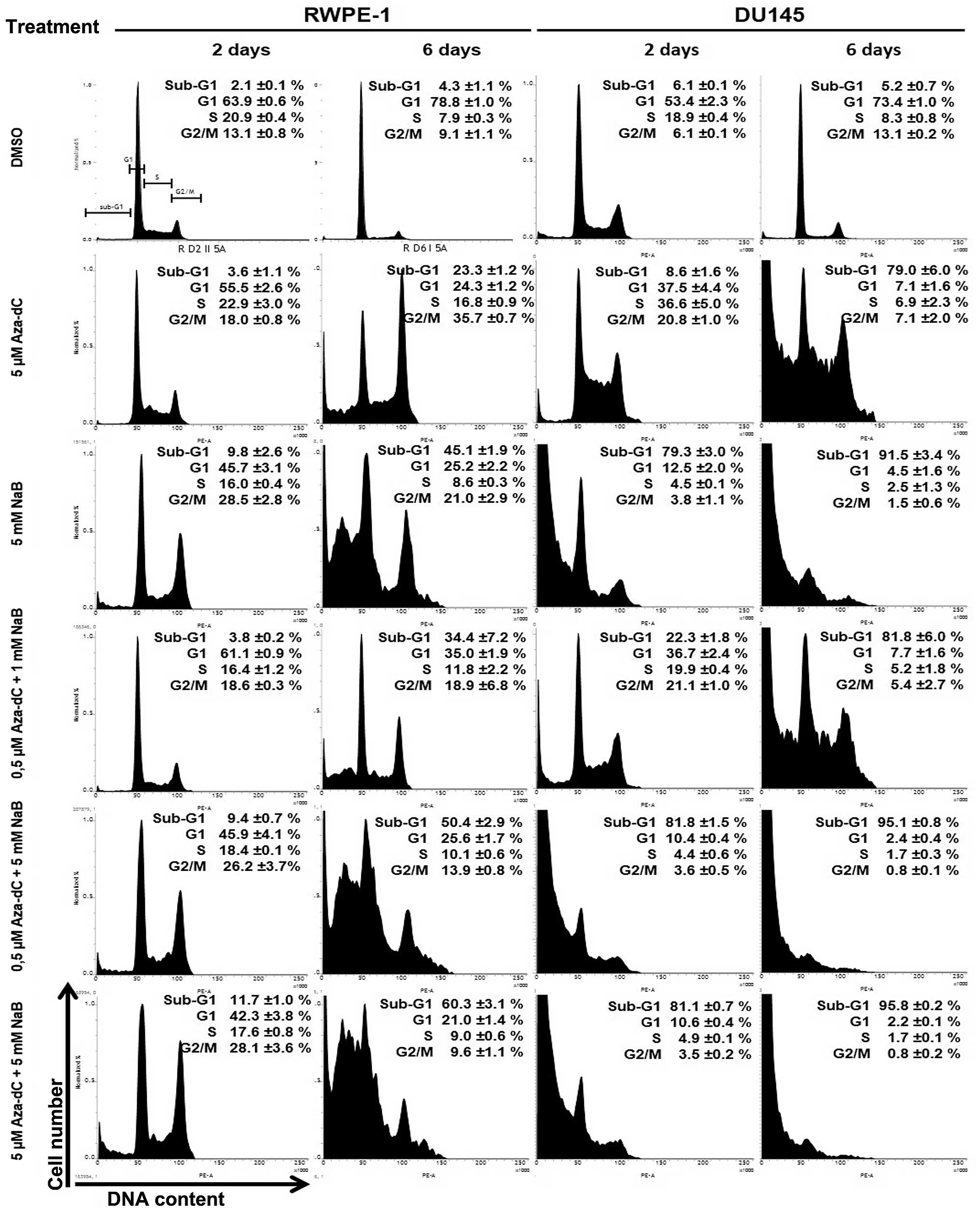
(Fig. 5).
By comparing the profiles of control (DMSO) and
Aza-dC treated cell cultures after 2 days, the accumulation of
cells in S and G2/M stages was shown moderately for RWPE-1 cells
and more prominent in DU145 cells. Prolonged Aza-dC treatment (6
days) revealed obvious difference in effectiveness of G2/M arrest
between compared cell lines. While significant and specific
accumulation of RWPE-1 cells in the G2/M compartment of the cell
cycle was observed, the DU145 cells massively died, yielding
predominant sub-G1 population reaching up to 80%. The treatment
with 5 mM NaB resulted in a much more conclusive contrast between
normal and cancer cells. The RWPE-1 cells survived 2-day NaB
treatment effectively, employing the G2/M arrest, while 79% of
DU145 cells was shifted to sub-G1 compartment, indicating cell
death. After 6 days of the NaB treatment, more than one half of
RWPE-1 cells were cycling, at the same time >90% of DU145 cells
were dead, belonging to sub-G1 cell population.
Combination treatment at lowest concentration (0.5
µM Aza-dC +1 mM NaB) was more efficient than the Aza-dC
treatment alone. G2/M accumulation was observed after 2-day
treatment, with appearance of the sub-G1 cell population
specifically in DU145 cells. Prolonged 6-day treatment led to
divergent response of both cell lines, revealing effective G2/M
arrest for RWPE-1 cell line and high degree of cell death for DU145
exhibiting <20% of cells in the cycle. Combination therapy with
higher concentrations (0.5 µM Aza-dC + 5 mM NaB and 5
µM Aza-dC + 5 mM NaB) resulted in generation of strong
sub-G1 population in DU145, reaching >80% within 2 days of
treatment. More than 95% of DU145 cells were shown to be sub-G1 for
both concentrations after 6 days, suggesting that at 0.5 µM
Aza-dC + 5 mM NaB combination treatment reached its plateau.
Highest concentrations of combination treatment resulted in
dose-dependent G2/M accumulation of RWPE-1 cells after 2 days,
followed by appearance of sub-G1 cells after day 6.
Discussion
Histone modification and DNA methylation are
associated with transcriptional repression and integrally linked
with following synergistic/additive effects (19). In our study we used two types of
epigenetic agents: sodium butyrate, a naturally occurring HDAC
inhibitor known for its selective cell toxicity, and
5′-Aza-2′-deoxycytidine, an inhibitor DNMT, and one of the most
promising and extensively used demethylating agents. The aim of
this study was to select optimal concentrations or combination of
concentrations of these inhibitors to achieve an epigenetic effect
leading to AR gene restoration in androgen-independent prostate
cancer cells.
The cancer DU145 cell line is androgen-independent
and characterized by strong DNA methylation of the AR gene
(17,20). This cell line is used as a model for
simulating the conditions found in CRPC patients with changed
genomic methylation patterns. We found that cancer line DU145
treated with the inhibitors used showed a substantial decrease in
cell viability especially after 5 mM NaB administration and on
6-day exposure. NaB toxicity is a problem as to be effective as an
inhibitor of histone deacetylases, NaB at the higher 5 mM
concentration is needed. Comparing the required levels of
cytotoxicity for NaB alone and NaB + Aza-dC co-treatments (0.5 and
5 µM Aza-dC with 5 mM NaB), it is clear that adding of the
Aza-dC could have the same or better anti-proliferative effect with
lower cell toxicity than 5 mM NaB alone in DU145 cells. The shorter
2-day treatments with used inhibitors had little effect, except for
the NaB treatment that showed slight cytotoxicity in DU145 cells,
and on the other hand growth stimulation in RWPE-1 cells. Similar
results were reported by Paskova et al (21), where NaB (in 0.5, 1, 2.5 or 5 mM
concentrations) had no toxic effect on normal RWPE-1 cells after
2-day treatment.
In accordance with the cell viability assay, the
cell cycle distribution results showed similar effects of the used
treatments, namely massive cell death upon 5 mM NaB treatment
(administered alone and in co-treatment with Aza-dC) in DU145 cells
compared to RWPE-1 cells. The finding that NaB inhibits cell
viability and proliferation in DU145 cells and in certain other
prostate cancer cell lines, in a time- and dose-dependent manner
has been described in several studies (22–25).
Pro-apoptotic activity induced by high doses of Aza-dC (~5
µM) has also been reported (26). In our study, Aza-dC alone also
induced cell death (79% cells in sub-G1 cell population) in DU145
cells, however the co-treatments did not impair the toxic effects
of NaB itself on the cell cycle. Although the cell death using a
single inhibitor appears to be high, the combination of the two
inhibitors might not have cumulative impact on prostate cancer
cells. The appearance of the sub-G1 population of cells is
apparently due to the DNA fragmentation of dead cells and could be
a result of the pro-apoptotic NaB activity in cancer DU145 cells
(24,25). A strong contrast of cell cycle
distribution between both cell lines is given also by features of
p-53 proficient RWPE-1 and p-53 deficient DU145 cells leading to
p53-dependent activation and maintenance of G2/M cell cycle arrest
and better survival in RWPE-1 cells compared to DU145 cells.
DU145 cells contain a methylated AR gene, thus the
cell line has very low or undetectable AR gene expression (no Ct
value in control DMSO treatment after 50 cycles was detected). The
Ct value was established arbitrarily as Ct 50 at both time-points.
Hence, the real re-expression of the AR gene could be considered
higher than that calculated here for DU145 cells. In DU145, the
most effective AR mRNA restoration was treatment with 0.5 µM
Aza-dC + 5 mM NaB for both time-points, while the RWPE-1 cells did
not show the same pattern and no significant difference between
individual treatments was found after day 6. Although we noted an
increased AR gene expression in RWPE-1, the level of mRNA was low
compared to AR expression in DU145 cells. We observed a tendency to
a decline in AR gene expression in a time-dependent manner apparent
for both cancer and normal cell lines. The declining trend was also
shown in histone H4 acetylation of the AR gene. As we changed the
medium after 2 days, the half-life of inhibitors and recovery of
remodulation enzymes, HDAC I and IIa that are targets of NaB
(27), could result in lower AR
gene re-expression and especially lower histone H4 acetylation.
We found significant re-acetylation of the histone
H4 compared to control DMSO in cancer DU145 cells treated with the
0.5 µM Aza-dC + 1 mM NaB combination, while the same
treatment induced significantly lower H4 acetylation levels in
normal RWPE-1 cells. On the other hand, 0.5 µM Aza-dC + 5 mM
NaB co-treatment had the same effect on both cell lines by
increased enrichment of histone H4 acetylation while the 5
µM Aza-dC + 5 mM NaB had only moderate effect. This implies
that low µM-concentration of Aza-dC together with
mM-concentration of NaB could have either an additive/synergistic
or antagonistic effect on chromatin remodeling.
The CRPC stadium harbours heterogeneous features
present with multiple alterations in the AR gene function. Besides
mutations, copy number changes, deregulation of coregulators, and
splice variants in AR gene, DNA methylation appears to be a minor
modification contributing to AR gene dysfunction. However, based on
a relatively recent study (12),
the AR gene promoter methylation is likely related to prostate
stem/progenitor traits and linked to enhanced castration
resistance. Prostate tumor consists of a mixture of normal/benign,
cancer and cancer stem cells with aberrant methylation profile. It
is noteworthy that AR gene re-expression using epigenetic therapy
leads to decreased proliferation and longer survival (13–15).
AR signaling does not aggravate the development of the disease. It
may act as a physiological regulator of AR downstream genes and
sensitizes the cancer cells to other phases of therapy. For this
reason, research efforts might be focused on keeping the active AR
gene at low expression level to preserve the native AR signaling
axis and to determine whether the restored AR gene could have
regulation capability. Although we showed a markedly increased AR
mRNA expression and significant re-acetylation of histone H4 around
AR gene promoter upon the co-treatment in DU145 cells, we assume
that the corresponding AR gene regulation was not restored as the
PSA level was not detected. Moreover, the high frequency of sub-G1
population of the dead cells was observed. To promote growth in
androgen-independent CaP, the active AR should have selective and
direct upregulation effect on M-phase cell cycle genes (28). Therefore, the cell cycle
distribution results appear to be solely a consequence of NaB and
Aza effects on cell death genes (24,25)
and not by activation/restoration of AR. Modulation of the AR
activity is mediated by the action of numerous coactivators and
corepressors [histone modifiers, splice proteins, proteins of RNA
metabolism and DNA repair, and cell cycle regulators (29)] and by phosphorylation of both AR and
the mentioned co-regulators (30).
It indicates further analysis is required aimed at mechanisms of
action of epigenetic inhibitors and their potential role leading to
completely active AR.
In conclusion, the impact of the combined treatment
shows cancer cell reduction of their proliferative activity and
changes in cell cycle distribution in comparison with normal cells
with time-dependent effect. Further, the combined treatment both
strongly increased histone H4 acetylation in the AR gene promoter
and induced the AR gene re-expression in cancer cells in comparison
with the normal cell line. Our results imply selective toxicity of
used inhibitors with a suggestion that appropriately chosen
inhibitor combination and concentration may have synergism/additive
potential for therapeutic procedures in CRPC patients.
Abbreviations:
|
AR
|
androgen receptor
|
|
Aza-dC
|
5′-Aza-2′-deoxycytidine
|
|
BCLT
|
bicalutamide
|
|
BPE
|
bovine pituitary extract
|
|
CaP
|
prostate cancer
|
|
ChIP
|
chromatin immunoprecipitation
|
|
CRPC
|
castration-resistant prostate
cancer
|
|
DMSO
|
dimethylsulfoxide
|
|
DNMTs
|
DNA methyltransferases
|
|
D-PBS
|
Dulbecco's phosphate-buffered
saline
|
|
FBS
|
fetal bovine serum
|
|
HDACs
|
histone deacetylases
|
|
MBD2
|
methyl-CpG binding domain protein
2
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
NaB
|
sodium butyrate
|
|
PBS
|
phosphate-buffered saline
|
|
PI
|
propidium iodide
|
|
PI3K/AKT
|
phosphatidylinositol-3-kinase/protein
kinase B
|
|
PSA
|
prostate specific antigen
|
|
TRAMP
|
transgenic adenocarcinoma mouse
prostate
|
Acknowledgments
The authors would like to thank Jana Holinková, Eva
Pimrová, Lenka Prokopová, Jana Fialová Kučerová and Kateřina
Čížková for technical assistance. Alexander Oulton is acknowledged
for critical reading of the manuscript. This study was supported by
grants IGA_LF_2016_013, PU I LO1304 and RVO: 61989592 from the
Czech Ministry of Education and The Kellner Family Foundation.
References
|
1
|
Han G, Foster BA, Mistry S, Buchanan G,
Harris JM, Tilley WD and Greenberg NM: Hormone status selects for
spontaneous somatic androgen receptor variants that demonstrate
specific ligand and cofactor dependent activities in autochthonous
prostate cancer. J Biol Chem. 276:11204–11213. 2001. View Article : Google Scholar
|
|
2
|
Steinkamp MP, O'Mahony OA, Brogley M,
Rehman H, Lapensee EW, Dhanasekaran S, Hofer MD, Kuefer R,
Chinnaiyan A, Rubin MA, et al: Treatment-dependent androgen
receptor mutations in prostate cancer exploit multiple mechanisms
to evade therapy. Cancer Res. 69:4434–4442. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li P, Yang R and Gao W-Q: Contributions of
epithelial-mesenchymal transition and cancer stem cells to the
development of castration resistance of prostate cancer. Mol
Cancer. 13:552014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Friedlander TW, Roy R, Tomlins SA, Ngo VT,
Kobayashi Y, Azameera A, Rubin MA, Pienta KJ, Chinnaiyan A, Ittmann
MM, et al: Common structural and epigenetic changes in the genome
of castration-resistant prostate cancer. Cancer Res. 72:616–625.
2012. View Article : Google Scholar
|
|
5
|
Berrevoets CA, Veldscholte J and Mulder E:
Effects of antiandrogens on transformation and transcription
activation of wild-type and mutated (LNCaP) androgen receptors. J
Steroid Biochem Mol Biol. 46:731–736. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McDonald S, Brive L, Agus DB, Scher HI and
Ely KR: Ligand responsiveness in human prostate cancer: Structural
analysis of mutant androgen receptors from LNCaP and CWR22 tumors.
Cancer Res. 60:2317–2322. 2000.PubMed/NCBI
|
|
7
|
Felgueiras J, Silva JV and Fardilha M:
Prostate cancer: The need for biomarkers and new therapeutic
targets. J Zhejiang Univ Sci B. 15:16–42. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schrecengost R and Knudsen KE: Molecular
pathogenesis and progression of prostate cancer. Semin Oncol.
40:244–258. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nelson WG, Yegnasubramanian S, Agoston AT,
Bastian PJ, Lee BH, Nakayama M and De Marzo AM: Abnormal DNA
methylation, epigenetics, and prostate cancer. Front Biosci.
12:4254–4266. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yegnasubramanian S, Haffner MC, Zhang Y,
Gurel B, Cornish TC, Wu Z, Irizarry RA, Morgan J, Hicks J, DeWeese
TL, et al: DNA hypomethylation arises later in prostate cancer
progression than CpG island hypermethylation and contributes to
metastatic tumor heterogeneity. Cancer Res. 68:8954–8967. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Scher HI and Sawyers CL: Biology of
progressive, castrationresistant prostate cancer: Directed
therapies targeting the androgen-receptor signaling axis. J Clin
Oncol. 23:8253–8261. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tian J, Lee SO, Liang L, Luo J, Huang CK,
Li L, Niu Y and Chang C: Targeting the unique methylation pattern
of androgen receptor (AR) promoter in prostate stem/progenitor
cells with 5-aza-2′-deoxycytidine (5-AZA) leads to suppressed
prostate tumorigenesis. J Biol Chem. 287:39954–39966. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
McCabe MT, Low JA, Daignault S, Imperiale
MJ, Wojno KJ and Day ML: Inhibition of DNA methyltransferase
activity prevents tumorigenesis in a mouse model of prostate
cancer. Cancer Res. 66:385–392. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zorn CS, Wojno KJ, McCabe MT, Kuefer R,
Gschwend JE and Day ML: 5-aza-2′-deoxycytidine delays
androgen-independent disease and improves survival in the
transgenic adenocarcinoma of the mouse prostate mouse model of
prostate cancer. Clin Cancer Res. 13:2136–2143. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gravina GL, Marampon F, Di Staso M,
Bonfili P, Vitturini A, Jannini EA, Pestell RG, Tombolini V and
Festuccia C: 5-Azacitidine restores and amplifies the bicalutamide
response on preclinical models of androgen receptor expressing or
deficient prostate tumors. Prostate. 70:1166–1178. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao L and Alumkal J: Epigenetic regulation
of androgen receptor signaling in prostate cancer. Epigenetics.
5:100–104. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fialova B, Smesny Trtkova K, Paskova L,
Langova K and Kolar Z: Effect of histone deacetylase and DNA
methyltransferase inhibitors on the expression of the androgen
receptor gene in androgen-independent prostate cancer cell lines.
Oncol Rep. 29:2039–2045. 2013.PubMed/NCBI
|
|
18
|
Trtkova K, Paskova L, Matijescukova N,
Strnad M and Kolar Z: Binding of AR to SMRT/N-CoR complex and its
co-operation with PSA promoter in prostate cancer cells treated
with natural histone deacetylase inhibitor NaB. Neoplasma.
57:406–414. 2010. View Article : Google Scholar
|
|
19
|
Walton TJ, Li G, Seth R, McArdle SE,
Bishop MC and Rees RC: DNA demethylation and histone deacetylation
inhibition co-operate to re-express estrogen receptor beta and
induce apoptosis in prostate cancer cell-lines. Prostate.
68:210–222. 2008. View Article : Google Scholar
|
|
20
|
Jarrard DF, Kinoshita H, Shi Y, Sandefur
C, Hoff D, Meisner LF, Chang C, Herman JG, Isaacs WB and Nassif N:
Methylation of the androgen receptor promoter CpG island is
associated with loss of androgen receptor expression in prostate
cancer cells. Cancer Res. 58:5310–5314. 1998.PubMed/NCBI
|
|
21
|
Paskova L, Smesny Trtkova K, Fialova B,
Benedikova A, Langova K and Kolar Z: Different effect of sodium
butyrate on cancer and normal prostate cells. Toxicol In Vitro.
27:1489–1495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim J, Park H, Im JY, Choi WS and Kim HS:
Sodium butyrate regulates androgen receptor expression and cell
cycle arrest in human prostate cancer cells. Anticancer Res.
27A:3285–3292. 2007.
|
|
23
|
Pajak B, Orzechowski A and Gajkowska B:
Molecular basis of sodium butyrate-dependent proapoptotic activity
in cancer cells. Adv Med Sci. 52:83–88. 2007.
|
|
24
|
Qiu J, Gao Z and Shima H: Growth of human
prostate cancer cells is significantly suppressed in vitro with
sodium butyrate through apoptosis. Oncol Rep. 27:160–167. 2012.
|
|
25
|
Mu D, Gao Z, Guo H, Zhou G and Sun B:
Sodium butyrate induces growth inhibition and apoptosis in human
prostate cancer DU145 cells by up-regulation of the expression of
Annexin A1. PLoS One. 8:e749222013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Patra A, Deb M, Dahiya R and Patra SK:
5-Aza-2′-deoxycytidine stress response and apoptosis in prostate
cancer. Clin Epigenetics. 2:339–348. 2011. View Article : Google Scholar
|
|
27
|
Zhang J and Zhong Q: Histone deacetylase
inhibitors and cell death. Cell Mol Life Sci. 71:3885–3901. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J,
Chen Z, Beroukhim R, Wang H, Lupien M, et al: Androgen receptor
regulates a distinct transcription program in androgen-independent
prostate cancer. Cell. 138:245–256. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Heemers HV and Tindall DJ: Androgen
receptor (AR) coregulators: A diversity of functions converging on
and regulating the AR transcriptional complex. Endocr Rev.
28:778–808. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Heinlein CA and Chang C: Androgen receptor
in prostate cancer. Endocr Rev. 25:276–308. 2004. View Article : Google Scholar : PubMed/NCBI
|